

The involvement of Zn2+ ions in the structure of ubiquitous protein zinc fingers gives this metal a particular position in the biology of the cell. Zinc has long been known as a trace element crucial for the proper folding and functioning of numerous proteins in the cell, including enzymes, signal transduction proteins, and transcription factors. However, deeper insights into the exact mechanistic role of zinc in different cellular processes, especially the physiological role of free-Zn2+ ions present in the cell, were limited, while the key elements of the cellular system regulating Zn2+ trafficking and storage remained unidentified. The discovery of the zinc binding metallothioneins (43) and the subsequent identification of the first zinc transporters in the mid-1990s (53) constituted milestones in this field, leading to a much better understanding of zinc biochemistry. Nevertheless, because of extremely low concentrations of unbound free Zn2+ in the cell, progress was long hampered by the lack of highly sensitive and specific Zn2+ indicators. Admittedly, it has been known that apart from their role in maintaining the proper zinc finger structure, free-zinc ions somehow affect a variety of signaling pathways, but only very recently has it been demonstrated that free-Zn2+ ions can serve not only as modulators of signal transduction but also as classical cellular second messengers (33, 73).
Uncovering these molecular details of zinc homeostasis in the cell has unexpectedly opened new avenues in the field of virology, shedding new light on host-virus interactions. It has long been recognized that Zn2+ is an important cofactor not only of cellular proteins but of many viral proteins as well. Recent studies demonstrated that the cellular environment itself, with its extremely small and tightly controlled pool of free zinc, may represent a limiting factor. Viruses rely on the intracellular store of zinc ions and use cellular Zn2+ for their newly synthesized proteins. Consequently, the cellular systems controlling zinc balance might constitute a natural protective barrier that limits the accessibility of zinc and thus interferes with virus replication. Indeed, disruption of this barrier, an event that naturally takes place in patients suffering from a rare genetic disease, epidermodysplasia verruciformis (EV), leads to a zinc imbalance in the cell (37) and confers an unusual sensitivity to infections by cutaneous human papillomaviruses (EV-HPVs) (51). Intriguingly, this protective barrier appears to constitute an evolutionarily conserved cellular target for viral proteins, and some viruses have developed an active mechanism that allows them to invade the cellular system managing zinc fluxes in order to maintain the desired free-zinc concentration (37, 49). These findings define a previously unknown type of host-virus interaction, further stressing the importance of cellular Zn2+ ions to the virus. However, to what extent this mechanism represents a more general phenomenon in virology remains an open question.
Here, we first present the general basis of the significance of zinc to the virus, and thereafter we discuss the molecular mechanisms of this enigmatic interplay between viruses and the cellular systems that manage Zn2+ flux, with particular emphasis on this newly discovered type of host-virus interaction.
ZINC HOMEOSTASIS IN THE CELL
Two distinct, although exchangeable, pools of intracellular zinc can be defined: (i) protein-bound zinc and (ii) the unbound or only loosely bound Zn2+, so-called “free” zinc. Due to the toxicity of zinc to the cell, the vast majority of intracellular zinc belongs to the first pool, and only an extremely small amount of zinc is present as free-zinc ions. The amount of Zn2+ might be as much as 103 atoms per cell, although the exact concentration and subcellular zinc distribution are still controversial (12). The level of free Zn2+ in the cell is tightly controlled, being maintained at a relatively stable level by two complementary mechanisms (Fig. 1). First, an overload of free zinc is rapidly buffered by binding to cellular proteins, in particular to metallothioneins. Metallothioneins constitute a family of cysteine-rich, low-molecular-weight metalloproteins. The main physiological role of metallothioneins in the cell still remains elusive, although it has been demonstrated that metallothioneins protect the cell from metal-induced toxicity. Their expression is induced by different metals, including zinc, and interestingly, they might also be induced in response to viral infections (i.e., coxsackievirus type B and measles virus) (31, 77). The second mechanism involves zinc transporters belonging mainly to two families: ZIP (SLC39) and CDF/ZnT (SLC30) (12, 40, 62). ZIP family members are the proteins that transport zinc from the extracellular space or organellar lumen into the cytoplasm. On the other hand, ZnT family transporters not only exchange Zn2+ between different cellular compartments, modulating its local concentration, but also mediate zinc efflux, decreasing the total intracellular zinc content. In addition, the increased intracellular Zn2+ level stimulates the expression of zinc transporters through the activation of a zinc-inducible transcription factor named metal-responsive transcription factor 1 (MTF-1) (52, 55), imposing a self-regulating feedback loop. Collectively, metallothioneins and zinc transporters, together with sensing elements like MTF-1, control Zn2+ trafficking and storage, maintaining cellular zinc balance. Such a balance is crucial for proper cell functioning, and even small alterations in the free-zinc concentration exert a pleiotropic influence on a variety of cellular processes (i.e., cell signaling and proliferation), while excess zinc is toxic to the cell and induces apoptosis (63).
FIG. 1.

The system that manages cellular zinc homeostasis. Cellular Zn2+ uptake is conferred by ZIP transporters (shown in green), although L-type calcium channels (LTCC) might participate in this process. An excess of unbound cytoplasmic Zn2+ is transported outside the cell by ZnT/CDF family members (shown in red), mainly ZnT-1. However, the influence of ZnT-1 on Zn2+ transport may also result from indirect suppression of Zn2+ influx through L-type calcium channels. The exact role of other ZnTs (ZnT-4 and ZnT-5) and the Na+/Zn2+ exchanger in Zn2+ efflux is not yet fully clarified and might differ, depending on cellular background. It is noteworthy that the splice variant ZnT-5b (*) constitutes the only known exception, and in contrast to other ZnT members, it might also transport Zn2+ into the cytoplasm. The cytoplasmic Zn2+ is buffered by binding to cellular proteins, metallothioneins, in particular, or is redistributed among organelles. So far, several proteins transporting Zn2+ into the Golgi apparatus have been identified (ZnT-4, ZnT-5, ZnT-6, and ZnT-7). The transfer of Zn2+ into the ER in mammalian cells is much less clarified, although the EVER/ZnT-1 complex might be involved in this process. Furthermore, a mutual exchange of Zn2+ between the ER and Golgi apparatus through antero- and retrograde vesicular transport has been postulated. Zn2+ is transported into the “vesicular compartment” by different sets of ZnT proteins, depending on the tissue context. Zn2+ possibly crosses the outer membrane of mitochondria through porin channels; subsequently, it might be bound to the metallothioneins in the intermembrane space and/or further transported to the mitochondrial matrix by not-yet-defined proteins. Zn2+ is transported in the reverse direction, from organelles to the cytosol, by ZIP family members, and ZIP1, ZIP7 (ER and Golgi apparatus), and ZIP8 (vesicles) have been implicated in this process. The arrows represent the directions of Zn2+ transport (the dashed arrows correspond to the hypothetical routes). The names of nonmammalian ER-residing transporters have been italicized. The “vesicular compartment” comprises different types of cellular vesicles (endosomes, lysosomes, synaptic vesicles, and secretory vesicles) without further distinction, depending on the tissue context.
The significance of precise control of the Zn2+ level is further strengthened by recent observations that like Ca2+ or cyclic AMP, Zn2+ itself serves as a classical intracellular second messenger (73). It has been demonstrated that the activation of cell surface receptor FcɛRI leads to the rapid release of Zn2+ ions from the internal stores, most likely located in the endoplasmic reticulum (ER), in a calcium influx- and mitogen-activated protein kinase-dependent manner (73). This, in turn, changes the expression of cellular genes and modifies the duration and strength of signaling, possibly by suppressing phosphatase activity. However, in what form zinc is stored in the ER and the molecular mechanism of its release from the ER into the cytoplasm as free ions remain unknown.
VIRUS LIFE CYCLE AND ZINC
There is a growing body of evidence that zinc is important for the invasion of viruses into the cell. Nevertheless, the molecular basis of this interplay between viruses and cellular zinc is still largely unknown. By default, at least two possible, mutually nonexclusive mechanisms exist. First, Zn2+ is a well-known structural cofactor of viral proteins, and second, zinc ions, participating in signal transduction in the cell, could change the activities of different transcription factors and thus the expression patterns of cellular and viral genes.
ZINC AS A STRUCTURAL COFACTOR OF VIRAL PROTEINS
The role of zinc as a protein cofactor is certainly a quite common phenomenon among viruses. Zinc binding proteins have been described for a great variety of both RNA and DNA viruses, such as retroviruses (4), rotaviruses (7, 14), adenoviruses (8), herpesviruses (17), polyomaviruses (24, 70), and papillomaviruses (1, 26), to mention only a few better-studied examples. These viral proteins comprise zinc binding motifs that resemble zinc finger domains found in cellular proteins. Zinc fingers are small, independently folded domains that bind at least one zinc ion. In eukaryotic cells, the most abundant are the proteins with a classical Cys2His2 (CCHH) zinc finger motif (36), whereas viral zinc fingers often comprise distinct motifs, different from the classical motif (see below). Both viral and cellular zinc fingers can participate in protein-nucleic acid and protein-protein interactions, and these motifs are usually highly conserved, being critical for protein function.
In discussing the possible interactions between zinc and viral proteins, we focus on the human immunodeficiency virus (HIV), which is one of the best-studied viruses in this context and could serve as a general example. HIV belongs to the family Retroviridae and comprises three essential genes common to all retroviruses (gag, pol, and env) and several accessory genes present only in the genomes of some of the Retroviridae (e.g., tat, rev, nef, vpr, vpu, vif, and vpx), which are thought to adapt the virus to the host cell. Some of the proteins encoded by these genes contain cysteine-rich motifs constituting a putative zinc finger. The role of zinc finger domains has been the most extensively studied role of the structural proteins Gag precursor protein and its derivative, nucleocapsid protein.
The precursor Gag protein encoded by the gag gene is involved in binding and packaging viral genomic RNA. Thereafter, the Gag precursor is cleaved by viral protease to yield structural proteins whose numbers differ, depending on the class of retrovirus. At least three structural proteins are encoded by all of them: the matrix protein (MA), capsid protein (CA), and nucleocapsid protein (NC). The HIV NC protein contains two copies of the highly conserved zinc binding motifs, Cys-X2-Cys-X4-His-X4-Cys (CCHC), referred to as “anisotropic” zinc fingers to distinguish them from the canonical eukaryotic zinc fingers with the symmetrical CCHH motif (4). The Gag precursor binds Zn2+ ions, and this is necessary for the recognition and encapsidation of viral RNA (4, 25). Moreover, this zinc binding motif seems to be critical for the appropriate cellular trafficking of Gag, and a mutation in the zinc finger excludes the protein from the endosomal compartment, leading to the accumulation of Gag in the cell and interfering with the release of infectious virus (25).
The role of the Zn2+ ions bound to the mature NC protein is less clear (4, 67). This issue has raised a large amount of interest, as NC is a crucial protein for both the early and late stages of the virus life cycle and the NC zinc binding motif is mutationally intolerant, suggesting that it might serve as an interesting therapeutical target. Intriguingly, deleting the zinc binding motif in NC results in premature viral DNA synthesis and the generation of noninfectious DNA-containing viral particles (29). Therefore, removal of zinc from the NC zinc finger structure could represent an interesting antiviral strategy, interfering simultaneously with the Gag precursor and mature NC protein function. On top of that, as only very few eukaryotic proteins contain this type of zinc finger, removal of Zn2+ from the NC protein by compounds specific for the “anisotropic” finger might constitute a much more selective therapeutical approach than simple zinc chelation. Indeed, it has been demonstrated that several C-nitroso compounds can eject Zn2+ from NC zinc fingers without affecting significant cellular zinc binding proteins (58, 59). Interestingly, a similar strategy has been successfully employed to eject zinc from the papillomavirus major oncoprotein E6 (3).
Distinct types of zinc binding domains have been found in several nonstructural HIV proteins—integrase and accessory proteins such as Tat or Vif proteins. Apart from the involvement of viral zinc fingers in interactions with nucleic acids, the zinc binding motifs of integrase, Tat, and Vif are implicated in protein-protein binding. The association of zinc ions with their zinc finger-like structures confers at least three types of interactions: (i) Zn2+ allows maintenance of the conformation required for the self-aggregation of viral protein and thus might participate in viral protein multimerization; (ii) the zinc ion could be involved in the generation of metal-linked dimers, binding simultaneously to two distinct molecules of the same protein, thus serving as a sort of bridge; and (iii) zinc might be a critical factor for the interactions between viral proteins and their cellular partners.
A representative of the first type of zinc-dependent protein-protein interactions could, for instance, be retroviral integrase. HIV integrase comprises three distinct domains, the N-terminal, C-terminal, and central core domain. The N-terminal domain contains an invariant zinc finger motif (HHCC), structurally different from both the zinc binding motif of the NC protein and the canonical eukaryotic zinc fingers. The binding of zinc by this domain has been shown to stabilize the structure of the enzyme and to facilitate its multimerization. As the active form of integrase is a multimer, these zinc-mediated interactions also enhance its catalytic activity (38, 39, 46). In contrast, Tat protein might serve as an example of the second and third types of Zn2+-mediated interactions. Tat protein comprises a cysteine-rich region that was shown to bind two Zn2+ per one monomer. The unique feature of this structure is that metal serves as a linker between monomers, and binding to two Tat proteins, zinc ions cross-link them (18, 19, 30, 47). A similar mode of Zn2+-mediated cross-linking has recently been proposed for Vif protein (41, 54).
On the other hand, the association of zinc with viral proteins might lead to conformational changes and consequently to the exposure of domains involved in interactions with their cellular partners. In fact, in the cases of both Tat and Vif, zinc was shown to be important for binding by their cellular partners, cyclin T1 (21) and cullin 5 (41, 54), respectively. It has been demonstrated that the binding of Zn2+ by Vif protein changes its conformation, leading to its folding predominantly into a β-sheet, which allows it to interact with cullin 5. This association with cullin 5 is crucial for Vif-mediated hijacking of cellular proteasome machinery and the subsequent degradation of the cellular proteins with antiviral activity, in this case APOBEC3G. In the absence of Vif, this host cell cytidine deaminase is incorporated into the virion and targets neosynthesized viral cDNA. Massive deamination leads to hypermutation and finally to degradation, consequently reducing virus infectivity (23). It has been demonstrated that both the chelation of zinc and the deletion of this zinc finger-coding sequence in Vif protein suppress its activity and significantly reduces virus infectivity (41, 54, 72).
ZINC, CELL SIGNALING, AND THE VIRUS
The role of zinc signaling in the virus life cycle has been much less studied. Clearly, zinc ions change the global phosphorylation of cellular proteins, which obviously might affect the virus life cycle. It has been proposed that phosphatases (PTPs) constitute a cellular target for zinc. Almost all of the cellular PTPs contain a cysteine residue in the active site, which exists in a thiolate form. Zn2+ can react with these PTP cysteines and thus inhibit their function (2, 27). Moreover, an excess of zinc ions can induce the generation of radical oxygen species (20), which also react with PTP cysteines, suppressing their activity (2). In addition, radical oxygen species themselves may release zinc from proteins, in particular from metallothioneins, further increasing the pool of free-zinc ions. Thus, through an increase in the phosphorylation of cellular proteins, zinc might activate some signaling pathways, and in turn, it could alter the activities of transcription factors, changing the expression of target genes relevant to the virus.
Indeed, zinc was shown to activate, among others, the phosphoinositide 3-kinase/Akt and extracellular signal-regulated kinase/Jun N-terminal protein kinase (JNK) (2, 13, 32, 63) pathways, which could presumably affect the life cycles of numerous viruses. For instance, the zinc-mediated activation of JNK, a cellular kinase involved in the phosphorylation and activation of c-Jun, a constituent of AP-1 transcription factors, stimulates AP-1 transcriptional activity (32; our unpublished data). This might have an impact on the virus in at least two different ways. First, directly, as AP-1 is a cellular transcription factor, which is important, or even critical in some instances, as in the case of papillomaviruses, for the expression of viral genes (35, 75). On the other hand, through the modulation of AP-1 activity, free zinc changes the expression levels of multiple cellular genes, among which are those relevant to the virus, e.g., constituents of the natural antiviral response (for example, cytokines).
However, it needs to be emphasized that many of these studies on zinc signaling have been performed under conditions in which cells were exposed to supraphysiological concentrations of zinc and could reflect a physiological situation to only a limited extent. Nevertheless, recent investigations demonstrated that Zn2+ also inhibits phosphatase activity in the range of physiological concentrations (73). It has been shown that physiological oscillations of the intracellular pool of free zinc affect protein phosphorylation and alter the expression levels of cellular genes. A burning question is to what extent these physiological oscillations of free zinc in the cell, within the range of very low concentrations, could be important for the virus.
FREE-ZN2+ CONCENTRATIONS AND THE OPTIMAL ENVIRONMENT FOR THE VIRUS
Even though it seems obvious that deviations in zinc concentrations from the given, optimal level desired by the virus could exert an effect on its life cycle, a distinct issue is whether the physiological concentrations of cellular free Zn2+ fit the demands of the virus. This is still a matter of debate, and there are a few fundamental questions related to this issue. (i) What is the viral protein requirement for Zn2+ as a cofactor, and is the physiological concentration of free zinc in the infected cell optimal for the function of these proteins? (ii) How are Zn2+ ions delivered to the newly synthesized viral proteins? (iii) How does the cellular system controlling zinc homeostasis in the cell respond to the increased global demands for Zn2+ ions in the infected cell? (iv) What might be the consequences of such a virus-induced deficit of free zinc on cell function and the virus life cycle? (v) Is the steady-state level of cellular free zinc optimal for the virus in terms of the activities of cellular transcription factors, or is an increased free-Zn2+ pool needed for efficient viral gene expression and replication? (vi) Finally, could viruses actively regulate the cellular systems controlling zinc homeostasis, changing the “settings” of the system in order to adjust it to the optimal level for the virus's demands?
Even though many of these questions remain unanswered, recent studies have provided some breakthroughs. As a relative deficit of free zinc could negatively affect the structure and function of viral proteins, one might speculate that maintaining a higher concentration of free zinc in the cell could generally constitute an advantage for the virus. It seems that at least for some viruses, the physiological level of free zinc is suboptimal and that changing the zinc balance is beneficial for the virus. Such a zinc imbalance could be conferred by (i) an inherited defect in the cellular systems managing zinc fluxes or (ii) by some viral proteins taking control over Zn2+ trafficking in the cell in order to keep a higher concentration of free zinc. In fact, recent studies suggest that both situations take place. Intriguingly, it has recently been found that patients suffering from EV (OMIM accession number 226400), a rare genodermatosis characterized by unusual sensitivity to papillomavirus infections, display a dysfunction in one of the zinc-transporting complexes. On the other hand, we have found that both the E6 and E7 proteins of HPV are able to interact with metallothioneins (our unpublished data), presumably to release free-zinc ions. In this context, metallothioneins could serve as the proteins that deliver Zn2+ directly to these viral proteins (or simply release Zn2+ upon viral protein binding, increasing the local concentration of zinc ions in the closest neighborhood of E6 and E7), thus ensuring their proper conformation and function. There is also speculation that the interaction between viral proteins and metallothioneins favors the release of free zinc, which activates signaling pathways (13), leading to the synthesis of the transcription factors needed for viral gene expression.
EV AS A NATURAL MODEL OF ZINC DYSREGULATION IN HPV INFECTIONS
EV is a rare human skin disease (42, 50, 51) in which the sole identified primary defect is an abnormal, highly selective susceptibility to a specific group of ubiquitous cutaneous HPVs, the so-called EV-HPVs, but not to other closely related HPVs, including genital HPVs (15, 51). HPVs are small, nonenveloped DNA viruses which are classified into five different phylogenetic genera (10). Genital HPVs are classified into the Alphapapillomavirus genus, whereas EV-HPVs belong to the distinct Betapapillomavirus genus. The HPV genome is circular and contains open reading frames encoding late proteins (L1 and L2), which are structural capsid proteins, and early proteins (E1, E2, E4, E5, E6, and E7), which coordinate replication and expression of the viral genome and are involved in the adaptation of the functions of the cell to the requirements of the virus (15). Papillomaviruses infect the keratinocytes of humans and a wide range of animal species and induce a large variety of lesions, from benign skin warts to invasive cancers (15, 51, 61). Clinical HPV genital infections are the most common sexually transmitted diseases in the world. The mechanisms of HPV-induced malignant transformation have been extensively studied, and two major viral oncoproteins, E6 and E7, which are zinc binding proteins (1, 26), have been identified and characterized in detail (78).
In contrast to genital HPVs, EV-HPVs, although ubiquitous, are usually harmless in the general population, causing widespread, asymptomatic infections. However, in patients suffering from EV, oncogenic HPV5 and HPV8 replicate very efficiently and reveal their full transforming potential, inducing multiple nonmelanoma skin cancers (51), which strongly suggests the existence of a natural antiviral barrier in the host cells. The majority of EV cases are caused by mutations in one of the two adjacent genes EVER1 or EVER2 (51, 56, 57). The EVER1 and EVER2 genes are evolutionarily conserved and encode proteins belonging to the transmembrane channel-like (TMC) family (34). Among the eight TMC genes, EVER1 and EVER2 correspond to TMC6 and TMC8, respectively. In keratinocytes, EVER proteins are located mainly in the ER (37, 57), and it has been speculated that like other TMC members, these two proteins may play a role in ion transport or the modulation of signal transduction.
PHYSIOLOGICAL ROLE OF EVER PROTEINS
We have recently reported that in human keratinocytes, EVER proteins form a complex in the ER with ZnT-1 and participate in maintaining cellular zinc homeostasis. The ZnT-1/EVER complex is involved in the regulation of Zn2+ trafficking in the cell and seems to control Zn2+ influx into the nucleus (37). Since the molecular mechanism of Zn2+ transfer across the nuclear membrane is itself poorly understood, the mode of action of EVER proteins remains speculative. It has been demonstrated that the ER might constitute an important organelle where zinc is stored and from where, upon the triggering of the cell surface receptor, a reservoir of Zn2+ can be rapidly released into the cytoplasm and subsequently passed to the nucleus, in what has been designated a “zinc wave” (73). Most likely, the ER-residing ZnT-1/EVER complex could be involved in the transfer of Zn2+ in the reverse direction, from the cytoplasm into the ER lumen, interfering with the spreading of the zinc wave and the function of Zn2+ as a second messenger. Thus, by decreasing the free-zinc concentration in the cytoplasm, the ZnT-1/EVER complex might indirectly alter Zn2+ transport from the cytoplasm to the nucleus. The exact role of EVER proteins within the ZnT-1/EVER complex remains to be determined, and EVERs might either (i) only modulate the activity of ZnT-1, a known zinc transporter, or (ii) serve as Zn2+ transporters themselves, based on the homology of the structure with those of other TMC members.
Regardless of the exact mechanism, a mutation in one of the EVER genes and the lack of the corresponding protein disrupt the function of the whole ZnT-1/EVER complex. When human keratinocytes are loaded with a fluorescent indicator specific for free zinc, a bright fluorescence appears in the nucleoli and is further augmented when the cells are exposed to a high concentration of zinc (37) (Fig. 2). However, it has been shown that the fluorescence intensity of the nucleoli is significantly higher in EVER2−/− cells than in EVER2+/+ cells, demonstrating an abnormal distribution of intracellular free zinc in EVER-deficient cells that could be interpreted as a retention of zinc within the nucleus (37). Notably, this redistribution of the free-zinc pool is accompanied by enhanced transcriptional activity of MTF-1 (37). Interestingly, apart from an effect on the transcription factor that is directly zinc inducible, ZnT-1 and EVER proteins have been shown to suppress the activity of c-Jun, an important constituent of the AP-1 transcription factor. This effect might be mediated by the inhibition of the zinc-induced activation of JNK (13, 32) but does not necessarily involve glycogen synthase kinase 3β (37; our unpublished data), another c-Jun kinase that in contrast to JNK, suppresses the activity of c-Jun upon phosphorylation (5).
FIG. 2.
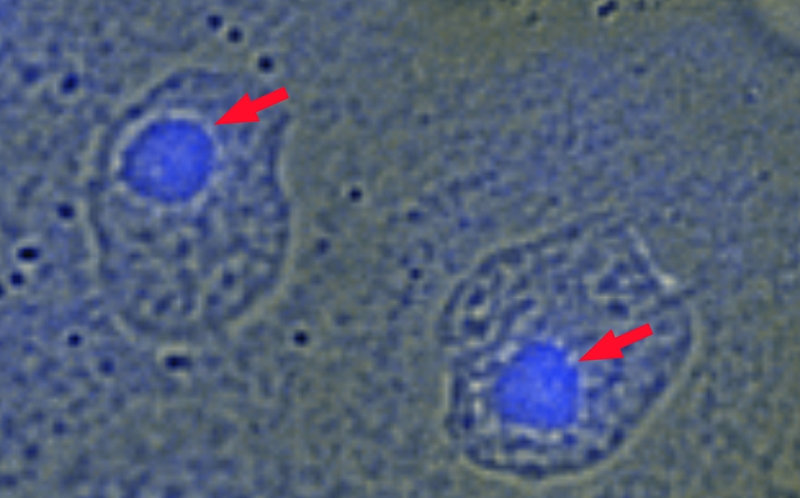
Free-zinc ions in keratinocytes. Keratinocytes with a mutated EVER gene (EVER2−/−) were loaded with Zinquin, a fluorescent indicator specific for free zinc; subsequently, the cells were incubated for 15 min in 100 μM zinc. A strong fluorescence, corresponding to places with a high concentration of free zinc, is visible mainly in the nucleoli (indicated by red arrows).
It needs to be emphasized that the influence of the EVER/ZnT-1 complex on the status of c-Jun phosphorylation, probably through changes in the level of Zn2+, certainly reflects much broader alterations in cell signaling and transcription factor activity (37; our unpublished data). Interestingly, even a transient increase in the intracellular concentration of Zn2+, which remains within the physiological range, confers a general suppression of cellular phosphatase activity and increases the phosphorylation of multiple cellular proteins (27, 73). Thus, cellular phosphatases constitute a presumptive target for the ZnT-1/EVER complex, and their disinhibition in response to the downregulation of free zinc by the ZnT-1/EVER complex might in turn globally affect cell signaling and modulate the activity of numerous transcription factors.
ROLE OF EVER PROTEINS IN INFECTED KERATINOCYTES
Although widely distributed in the general population, EV-HPVs are usually detected solely by a highly sensitive nested-PCR approach; in EV patients, the number of copies of viral DNA in keratinocytes is much higher. Thus, the existence of a natural protective mechanism in the host cells has to be assumed. Presumably, in EV patients this selective barrier might be disrupted or compromised as a consequence of EVER mutations (56, 57), imposing a host restriction on EV-HPV. These mutations that lead to an increase in cellular free zinc activate JNK-related pathways and induce the transcriptional activity of AP-1 (13, 32, 37). This might constitute an important promoting factor for the HPV life cycle, since AP-1 transcription factors are essential for the expression of the HPV genome (35, 75) and, notably, for virus-induced carcinogenesis (75), at least as far as alphapapillomaviruses are concerned. Thus, a massive expression of viral genes and efficient virus replication are induced in EVER-deficient keratinocytes, eventually conferring malignant transformation. However, a distinct HPV-independent mechanism of skin carcinogenesis in EV patients cannot be completely ruled out. It has previously been shown that forced overexpression of AP-1 itself can increase the proliferation rate and participate in cell transformation. In accordance with these findings, we have observed that the EVER2 mutation is also associated with the enhanced growth potential of keratinocytes in the absence of HPV (37).
Despite the recent advances in understanding of the role of EVER proteins in cell physiology, the intriguing selectivity of the EVER-based molecular barrier and the lack of protection against genital HPVs remain a mystery. Two possible scenarios can be proposed: (i) the EVER-based barrier could indeed be highly selective toward betapapillomaviruses (β-HPV), blocking some cellular processes that are crucial solely for these papillomaviruses, not for genital HPVs; and (ii) although the barrier controls processes equally important for the life cycles of both alpha- and β-HPVs, genital HPVs might have developed an evolutionary mechanism that enables them to bypass the EVER-based barrier. Recent observations seem to favor the latter possibility (37, 49). As the genomes of genital HPVs contain open reading frame E5, in contrast to those of EV-HPVs, the E5 protein emerged as the first presumable permissive factor that might allow genital HPVs to break the EVER barrier.
HPV AS AN ACTIVE ELEMENT IN THE MANAGEMENT OF CELLULAR ZINC FLUXES
HPV E5 is a highly hydrophobic transmembrane protein, localized in the cell predominantly in the ER, the Golgi apparatus, and nuclear membranes (69). The exact molecular mechanism of HPV E5 action is not fully elucidated. Initially, it was noted that the bovine papillomavirus (BPV) E5 protein can augment the transforming activity of the epithelial growth factor receptor (EGFR) (44), and thereafter it was postulated that HPV E5 might act similarly. Indeed, HPV type 16 (HPV16) E5 was shown to enhance epithelial growth factor (EGF)-triggered signaling, possibly due to the E5-mediated inhibition of EGFR cellular recycling and, in consequence, to the retention of EGFR at the cellular membrane (66, 69). Similarly to BPV E5, the HPV16 E5 protein binds to the 16-kDa subunit of the proton pump [(H+)-ATPase] (6), thereby possibly inhibiting vesicular acidification and preventing EGFR degradation. However, the binding of HPV E5 to (H+)-ATPase seems to be insufficient to confer HPV E5-mediated enhancement of EGF signaling (60), and HPV E5 can directly block the proteasomal degradation of EGFR by inhibiting the interaction between EGFR and ubiquitin ligase (76). Nevertheless, even though the possibility of a link between HPV E5 and EGF-related signaling has been raised by multiple studies from different laboratories and is generally well accepted, it must be emphasized that HPV E5 displays little or no homology with BPV E5 and that some of its biological effects are clearly EGFR independent (16).
A new insight into the molecular mechanism of action of the HPV E5 protein came from recent studies on the function of the EVER and ZnT-1 proteins. First, it was reported that HPV E5 protein interacts with ZnT-1, suggesting a possible involvement of HPV E5 in the disruption of the system controlling cellular zinc homeostasis (37, 49). Interestingly, a similar interaction with ZnT-1 was found in the case of cottontail rabbit papillomavirus E8 protein (49), a transmembrane oncoprotein structurally and functionally related to E5 (28). These findings further suggest that zinc homeostasis in the cell might represent a common cellular target for different papillomaviruses. Subsequently, we have shown that HPV E5 interacts with EVER proteins and colocalizes with the EVER/ZnT-1 complex in keratinocytes (37). More importantly, HPV16 E5 has been shown to influence the cellular system controlling zinc balance, leading to an increase in the concentration of intracellular free Zn2+ in keratinocytes, as measured by the activity of the zinc-inducible transcription factor MTF-1. In addition, HPV E5 interfered with EVER2-mediated inhibition of EGF-induced c-Jun activity in human keratinocytes (37). Thus, HPV E5 exerts an influence strikingly similar to that of EVER deficiency, functionally mimicking the effect of EVER mutations in EV patients. As zinc inhibits cellular phosphatases and was shown to increase the phosphorylation of EGFR as well (68), the influence of HPV E5 on the cellular system controlling zinc homeostasis might integrate the previously published results. On the one hand, it could explain the well-documented effect of HPV E5 on EGF-triggered signaling, and on the other hand, as changes in free zinc are likely to exert a rather global effect in the cell, it could also explain some of the EGFR-independent HPV E5 activities.
Although HPV E5 protein is not an indispensable factor for the virus, it does ensure proliferative competence in more-differentiated cells, and it augments viral genome amplification and regulates the expression of other viral genes (16, 22). Therefore, E5 is thought to be involved in the coordination of the HPV life cycle and is important mainly during the productive stages (11). We propose that EVER deficiency could play a similar role for EV-HPVs. Indeed, it has been demonstrated that a mutation in EVER2 in keratinocytes compensates for the lack of the functional E5 protein in β-HPVs, explaining the host restriction in the case of EV (37). On the other hand, the significance of the interaction between E5 protein and the EVER complex and modifications of cellular zinc balance could have some important implications not only for the virus but also for the host cell itself. Although HPV E5 does not display full transforming capacity, it can potentiate the oncogenic activity of E6/E7 proteins (65), which even though constituting more of a “side effect” for the virus, is important to fully understand the pathology of HPV-related cancers. Using a transgenic murine model, it has been demonstrated that although the HPV E5 protein is not a major factor involved in the initiation step of papilloma formation, it does contribute to the promotion and progression stages of carcinogenesis (45). Moreover, in this model, HPV E5 conferred an endophytic and highly penetrant type of cancer growth. It is tempting to speculate that this biological effect of HPV E5 action could be related to its influence on EVER/ZnT-1 function and in turn on cellular zinc homeostasis. Remarkably, a similar, highly penetrating and invasive mode of tumor growth with a propensity to metastasize in many cases has been observed in EV patients (9). We hypothesize that breaking the EVER barrier, regardless of whether it is due to a mutation in either of the EVER genes or to the effect of E5, could constitute a tumor-promoting factor, which changes the natural evolution of the developing cancer.
All these basic findings point to the deregulation of the cellular system controlling zinc homeostasis as a crucial step in the HPV life cycle, both for alphapapillomaviruses and β-HPVs (genital HPVs and EV-HPVs). However, two distinct molecular mechanisms could be involved: E5-mediated suppression of EVER/ZnT-1 complex activity for genital HPVs and an EVER mutation for β-HPVs. One might pose the question, why did EV-HPVs evolve to be devoid of an apparently important mechanism facilitating their replication? The high viral load found in some EV patients suggests that inhibition of the EVER complex might indeed be helpful for β-HPV genome amplification. This can be understood at least in the context of oncogenic EV-HPVs when one considers the clinical phenotype of EV patients. In contrast to the oncogenic genital HPVs like HPV16 or HPV18, in the case of which the majority of infections do not confer a clinically relevant malignancy, β-HPVs consistently lead to cancerous transformation, as only they infect EVER-deficient cells. Possibly, important qualitative differences between the alphapapillomaviruses and β-HPVs in the set of early genes other than E5 could explain this striking contrast observed in cells with disrupted EVER complexes. Thus, taking into account the whole repertoire of viral genes, the existence of a mechanism that disrupts zinc balance could dramatically shorten the life expectancy of the infected host and eventually might not necessarily constitute an advantage for EV-HPVs. Therefore, β-HPVs appear to be viruses that are remarkably well adapted to their hosts and do not need to deregulate the cellular zinc balance in order to efficiently spread in the human population. On the other hand, genital HPVs represent the first known examples of the virus, which evolved to actively control the cellular zinc balance, which could be beneficial for the virus life cycle and, in most cases, will not result in fatal consequences for the host.
CONCLUDING REMARKS
A major advance in the understanding of host-microbe interactions often originates at the frontiers of distinct fields. A better understanding of the molecular mechanisms controlling cellular zinc homeostasis has enabled more mechanistic studies on the functions of the proteins encoded by the EVER genes, shedding new light on the molecular foundation of EV. In turn, this important insight into the pathogenesis of EV, an extremely rare genetic disease, brought new clues to the biology of the widespread oncogenic papillomaviruses. Finally, the integration of all of these findings from seemingly distinct fields led to the discovery of a previously unknown type of host-virus interaction, showing that the mechanisms controlling cellular Zn2+ trafficking constitute natural antiviral barriers. This, together with the previously gathered broad knowledge of the role of Zn2+ as a cofactor of viral proteins, suggests that the accessibility of zinc ions in infected cells could be a potentially limiting factor in the virus life cycle.
It remains an intriguing question whether this type of interplay between viruses and cellular zinc might be a more-universal phenomenon, conserved among distinct groups of viruses. Interestingly, an increase in c-Jun phosphorylation and the constitutive activation of AP-1 have been reported as important viral strategies in the case of polyomaviruses, for example (64), and inhibition of AP-1 activity can interfere with the transforming potential of viral proteins (71). In this context, it is possible that the deregulation of cellular zinc homeostasis by the virus, which leads to persistent activation of AP-1, might simply constitute yet another evolutionarily selected viral strategy dedicated to maintaining high levels of activity of the desired transcription factors.
Moreover, the recent findings have some general implications for the specific field of papillomavirus-associated diseases, including EV itself. Specifically, the novel insight into the pathogenesis of HPV infections defines new directions in the search for future therapeutic strategies. One such strategy might be an attempt to increase EVER expression in order to prevent cellular zinc imbalance. Overexpression of ZnT-1 and EVER proteins has already been shown to suppress the activities of AP-1 and MTF-1 (37). More importantly, in transfection experiments, an EVER2 mutation leading to the lack of the functional EVER2 protein can be compensated by overexpression of the EVER1 or ZnT-1 protein (our unpublished data). All EV patients carry a wild-type copy of one of the EVER genes (either the EVER2 wild-type for EVER1−/− individuals or the EVER1 wild-type for EVER2−/− individuals) in their genomes, which implies that the “pharmacological” enhancement of expression of the remaining wild-type gene might in general constitute an interesting therapeutic approach for these patients. Conceptually similar approaches in which a missing protein is replaced by the overexpression of the gene encoding the other protein have already been proposed in different fields, for muscular dystrophies, for example (48). However, currently nothing is known about the physiological mechanisms controlling the expression of the EVER genes, and a detailed analysis on that subject is more than desirable. It would also be of interest to verify whether such an increased expression of EVER proteins could prevent the HPV E5-induced increase in cellular free zinc in infected keratinocytes, thus conferring a rather generally augmented protection against papillomaviruses. If so, it could open completely new avenues for preventive and/or therapeutic strategies in the field of genital HPVs as well. On the other hand, as we postulated that the clinical phenotype in EV patients could be causatively associated with the increased activity of JNK and c-Jun, pharmacological suppression of this pathway might also represent an attractive approach for these patients. The successful therapeutical employment of a JNK inhibitor (SP600125) in vivo has recently been reported (74).
Acknowledgments
The authors have no conflicting financial interests.
We thank Simon Wain-Hobson for critical reading of the manuscript.
This work was supported by grants from the Contrats de la Ligue Nationale contre le Cancer (R05/75-129 and RS07/75-75) and the Association de la Recherche contre le Cancer (3731XA0531F and 4867). M.L. was supported by postdoctoral fellowships from the Association pour la Recherche sur le Cancer (ARC) and the Foundation for Polish Science (FNP).
Footnotes
Published ahead of print on 10 September 2008.
REFERENCES
- 1.Barbosa, M. S., D. R. Lowy, and J. T. Schiller. 1989. Papillomavirus polypeptides E6 and E7 are zinc-binding proteins. J. Virol. 631404-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barthel, A., E. A. Ostrakhovitch, P. L. Walter, A. Kampkötter, and L. O. Klotz. 2007. Stimulation of phosphoinositide 3-kinase/Akt signaling by copper and zinc ions: mechanisms and consequences. Arch. Biochem. Biophys. 463175-182. [DOI] [PubMed] [Google Scholar]
- 3.Beerheide, W., H. U. Bernard, Y. J. Tan, A. Ganesan, W. G. Rice, and A. E. Ting. 1999. Potential drugs against cervical cancer: zinc-ejecting inhibitors of the human papillomavirus type 16 E6 oncoprotein. J. Natl. Cancer Inst. 911211-1220. [DOI] [PubMed] [Google Scholar]
- 4.Bess, J. W., P. J. Powell, H. J. Issaq, L. J. Schumack, M. K. Grimes, L. E. Henderson, and L. O. Arthur. 1992. Tightly bound zinc in human immunodeficiency virus type 1, human T-cell leukemia virus type I, and other retroviruses. J. Virol. 66840-847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyle, W. J., T. Smeal, L. H. Defize, P. Angel, J. R. Woodgett, M. Karin, and T. Hunter. 1991. Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell 64573-584. [DOI] [PubMed] [Google Scholar]
- 6.Briggs, M. W., J. L. Adam, and D. J. McCance. 2001. The human papillomavirus type 16 E5 protein alters vacuolar H+-ATPase function and stability in Saccharomyces cerevisiae. Virology 280169-175. [DOI] [PubMed] [Google Scholar]
- 7.Brottier, P., P. Nandi, M. Bremont, and J. Cohen. 1992. Bovine rotavirus segment 5 protein expressed in the baculovirus system interacts with zinc and RNA. J. Gen. Virol. 731931-1938. [DOI] [PubMed] [Google Scholar]
- 8.Culp, J. S., L. C. Webster, D. J. Friedman, C. L. Smith, W. J. Huang, F. Y. Wu, M. Rosenberg, and R. P. Ricciardi. 1988. The 289-amino acid E1A protein of adenovirus binds zinc in a region that is important for trans-activation. Proc. Natl. Acad. Sci. USA 856450-6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Oliveira, W. R., C. Festa Neto, P. L. Rady, and S. K. Tyring. 2003. Clinical aspects of epidermodysplasia verruciformis. J. Eur. Acad. Dermatol. Venereol. 17394-398. [DOI] [PubMed] [Google Scholar]
- 10.de Villiers, E. M., C. Fauquet, T. R. Broker, H. U. Bernard, and H. zur Hausen. 2004. Classification of papillomaviruses. Virology 32417-27. [DOI] [PubMed] [Google Scholar]
- 11.Doorbar, J. 2006. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. (London) 110525-541. [DOI] [PubMed] [Google Scholar]
- 12.Eide, D. J. 2006. Zinc transporters and the cellular trafficking of zinc. Biochim. Biophys. Acta 1763711-722. [DOI] [PubMed] [Google Scholar]
- 13.Eom, S. J., E. Y. Kim, J. E. Lee, H. J. Kang, J. Shim, S. U. Kim, B. J. Gwag, and E. J. Choi. 2001. Zn2+ induces stimulation of the c-Jun N-terminal kinase signaling pathway through phosphoinositide 3-kinase. Mol. Pharmacol. 59981-986. [DOI] [PubMed] [Google Scholar]
- 14.Erk, I., J. C. Huet, M. Duarte, S. Duquerroy, F. Rey, J. Cohen, and J. Lepault. 2003. A zinc ion controls assembly and stability of the major capsid protein of rotavirus. J. Virol. 773595-3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Favre, M., N. Ramoz, and G. Orth. 1997. Human papillomaviruses: general features. Clin. Dermatol. 15181-198. [DOI] [PubMed] [Google Scholar]
- 16.Fehrmann, F., D. J. Klumpp, and L. A. Laimins. 2003. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J. Virol. 772819-2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraefel, C., J. Zeng, Y. Choffat, M. Engels, M. Schwyzer, and M. Ackermann. 1994. Identification and zinc dependence of the bovine herpesvirus 1 transactivator protein BICP0. J. Virol. 683154-3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frankel, A. D., D. S. Bredt, and C. O. Pabo. 1988. Tat protein from human immunodeficiency virus forms a metal-linked dimer. Science 24070-73. [DOI] [PubMed] [Google Scholar]
- 19.Frankel, A. D., L. Chen, R. J. Cotter, and C. O. Pabo. 1988. Dimerization of the tat protein from human immunodeficiency virus: a cysteine-rich peptide mimics the normal metal-linked dimer interface. Proc. Natl. Acad. Sci. USA 856297-6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frazzini, V., E. Rockabrand, E. Mocchegiani, and S. L. Sensi. 2006. Oxidative stress and brain aging: is zinc the link? Biogerontology 7307-314. [DOI] [PubMed] [Google Scholar]
- 21.Garber, M. E., P. Wei, V. N. KewalRamani, T. P. Mayall, C. H. Herrmann, A. P. Rice, D. R. Littman, and K. A. Jones. 1998. The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 123512-3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Genther, S. M., S. Sterling, S. Duensing, K. Münger, C. Sattler, and P. F. Lambert. 2003. Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J. Virol. 772832-2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goff, S. P. 2003. Death by deamination: a novel host restriction system for HIV-1. Cell 114281-283. [DOI] [PubMed] [Google Scholar]
- 24.Goswami, R., B. Turk, K. Enderle, A. Howe, and K. Rundell. 1992. Effect of zinc ions on the biochemical behavior of simian virus 40 small-t antigen expressed in bacteria. J. Virol. 661746-1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grigorov, B., D. Décimo, F. Smagulova, C. Péchoux, M. Mougel, D. Muriaux, and J. L. Darlix. 2007. Intracellular HIV-1 Gag localization is impaired by mutations in the nucleocapsid zinc fingers. Retrovirology 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grossman, S. R., and L. A. Laimins. 1989. E6 protein of human papillomavirus type 18 binds zinc. Oncogene 41089-1093. [PubMed] [Google Scholar]
- 27.Haase, H., and W. Maret. 2003. Intracellular zinc fluctuations modulate protein tyrosine phosphatase activity in insulin/insulin-like growth factor-1 signaling. Exp. Cell Res. 291289-298. [DOI] [PubMed] [Google Scholar]
- 28.Han, R., N. M. Cladel, C. A. Reed, and N. D. Christensen. 1998. Characterization of transformation function of cottontail rabbit papillomavirus E5 and E8 genes. Virology 251253-263. [DOI] [PubMed] [Google Scholar]
- 29.Houzet, L., Z. Morichaud, L. Didierlaurent, D. Muriaux, J. L. Darlix, and M. Mougel. 2008. Nucleocapsid mutations turn HIV-1 into a DNA-containing virus. Nucleic Acids Res. 362311-2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang, H. W., and K. T. Wang. 1996. Structural characterization of the metal binding site in the cysteine-rich region of HIV-1 Tat protein. Biochem. Biophys. Res. Commun. 227615-621. [DOI] [PubMed] [Google Scholar]
- 31.Ilbäck, N. G., A. W. Glynn, L. Wikberg, E. Netzel, and U. Lindh. 2004. Metallothionein is induced and trace element balance changed in target organs of a common viral infection. Toxicology 199241-250. [DOI] [PubMed] [Google Scholar]
- 32.Kim, Y. M., W. Reed, W. Wu, P. A. Bromberg, L. M. Graves, and J. M. Samet. 2006. Zn2+-induced IL-8 expression involves AP-1, JNK, and ERK activities in human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 290L1028-L1035. [DOI] [PubMed] [Google Scholar]
- 33.Kitamura, H., H. Morikawa, H. Kamon, M. Iguchi, S. Hojyo, T. Fukada, S. Yamashita, T. Kaisho, S. Akira, M. Murakami, and T. Hirano. 2006. Toll-like receptor-mediated regulation of zinc homeostasis influences dendritic cell function. Nat. Immunol. 7971-977. [DOI] [PubMed] [Google Scholar]
- 34.Kurima, K., Y. Yang, K. Sorber, and A. J. Griffith. 2003. Characterization of the transmembrane channel-like (TMC) gene family: functional clues from hearing loss and epidermodysplasia verruciformis. Genomics 82300-308. [DOI] [PubMed] [Google Scholar]
- 35.Kyo, S., D. J. Klumpp, M. Inoue, T. Kanaya, and L. A. Laimins. 1997. Expression of AP1 during cellular differentiation determines human papillomavirus E6/E7 expression in stratified epithelial cells. J. Gen. Virol. 78401-411. [DOI] [PubMed] [Google Scholar]
- 36.Laity, J. H., B. M. Lee, and P. E. Wright. 2001. Zinc finger proteins: new insights into structural and functional diversity. Curr. Opin. Struct. Biol. 1139-46. [DOI] [PubMed] [Google Scholar]
- 37.Lazarczyk, M., C. Pons, J. A. Mendoza, P. Cassonnet, Y. Jacob, and M. Favre. 2008. Regulation of cellular zinc balance as a potential mechanism of EVER-mediated protection against pathogenesis by cutaneous oncogenic human papillomaviruses. J. Exp. Med. 20535-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee, S. P., and M. K. Han. 1996. Zinc stimulates Mg2+-dependent 3′-processing activity of human immunodeficiency virus type 1 integrase in vitro. Biochemistry 353837-3844. [DOI] [PubMed] [Google Scholar]
- 39.Lee, S. P., J. Xiao, J. R. Knutson, M. S. Lewis, and M. K. Han. 1997. Zn2+ promotes the self-association of human immunodeficiency virus type-1 integrase in vitro. Biochemistry 36173-180. [DOI] [PubMed] [Google Scholar]
- 40.Liuzzi, J. P., and R. J. Cousins. 2004. Mammalian zinc transporters. Annu. Rev. Nutr. 24151-172. [DOI] [PubMed] [Google Scholar]
- 41.Luo, K., Z. Xiao, E. Ehrlich, Y. Yu, B. Liu, S. Zheng, and X. F. Yu. 2005. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc. Natl. Acad. Sci. USA 10211444-11449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Majewski, S., S. Jablonska, and G. Orth. 1997. Epidermodysplasia verruciformis. Immunological and nonimmunological surveillance mechanisms: role in tumor progression. Clin. Dermatol. 15321-334. [DOI] [PubMed] [Google Scholar]
- 43.Margoshes, M., and B. L. Vallee. 1957. A cadmium protein from equine kidney cortex J. Am. Chem. Soc. 794813-4814. [Google Scholar]
- 44.Martin, P., W. C. Vass, J. T. Schiller, D. R. Lowy, and T. J. Velu. 1989. The bovine papillomavirus E5 transforming protein can stimulate the transforming activity of EGF and CSF-1 receptors. Cell 5921-32. [DOI] [PubMed] [Google Scholar]
- 45.Maufort, J. P., S. M. Williams, H. C. Pitot, and P. F. Lambert. 2007. Human papillomavirus 16 E5 oncogene contributes to two stages of skin carcinogenesis. Cancer Res. 676106-6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McEuen, A. R., B. Edwards, K. A. Koepke, A. E. Ball, B. A. Jennings, A. J. Wolstenholme, M. J. Danson, and D. W. Hough. 1992. Zinc binding by retroviral integrase. Biochem. Biophys. Res. Commun. 189813-818. [DOI] [PubMed] [Google Scholar]
- 47.Misumi, S., N. Takamune, Y. Ohtsubo, K. Waniguchi, and S. Shoji. 2004. Zn2+ binding to cysteine-rich domain of extracellular human immunodeficiency virus type 1 Tat protein is associated with Tat protein-induced apoptosis. AIDS Res. Hum. Retrovir. 20297-304. [DOI] [PubMed] [Google Scholar]
- 48.Miura, P., and B. J. Jasmin. 2006. Utrophin upregulation for treating Duchenne or Becker muscular dystrophy: how close are we? Trends Mol. Med. 12122-129. [DOI] [PubMed] [Google Scholar]
- 49.Nonnenmacher, M., J. Salmon, Y. Jacob, G. Orth, and F. Breitburd. 2006. Cottontail rabbit papillomavirus E8 protein is essential for wart formation and provides new insights into viral pathogenesis. J. Virol. 804890-4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orth, G. 1987. Epidermodysplasia verruciformis, p. 199-243. In N.P. Salzman and P.M. Howley (ed.), The Papovaviridae: the papillomaviruses. Plenum Press, New York, NY.
- 51.Orth, G. 2006. Genetics of epidermodysplasia verruciformis: insights into host defense against papillomaviruses. Semin. Immunol. 18362-374. [DOI] [PubMed] [Google Scholar]
- 52.Palmiter, R. D. 1994. Regulation of metallothionein genes by heavy metals appears to be mediated by a zinc-sensitive inhibitor that interacts with a constitutively active transcription factor, MTF-1. Proc. Natl. Acad. Sci. USA 911219-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palmiter, R. D., and S. D. Findley. 1995. Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. 14639-649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paul, I., J. Cui, and E. L. Maynard. 2006. Zinc binding to the HCCH motif of HIV-1 virion infectivity factor induces a conformational change that mediates protein-protein interactions. Proc. Natl. Acad. Sci. USA 10318475-18480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Radtke, F., R. Heuchel, O. Georgiev, M. Hergersberg, M. Gariglio, Z. Dembic, and W. Schaffner. 1993. Cloned transcription factor MTF-1 activates the mouse metallothionein I promoter. EMBO J. 121355-1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramoz, N., A. Taïeb, L. A. Rueda, L. S. Montoya, B. Bouadjar, M. Favre, and G. Orth. 2000. Evidence for a nonallelic heterogeneity of epidermodysplasia verruciformis with two susceptibility loci mapped to chromosome regions 2p21-p24 and 17q25. J. Investig. Dermatol. 1141148-1153. [DOI] [PubMed] [Google Scholar]
- 57.Ramoz, N., L. A. Rueda, B. Bouadjar, L. S. Montoya, G. Orth, and M. Favre. 2002. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat. Genet. 32579-581. [DOI] [PubMed] [Google Scholar]
- 58.Rice, W. G., C. A. Schaeffer, B. Harten, F. Villinger, T. L. South, M. F. Summers, L. E. Henderson, J. W. Bess, L. O. Arthur, J. S. McDougal, S. L. Orloff, J. Mendeleyev, and E. Kun. 1993. Inhibition of HIV-1 infectivity by zinc-ejecting aromatic C-nitroso compounds. Nature 361473-475. [DOI] [PubMed] [Google Scholar]
- 59.Rice, W. G., C. A. Schaeffer, L. Graham, M. Bu, J. S. McDougal, S. L. Orloff, F. Villinger, M. Young, S. Oroszlan, M. R. Fesen, Y. Pomier, J. Mendeleyev, and E. Kun. 1993. The site of antiviral action of 3-nitrosobenzamide on the infectivity process of human immunodeficiency virus in human lymphocytes. Proc. Natl. Acad. Sci. USA 909721-9724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodríguez, M. I., M. E. Finbow, and A. Alonso. 2000. Binding of human papillomavirus 16 E5 to the 16 kDa subunit c (proteolipid) of the vacuolar H+-ATPase can be dissociated from the E5-mediated epidermal growth factor receptor overactivation. Oncogene 193727-3732. [DOI] [PubMed] [Google Scholar]
- 61.Schiffman, M., P. E. Castle, J. Jeronimo, A. C. Rodriguez, and S. Wacholder. 2007. Human papillomavirus and cervical cancer. Lancet 370890-907. [DOI] [PubMed] [Google Scholar]
- 62.Sekler, I., S. L. Sensi, M. Hershfinkel, and W. F. Silverman. 2007. Mechanism and regulation of cellular zinc transport. Mol. Med. 13337-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seo, S. R., S. A. Chong, S. I. Lee, J. Y. Sung, Y. S. Ahn, K. C. Chung, and J. T. Seo. 2001. Zn2+-induced ERK activation mediated by reactive oxygen species causes cell death in differentiated PC12 cells. J. Neurochem. 78600-610. [DOI] [PubMed] [Google Scholar]
- 64.Srinivas, S., A. Schönthal, and W. Eckhart. 1994. Polyomavirus middle-sized tumor antigen modulates c-Jun phosphorylation and transcriptional activity. Proc. Natl. Acad. Sci. USA 9110064-10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stöppler, M. C., S. W. Straight, G. Tsao, R. Schlegel, and D. J. McCance. 1996. The E5 gene of HPV-16 enhances keratinocyte immortalization by full-length DNA. Virology 223251-254. [DOI] [PubMed] [Google Scholar]
- 66.Straight, S. W., P. M. Hinkle, R. J. Jewers, and D. J. McCance. 1993. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J. Virol. 674521-4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Surovoy, A., D. Waidelich, and G. Jung. 1992. Nucleocapsid protein of HIV-1 and its Zn2+ complex formation analysis with electrospray mass spectrometry. FEBS Lett. 311259-262. [DOI] [PubMed] [Google Scholar]
- 68.Tal, T. L., L. M. Graves, R. Silbajoris, P. A. Bromberg, W. Wu, and J. M. Samet. 2006. Inhibition of protein tyrosine phosphatase activity mediates epidermal growth factor receptor signaling in human airway epithelial cells exposed to Zn2+. Toxicol. Appl. Pharmacol. 21416-23. [DOI] [PubMed] [Google Scholar]
- 69.Tsai, T. C., and S. L. Chen. 2003. The biochemical and biological functions of human papillomavirus type 16 E5 protein. Arch. Virol. 1481445-1453. [DOI] [PubMed] [Google Scholar]
- 70.Turk, B., A. Porras, M. C. Mumby, and K. Rundell. 1993. Simian virus 40 small-t antigen binds two zinc ions. J. Virol. 673671-3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Winnischofer, S. M., M. L. de Oliveira, and M. C. Sogayar. 2003. Suppression of AP-1 constitutive activity interferes with polyomavirus MT antigen transformation ability. J. Cell. Biochem. 90253-266. [DOI] [PubMed] [Google Scholar]
- 72.Xiao, Z., E. Ehrlich, K. Luo, Y. Xiong, and X. F. Yu. 2007. Zinc chelation inhibits HIV Vif activity and liberates antiviral function of the cytidine deaminase APOBEC3G. FASEB J. 21217-222. [DOI] [PubMed] [Google Scholar]
- 73.Yamasaki, S., K. Sakata-Sogawa, A. Hasegawa, T. Suzuki, K. Kabu, E. Sato, T. Kurosaki, S. Yamashita, M. Tokunaga, K. Nishida, and T. Hirano. 2007. Zinc is a novel intracellular second messenger. J. Cell Biol. 177637-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yoshimura, K., H. Aoki, Y. Ikeda, A. Furutani, K. Hamano, and M. Matsuzaki. 2006. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase in mice. Ann. N. Y. Acad. Sci. 108574-81. [DOI] [PubMed] [Google Scholar]
- 75.Young, M. R., L. Farrell, P. Lambert, P. Awasthi, and N. H. Colburn. 2002. Protection against human papillomavirus type 16-E7 oncogene-induced tumorigenesis by in vivo expression of dominant-negative c-jun. Mol. Carcinog. 3472-77. [DOI] [PubMed] [Google Scholar]
- 76.Zhang, B., A. Srirangam, D. A. Potter, and A. Roman. 2005. HPV16 E5 protein disrupts the c-Cbl-EGFR interaction and EGFR ubiquitination in human foreskin keratinocytes. Oncogene 242585-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zilliox, M. J., G. Parmigiani, and D. E. Griffin. 2006. Gene expression patterns in dendritic cells infected with measles virus compared with other pathogens. Proc. Natl. Acad. Sci. USA 1033363-3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.zur Hausen, H. 2000. Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J. Natl. Cancer Inst. 92690-698. [DOI] [PubMed] [Google Scholar]