

Abstract
The varicella-zoster virus (VZV) origin of DNA replication (oriS) contains a 46-bp AT-rich palindrome and three consensus binding sites for the VZV origin binding protein (OBP) encoded by VZV ORF51. All three OBP binding sites are upstream of the palindrome in contrast to the sequence of the herpes simplex virus oriS, which has required OBP binding sites upstream and downstream of the AT-rich region. We are investigating the roles that sequences downstream of the palindrome play in VZV oriS-dependent DNA replication. Computer analysis identified two GC boxes, GC box 1 and GC box 2, in the downstream region which were predicted to be binding sites for the cellular transcription factor Sp1. Electrophoretic mobility shift assay and supershift assays showed that two members of the Sp family (Sp1 and Sp3) stably bind to GC box 1, but not to GC box 2. A predicted binding site for the cellular factor Yin Yang 1 (YY1) that overlaps with GC box 2 was also identified. Supershift and mutational analyses confirmed the binding of YY1 to this site. Mutation of GC box 1 resulted in loss of Sp1 and Sp3 binding and an increase in origin-dependent replication efficiency in DpnI replication assays. In contrast, mutation of the YY1 site had a statistically insignificant effect. These results suggest a model where origin-dependent DNA replication and viral transcription are coupled by the binding of Sp1 and Sp3 to the downstream region of the VZV replication origin during lytic infection. They may also have implications regarding establishment or reactivation of viral latency.
Varicella-zoster virus (VZV) is a member of the Alphaherpesvirus subfamily of the Herpesviridae family. VZV is the causative agent of two types of disease, chicken pox (varicella) upon primary infection and shingles (zoster) after reactivation from latency (1, 2). The VZV genome consists of 125 kbp which encodes approximately 69 open reading frames (ORFs). The viral genome is made up of long and short unique segments designated UL and US, respectively, both of which are bounded by inverted repeat sequences (11). The linear sequence of VZV genes is similar to that of herpes simplex virus type 1 (HSV-1) and includes the coding sequences for orthologues of the seven HSV-1 proteins required for origin-dependent DNA replication (9).
The VZV genome contains two origins of DNA replication (oriS) within the internal repeats (IRs) and terminal repeats (TRs) bounding the US segment at sites analogous to those within the HSV-1 genome. VZV, however, lacks the third HSV-1 origin (oriL), which is located near the center of the HSV-1 UL region (10, 11, 48, 49). HSV-1 oriL has been shown to be dispensable for replication of HSV DNA (3, 53) but has been implicated in HSV pathogenesis and reactivation from latency (4). The equivalent region in the VZV genome is comprised of a bidirectional promoter that regulates the transcription of the VZV DNA polymerase catalytic subunit (ORF28) and DNA binding protein (ORF29) genes (32, 54).
The architecture of the VZV and HSV-1 oriS regions differs significantly (Fig. 1). The two VZV oriS regions contain an AT-rich palindrome and three consensus 10-bp binding sites [5′-C(G/A)TTCGCACT-3′] for the VZV origin binding protein (OBP) encoded by VZV ORF51 located upstream of the AT-rich palindrome (48, 50). These binding sites, designated boxes A, B, and C are identical or nearly identical to the consensus site for the HSV-1 UL9 OBP with which the VZV ORF51 OBP shares 54.8% similarity and 46.5% identity (8, 50). All three OBP binding sites in the VZV origin are oriented in the same direction and are present on the same strand of the viral DNA. Origin-dependent DNA replication in VZV requires the AT-rich region and box A. Box C is not essential, but its presence increases replication efficiency, while box B appears to be completely dispensable (48, 50). Nothing is currently known regarding the role played by sequences downstream of the VZV minimal origin in viral DNA replication. A partial (7 of 10 bp) OBP binding site is present downstream of the AT-rich palindrome with the same orientation and on the same strand as the upstream OBP boxes. However, gel shift and DNase I protection assays failed to demonstrate VZV OBP binding to this sequence (8, 50). In contrast, in the HSV-1 oriS, binding sites for the UL9 OBP (boxes I, II, and III) occur both upstream and downstream of the AT-rich region. Boxes I and II are located upstream and downstream on opposite strands of the DNA and are oriented in opposite directions. Box III is located upstream of box I but is oriented in the same direction and occurs on the same DNA strand as box II. Mutational analysis has shown that the minimal HSV-1 requires the presence of both box I and box II and the central AT-rich region (4, 12, 49, 52).
FIG. 1.

(A) Comparison of the architecture of the VZV and HSV-1 oriS origins of DNA replication. The positions of the OBP binding site boxes on the two strands and their orientations are indicated. (B) Nucleotide sequence of VZV oriS showing the positions of the OBP binding sites and potential binding sites for cellular factors in the region downstream of the AT repeats. The consensus Sp1 site within GC box 1 is shown in bold type, and the YY1 site overlapping GC box 2 is underlined. min, minimal.
The structural differences between the VZV and HSV-1 oriS in terms of the positioning and orientation of the OBP binding sites suggest that the mechanism and/or transacting factors involved in initiation or regulation of origin-dependent DNA replication may differ between HSV-1 and VZV. One possibility raised by Stow et al. (50) is that cellular factors or as yet unidentified viral proteins could bind downstream of the AT-rich region in the VZV oriS to facilitate unwinding of the origin and assembly of the viral DNA replication machinery.
There is precedence for this possibility based on work with Epstein-Barr virus (EBV), HSV-1, simian virus 40 (SV40), and human papillomavirus (HPV). The cellular transcription factor Sp1 binds to the downstream region of EBV ORILyt and physically interacts with the EBV DNA polymerase catalytic and accessory subunits, increasing their recruitment and resulting in a positive effect on DNA replication (5, 15). Sp1 and the related factor Sp3 bind to the upstream region of the HSV-1 oriS at a cis element, designated OscarL, and also have a positive effect on origin-dependent DNA replication, although the specific mechanism remains unknown (37). Several transcription factors were found to stimulate SV40 DNA replication, including NF1, AP1, and Sp1 (16). Sp1 has also been reported (13) to enhance replication of human papillomavirus type 18 (HPV18). In contrast, the transcription factor Yin Yang 1 (YY1) has been shown to suppress HPV DNA replication through interference with the function of the HPV E2 protein (25).
In the work presented here, we identified and authenticated the presence of binding sites for cellular transcription factors within the downstream region of the VZV oriS and tested the effects of mutations of those sequences on VZV origin-dependent replication. Our data show that the cellular factors Sp1, Sp3, and YY1 stably bind to different portions of the downstream region of the VZV oriS. YY1 appears to have no effect on origin-dependent replication. However, mutation of the Sp1/Sp3 site that ablates binding of these factors resulted in an increase in replication in in situ DpnI replication assays. This indicates that Sp1 and Sp3 act to suppress VZV oriS-dependent replication in contrast to what has been observed, as described above, in other virus systems. Further, this represents the first documentation of a role for Sp3 in VZV infection.
MATERIALS AND METHODS
Cells and viruses.
MeWo cells, a human melanoma cell line that supports the replication of VZV, were grown in Eagle's minimal essential medium supplemented with 10% fetal bovine serum as previously described (47). VZV strain MSP was propagated in MeWo cell monolayers as described by Lynch et al. (29) and Peng et al. (39).
Nuclear and whole-cell lysates.
Nuclear extracts of VZV-infected and uninfected MeWo cells were prepared as previously described (29). Briefly, pelleted cells were washed once with 30 volumes of phosphate-buffered saline. Packed cells were resuspended in one packed cell volume of buffer A (10 mM HEPES [pH 7.9], 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol) at 4°C and allowed to swell on ice for 15 min. Cells were then lysed by 10 rapid passages through a 25-gauge hypodermic syringe, and the homogenate was sedimented briefly at 12,000 × g. The crude nuclear pellet was resuspended in two-thirds of one packed cell volume (determined at the time of cell harvest) of buffer C (20 mM HEPES [pH 7.9], 25% [vol/vol] glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride, 0.5 mM dithiothreitol), and the resulting solution was incubated on ice with stirring for 30 min. The nuclear debris was pelleted by centrifugation for 5 min at 12,000 × g, and the supernatant (nuclear extract) was dialyzed against buffer D (20 mM HEPES [pH 7.9], 20% [vol/vol] glycerol, 0.1 M KCl, 0.2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride, 0.5 mM dithiothreitol) for 2 h. The dialyzed extract was then quick-frozen in liquid nitrogen and stored at −70°C. Whole-cell lysates of VZV-infected and uninfected MeWo cells were prepared as previously described (39). Cells were grown to confluence in 100-mm petri dishes, washed with phosphate-buffered saline buffer, and then suspended in lysis buffer (50 mM Tris-HCl [pH 7.5], 0.15 M NaCl, 1 mM EDTA, 0.1% Triton X-100, and protease inhibitor cocktail [Roche, Mannheim, Germany] added per the manufacturer's instruction). The lysates were collected and centrifuged for 5 min, and the supernatants were stored at −70°C for subsequent use.
Immunoblot analyses.
Whole-cell lysates of VZV-infected and uninfected MeWo cells were analyzed by sodium dodecyl sulfate-10% polyacrylamide gel electrophoresis and immunoblotting for the presence of Sp family members and YY1 as described by Yang et al. (55). Antibodies against Sp1, Sp2, Sp3, Sp4, and β-tubulin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody against Sp1 was also obtained from Upstate (Temecula, CA). Rabbit polyclonal antibody against YY1 was generously supplied by Te-Chung Lee (University at Buffalo). Reactive bands were visualized using goat anti-rabbit immunoglobulin G conjugated with horseradish peroxidase (Chemicon, Temecula, CA) in conjunction with Supersignal West Pico chemiluminescence substrate (Pierce, Rockford, IL). Quantification of the relative amounts of these transcription factors normalized to β-tubulin in loading controls was performed using a Bio-Rad GS700 imagining densitometer (Bio-Rad, Hercules, CA). Statistical significance was determined by a one-way analysis of variance, followed by Tukey's post hoc test.
Plasmids.
The pVO2 plasmid containing a 259-bp fragment encompassing nucleotides 5502 to 5760 of the TRs/IRs sequence of the VZV Dumas strain (10) was generously supplied by Nigel Stow (MRC Virology Unit, Institute of Virology, University of Glasgow). This fragment contains the VZV oriS region including OBP boxes C and A, the AT-rich palindrome, and 125 bp of downstream sequence. The pVO2 parental plasmid, pAT153, was purchased from MoBiTec (Goettingen, Germany).
Plasmids containing GC box 1 and YY1 and box A substitution mutations within the oriS region were generated by mutation of pVO2 using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) using the following primer sets: primers 5′-ATGTCGCGGTTTTATGGGGTGTTCGCGGGCTTTTCACAGAATATA-3′ and 5′-TATATTCTGTGAAAAGCCCGCGAACACCCCATAAAACCGCGACAT-3′ for GC box 1, primers 5′-ACAGAATATATATATTCCAAATTTAGCGGCAGGCTTTTTAAAATC-3′ and 5′-GATTTTAAAAAGCCTGCCGCTAAATTTGGAATATATATATTCTGT-3′ for YY1, and primers 5′-GGCATGTGTCCAACCACCGTTAAAACTTTCTTTCTATATATATAT-3′ and 5′-ATATATATATAGAAAGAAAGTTTTAACGGTGGTTGGACACATGCC-3′ for box A. All primers were synthesized by IDT (Coralville, IA). The positions and sequences of the mutations in the VZV oriS sequence contained within the pVO2 plasmid used in transfections were verified by sequencing at the Roswell Park sequencing facility.
DpnI replication assays.
MeWo cells were transfected with Lipofectamine reagent (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. Four microliters of Lipofectamine reagent was used per microgram of transfected DNA in each transfection. In transfections performed in 100-mm-diameter petri dishes, 2.1 × 106 MeWo cells per dish were seeded in 12 ml of complete growth medium. The cells were 80% confluent at the time of transfection. Three hours before transfection, the medium was replaced with fresh medium. Replication experiments were performed as described by Stow and McMonagle (49) and Stow and Davison (48). The cells were transfected with 5 μg of wild-type or mutant pVO2 plasmids. At 6 h posttransfection, cells were superinfected with VZV strain MSP. VZV superinfections were performed by adding a ratio of 0.4 infected cell per 1 uninfected cell of the infected cells to each monolayer. Total cellular DNA was prepared 48 h after superinfection by the addition of 5 ml of lysis buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 0.5% sodium dodecyl sulfate, 20 μg/ml RNase I) per dish. Lysates were transferred to 15-ml conical test tubes and incubated for 1 h at 37°C. Proteinase K in TE buffer (10 mM Tris-HCl and 0.001 M EDTA) was added to a final concentration of 100 μg per ml, and the NaCl concentration was adjusted to 0.15 M. The mixture was then incubated at 50°C for 3 h. The DNA was isolated by phenol chloroform extraction, followed by ethanol precipitation. The DNA was digested with DpnI and EcoRI as described by Stow and Davison (48) and analyzed by Southern blot hybridization. Transfers were done using TurboBlotter kits obtained from Whatman, Inc. (Sanford, ME). The blots were probed with a 400-bp PCR product prepared from pVO2 using primers 5′-GTGCTCCTGTCGTTGAGGACCCGG-3′ and 5′-CCTCTGACTTGAGCGTCGATTTTT-3′ and end labeled with[α-32P]ATP using T4 kinase (Invitrogen, Carlsbad, CA). This fragment includes a portion (nucleotide positions 1465 to 1864) of the pVO2 plasmid containing the ColE1 origin of replication and was designed to detect both intact DpnI-resistant linearized pVO2 (3.9 kb) resulting from replication of the input plasmid and an 872-bp fragment resulting from the DpnI-sensitive unreplicated input plasmid. The resulting bands were quantified by PhosphorImager (Molecular Dynamics, Sunnyvale, CA) analysis. The ratio of replicated plasmid to input plasmid represented the replication efficiency of the test plasmid. The data from representative experiments are presented as means of triplicate DpnI replications. Error bars indicate standard errors. Statistical significance was determined by a one-way analysis of variance followed by Tukey's post hoc test.
EMSA and supershift analyses.
Thirty-base-pair oligonucleotide probes (IDT, Coralville, IA) containing wild-type and mutant GC box 1 elements and 40-bp probes containing wild-type and mutant YY1 and GC box 2 binding site elements were used in electrophoretic mobility shift assays (EMSAs). Probes were end labeled with [α-32P]ATP using T4 kinase (Invitrogen, Carlsbad, CA). For the GC box 1 probes, 100 femtomoles of the 30-bp labeled probes containing wild-type and mutant GC box 1 (∼1 × 105 dpm) were incubated with 12 μg of uninfected or infected MeWo cell nuclear extract in a 10-μl reaction mixture in binding buffer A [40 mM HEPES (pH 7.9), 100 mM NaCl, 10 mM MgCl2, 200 μg/ml bovine serum albumin, 12% glycerol, 0.05% NP-40, 1 mM dithiothreitol, and 3 μg poly(dI-dC)]. For the GC box 2/YY1 probes, 100 femtomoles of 40-bp labeled probes containing wild-type and mutant GC box 2 sequences or wild-type and mutant YY1 binding sites (∼1 × 105 dpm) were incubated with 12 μg of MeWo cell nuclear extracts in a 10-μl reaction mixture in binding buffer B [10 mM Tris-HCl (pH 8.0), l mM EDTA, 100 mM NaCl, 3 μg poly(dI-dC), 0.1% NP-40, 1 mM dithiothreitol, and 50 μg/ml bovine serum albumin]. The samples were analyzed by electrophoresis on a 5% polyacrylamide (37.5:1 acrylamide/bisacrylamide) gel and then autoradiography.
In the supershift assays, anti-Sp family antibodies (antibodies against the four members of the Sp family [Sp1, Sp2, Sp3, and Sp4]) obtained from Santa Cruz Biologicals (Santa Cruz, CA) or anti-Sp1 antibodies (Upstate, Temecula, CA) and rabbit polyclonal YY1 antiserum, generously supplied by Te-Chung Lee (University at Buffalo) were added in 4-μg (Sp family) or 2-μl (YY1) aliquots to reaction mixtures containing infected cell nuclear extracts and either GC box 1 or YY1/GC box 2 probes. All of the antibodies directed against the Sp family members were described by the commercial suppliers as being positive in supershift assays. This was confirmed in control experiments using consensus GC box-containing duplex probes (data not shown). The samples were then analyzed by electrophoresis on a 5% polyacrylamide (37.5:1 acrylamide/bisacrylamide) gel and autoradiography.
RESULTS
Identification of potential binding sites for cellular factors in the downstream region of VZV oriS.
The 125 bp immediately downstream of the AT-rich palindrome of the VZV oriS, located within the IRs region of the genome, were analyzed for the presence of cellular transcription factor binding sites using the AliBaba2.1 and Patch programs (http://www.gene-regulation.com/pub/programs.html; Biobase). Two GC-rich sequences, designated GC box 1 and GC box 2, were identified 57 to 72 bp and 99 to 110 bp, respectively, 3′ to the final TA pair in the minimal origin (Fig. 1B). Both contained predicted binding sites for the ubiquitous cellular transcription factor Sp1. GC box 1 contains the canonical Sp1 binding sequence (5′-GGGCGGG-3′) first identified by Dynan and Tjian (14) in the SV40 early promoter. GC box 2 contains an alternative site (5′-GGAGCGG-3′) predicted to bind Sp1. Sp1 is well documented to be involved in VZV replication based on studies describing its role in several important VZV promoters, including those regulating expression of VZV gI, gE, the VZV major single-strand binding protein, and the VZV DNA polymerase catalytic subunit (6, 19, 39, 43, 54). Sp1 has also been shown to interact physically and functionally with the VZV major transcriptional activator, IE62 (36, 39). In addition, there is a high incidence of predicted Sp1 binding sites within VZV promoters (a minimum of 18) beyond those already authenticated (44).
A potential binding site (5′-CAAATGGAG-3′) for the cellular transcription factor YY1 was also identified partially overlapping GC box 2. This sequence is highly homologous to a YY1 site identified in the HPV8 E6 promoter (5′-CAAATGGAC-3′) and is also very similar to YY1 sites identified in adeno-associated virus P5 promoter and the murine leukemia virus long terminal repeat (26, 38). YY1 can act either as an activator or a repressor and has been shown to be capable of interacting with Sp1 (24, 45, 46). In contrast to Sp1, the role or roles that YY1 plays in VZV transcription and the viral life cycle are currently unknown. Based on the results from the computer analysis, EMSA, supershift, and mutational analyses were then performed in order to determine whether GC box 1, GC box 2, and the YY1 site were bona fide binding targets for the specific transcription factors.
Sp1 and Sp3 bind to GC box 1.
In the first series of experiments, EMSAs were performed using a 30-bp duplex oligonucleotide containing the 15-bp GC box 1 and upstream and downstream flanking sequences (Fig. 2A). Shift patterns were obtained using uninfected and VZV-infected MeWo cell nuclear extracts. Two complexes, one major and one minor, were consistently observed with both infected and uninfected nuclear extracts, with the major complex being the slower migrating of the two (Fig. 2B). There was no difference in the shift pattern, qualitatively or quantitatively, obtained with the two nuclear extracts, suggesting that viral proteins and/or cellular proteins whose expression or DNA binding properties were significantly modified by VZV infection did not play a role in the formation of the observed complexes. To test the specificity of the formation of these two complexes, competition EMSA experiments were performed using the unlabeled 30-bp GC box 1 probe as the specific competitor and a 30-bp oligonucleotide containing the OBP box A sequence as the nonspecific competitor. As shown in Fig. 2C, the cold specific competitor efficiently competed away the formation of the two complexes, while the presence of the nonspecific competitor had no effect on the pattern of the shifted bands.
FIG. 2.

EMSA and competition assays using a 30-bp oligonucleotide containing GC box 1 and the native flanking region in the presence of uninfected and VZV-infected MeWo cell nuclear extracts. (A) Sequences of the two oligonucleotides used in the assays. NSC, nonspecific competitor. (B) Typical data from EMSA experiments using the GC box 1 probe and uninfected and infected cell extracts. Lanes: FO, free oligonucleotide; UI, uninfected nuclear extract; I, VZV-infected nuclear extract. (C) Competition assays using a 50-fold excess of cold GC box 1-specific competitor (SC) and nonspecific competitor (NSC). The nonspecific competitor included the upstream box A site and attendant flanking sequence. The positions of the two consistently observed complexes in panels B and C are indicated by asterisks.
In the next series of experiments, EMSAs were performed using oligonucleotides containing mutations in the consensus Sp1 binding site in the GC box 1 sequence. These included deletion of the 7-base-pair Sp1 binding site and site-specific mutagenesis which substituted the first two G residues with T and C, respectively (Fig. 3A). Both of these mutations were predicted to abolish Sp1 binding using the same computer analysis that had initially identified the site. As shown in Fig. 3B, both the deletion and substitution mutations ablated formation of both of the complexes formed with the wild-type sequence.
FIG. 3.

EMSA and supershift assays using wild-type, GC box 1 deletion, and substitution mutant duplex oligonucleotides. (A) Sequences of the oligonucleotides containing deletions and site-specific mutations. The positions and limits of GC box 1 are boxed. The deleted sequence is indicated, and additional flanking sequence added to maintain the 30-bp length for all probes is underlined. The site-specific mutations are indicated in italics. (B) Typical EMSA results showing loss of binding to the two mutant probes. Lanes: FO, free oligonucleotide; I, VZV-infected nuclear extract. (C) Supershift assays using the wild-type GC box 1 probe in the presence of infected cell nuclear extracts. Polyclonal antibodies against Sp1, Sp2, Sp3, Sp4, and YY1 were used in the supershifts as indicated. The positions of the original complexes are shown by asterisks, and the positions of the supershifted bands are shown by small black arrows. αSp1, anti-Sp1 antibody.
Antibody supershift assays were then performed in order to confirm that Sp1 was, in fact, present in the observed complexes and to investigate whether any of the other three major members of the Sp family of transcription factors (Sp2, Sp3, and Sp4), all of which are capable of binding to GC-rich sequences (17, 42), were also present. The results are presented in Fig. 3C and show that a significant fraction of the major, slower-migrating complex was supershifted only in the presence of anti-Sp1 antibody. Two anti-Sp1-specific supershifted complexes were consistently observed in these experiments. One migrated slightly more slowly than the major complex did, and the second, representing the majority of the supershifted material, failed to enter the gel. The less-abundant faster-migrating complex was completely supershifted only in the presence of anti-Sp3 antibody. In the data presented in Fig. 3C, a small amount of material failed to enter the gel in the presence of anti-Sp4 antibody. This occurred in three of five separate EMSA experiments, whereas the shifts observed with the Sp1 and Sp3 antibodies occurred in all five experiments. In contrast, no supershifts or losses of complexes were observed in the presence of anti-Sp2 antibodies. Anti-YY1 antibody, used here as a negative control, also showed no effect on complex formation or migration. Thus, Sp1 is present in at least one complex within the major supershifted band. Sp3 appears to be present exclusively in the complex represented by the less-abundant, faster-migrating band. Sp4 may also be associated with GC box 1 but to a much lower and less stable extent than Sp1 and Sp3 under these experimental conditions.
YY1 binds to the predicted YY1 binding site.
In the next series of experiments, we wished to determine whether protein complexes were formed with duplex oligonucleotides containing GC box 2 sequence and the predicted overlapping YY1 binding site. EMSAs were performed using a 40-bp probe containing both sequences and 8-bp upstream and 15-bp downstream flanking sequences (Fig. 4A). Shifts were performed with both uninfected and infected MeWo cell extracts. Three major complexes were formed in the presence of both uninfected and infected cell nuclear extracts and displayed identical migration positions (Fig. 4B). A decrease in the level of the intermediate migrating complex with infected protein extracts compared to uninfected protein extracts was frequently observed in these experiments, suggesting that this complex is less stable in the case of the infected cell extracts.
FIG. 4.

Results of EMSA experiments using a 40-bp probe containing GC box 2 and the YY1 site in the presence of uninfected and VZV-infected cell nuclear extracts. (A) Sequence of the 40-bp probe showing the positions of GC box 2 and the YY1 site. The YY1 site is underlined. (B) Typical results showing the presence of three shifted complexes. Lanes: FO, free oligonucleotide; UI, uninfected nuclear extract; I, VZV-infected nuclear extract. The positions of the three shifted complexes are indicated by asterisks.
We next wished to determine whether any of the Sp family members or YY1 bound to this sequence. Supershift experiments with antibodies against Sp1, Sp2, Sp3, and Sp4 indicated that, in contrast to our results with GC box 1, none of these factors bound to the probe containing GC box 2 (data not shown). Supershifts with anti-YY1, however, consistently resulted in loss of the fastest-migrating band in the case of the wild-type sequence and with probes containing deletion and substitution mutations predicted to disrupt the binding of Sp1 (Fig. 5). In each of these cases, signal was present in the well, suggesting that the supershifted complex could not enter the gel. Finally, site-specific mutation of the YY1 site led to loss of the fastest-migrating band, and here, no signal was seen in the well in the presence of anti-YY1 antibody.
FIG. 5.

EMSA and supershift assays using wild-type, GC box 2 deletion, GC box 2, and YY1 substitution mutant probes. (A) Sequences of the oligonucleotides containing deletions and site-specific mutations. The positions and limits of GC box 2 are boxed. The YY1 site is underlined. The deleted sequence in GC box 2 is indicated, and additional flanking sequence added to maintain the 40-bp length for all probes is underlined. (B) Results of EMSA and supershift assays using wild-type, GC box 2 deletion, GC box 2, and YY1 substitution mutant probes. Lanes: FO, free oligonucleotide; I, VZV-infected nuclear extract. αYY1, anti-YY1 antibody. The YY1-specific band that is lost with both anti-YY1 antibody supershifts and the probe containing the YY1 site substitution mutation are indicated by asterisks.
Thus, our results show that YY1 binds to the predicted YY1 binding site, whereas there is no evidence either by supershift assays or by mutational analysis that any members of the Sp family bind to this portion of the downstream region of VZV oriS. The site-specific and deletion mutations narrow the YY1 site to 5′-CCAAATGG-3′ and indicate that the two 3′-terminal guanine residues are critical for YY1 binding. The persistence of the two slower-migrating bands suggests that their formation involves the flanking regions, rather than the core elements mutated. This was confirmed in competition experiments where the cold 40-bp probe successfully competed away all three complexes (data not shown).
VZV infection lowers the level of Sp4 present in MeWo cells.
We next examined the levels of Sp1, Sp2, Sp3, and Sp4 and YY1 present in uninfected and infected whole-cell extracts by immunoblotting. Our results (Fig. 6) show that all four Sp family members and YY1 are expressed in MeWo cells. Quantification of the relative amounts of these factors using three sets of paired uninfected and infected cell extract preparations showed that there was no statistically significant difference in the levels of Sp1 (P = 0.373), Sp2 (P = 0.254), Sp3 (P = 0.563), and YY1 (P = 0.373) in infected versus uninfected MeWo cell extracts. Also, infection did not alter the levels of the two shorter forms of Sp3 of very similar molecular weights that are generated via internal translation initiation start sites (20). In contrast, a highly significant (P = 0.002) 40 to 50% decrease in the level of Sp4 in infected versus uninfected cell extracts was observed.
FIG. 6.
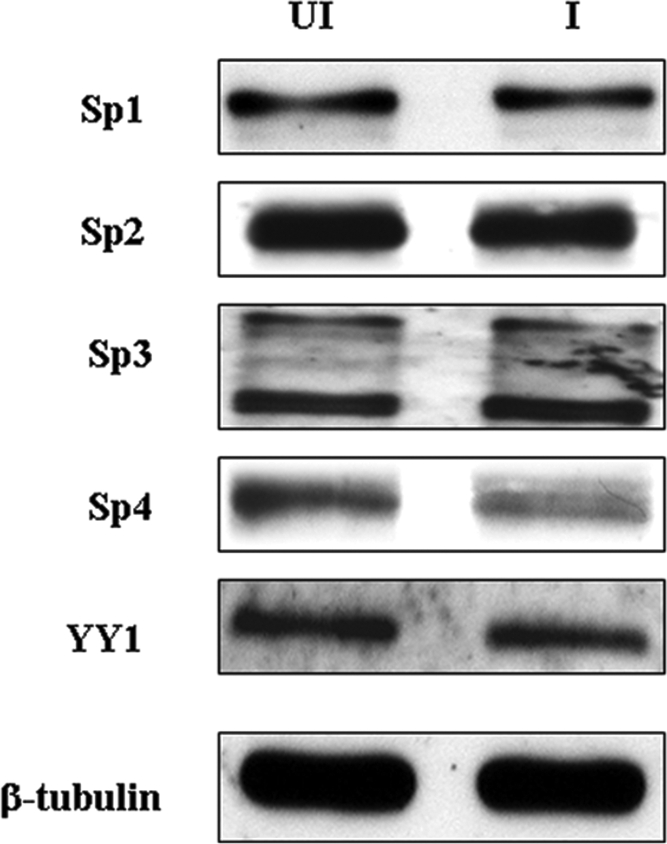
Immunoblot analysis examining the relative levels of Sp1, Sp2, Sp3, Sp4, and YY1 present in uninfected (UI) and infected (I) whole-cell extracts. Equivalent amounts of protein were added in the uninfected and infected lanes. β-Tubulin was used as a loading control.
Mutation of the Sp1/Sp3 site results in an increase in origin-dependent DNA replication.
Previous work from the Schaffer laboratory (37) showed that mutation of upstream binding sites for Sp1 and Sp3 resulted in an increase in origin-dependent DNA replication in DpnI resistance replication assays using a plasmid containing the HSV oriS. Analogous experiments were performed in the work reported here in order to determine the effects of mutation of the Sp1/Sp3 site in GC box 1 and the downstream YY1 site. These experiments were performed by transfection of the pVO2 plasmid (48, 50) containing the VZV oriS sequence (OBP boxes C and A, the AT-rich palindrome, and the 125-bp downstream region) into MeWo cells followed by VZV superinfection at a ratio of 0.4/1 infected to uninfected cells. The cells were harvested 48 h after superinfection, and the state of the oriS-containing plasmid DNA was analyzed by DpnI digestion, followed by separation on agarose gels and Southern blot hybridization. Blots were probed with a 400-bp pVO2 fragment that allowed clear differentiation between replicated (unmethylated, DpnI-resistant) progeny DNA and unreplicated (methylated DpnI-sensitive) input plasmids.
Control DpnI replication assays using pVO2 containing the wild-type VZV oriS showed a clear and readily detectable signal at the position predicted for the replicated DNA (Fig. 7A). In contrast, assays performed under the same conditions using the parent pAT153 plasmid lacking the VZV oriS resulted in no signal at the position of the replicated DNA. For a second negative control, pVO2 containing a three-base substitution in the box A OBP binding site was used. This also resulted in a loss of the replicated DNA band in agreement with the results of Stow et al. (50) who showed via deletion that box A is required for oriS-dependent DNA replication. Individual point mutations at the CGC positions were shown by Chen and Olivo (8) to eliminate OBP binding. Thus, these data confirm the long-held supposition that OBP binding to box A is required for VZV oriS-dependent DNA replication even in the presence of box C.
FIG. 7.

Results of DpnI oriS-dependent replication assays. (A) Southern blot analysis of control assays with wild-type pVO2, pVO2 containing a triple point mutation of the core box A CGC triplet, and the parental pAT153 plasmid lacking VZV oriS. The upper band (R) indicates the position of DpnI-resistant DNA resulting from replication within the MeWo cells. The lower band (U) indicates the position of unreplicated input plasmid. (B) Typical results showing Southern blot analysis of the effects of site-specific mutation of GC box 1 and the YY1 site. (C) Histogram summarizing the data from three independent DpnI replication assays analyzed 48 h after VZV superinfection. Error bars indicate standard errors. Statistical significance was determined by a one-way analysis of variance followed by Tukey's post hoc test. Mut, mutant.
In the next series of experiments, triplicate DpnI assays were performed using wild-type pVO2 and pVO2 plasmids containing the site-specific mutations within GC box 1 and the YY1 site which ablated Sp1/Sp3 and YY1 binding, respectively. As shown in Fig. 7B and C, mutation of GC box 1 increased the level of origin-dependent replication two- to threefold, and this difference was statistically significant (P = 0.035). In contrast, the YY1 substitution mutation had no statistically significant effect (P = 0.162) on the efficiency of origin-dependent replication under these conditions. Similar results were obtained in experiments where cells were harvested 36 h after VZV superinfection. A statistically significant (P = 0.011) 2.5- to 3.5-fold increase in replication efficiency was observed with the GC box 1 mutation, whereas variation from wild-type levels observed with the YY1 mutation was not significant (P = 0.103).
DISCUSSION
We have examined the roles of specific cellular transcription factor binding sites on the efficiency of replication from the VZV DNA replication origin. This replication origin designated oriS, is present within the inverted repeat sequences surrounding the unique short (US) region of the VZV genome. The structure of the VZV oriS is distinctly different from that of the oriS of HSV-1. While both contain an AT-rich palindrome 45 to 46 bp in length, the arrangement of binding sites for the HSV and VZV OBPs differs significantly. Specifically, the HSV oriS contains experimentally verified OBP binding sites oriented in opposite directions upstream and downstream of the AT-rich region (12, 49, 52, 53). In contrast, the three experimentally verified OBP binding sites within the VZV oriS are all located upstream of the AT-rich palindrome (8, 50). Furthermore, Stow and Davison (48) showed that a plasmid containing only the proximal OBP binding site, the AT-rich palindrome, and 125 bp of sequence downstream from the AT-rich sequence were sufficient for origin-dependent replication in DpnI replication assays.
Our results show that the cellular transcription factors Sp1 and Sp3 and possibly Sp4 interact with a GC-rich site (GC box 1) downstream of the AT-rich palindrome. This is true despite the fact that all four Sp family members are expressed in MeWo cells. Two complexes reproducibly formed with GC box 1 were identified by EMSA analysis and antibody supershifts, with one being specific for Sp1 and the other specific for Sp3. Partial deletion and site-specific mutation of the GC box 1 abolished formation of both complexes, providing further proof of specific binding. No difference in complex formation was observed using infected and uninfected cell extracts, suggesting that upon infection, no additional viral or cellular proteins were recruited to these complexes. Sp1 and Sp3 belong to the growing family of Sp/XKLF cellular transcription factors of which Sp1 is the prototype. Some 23 members of this family have been identified in the human genome based on the presence of a combination of three conserved Cys2His2 zinc fingers that form a DNA recognition motif (7). However, only Sp1, Sp2, Sp3, and Sp4 have been extensively characterized as to their tissue specificity and role(s) in transcription (7, 28, 42). Sp1 through Sp4 all show a modular structure with activation and, in some cases inhibitory, domains present in their N-terminal regions and the characteristic set of zinc fingers present near their C termini. This similar modular structure has resulted in their being designated a subgroup of the larger Sp/XKLF family (7). Sp1 and Sp3 are believed to be ubiquitously expressed in all tissues, and both are capable of activating expression of mammalian genes via recognition of GC- and GT-rich motifs (28, 42). However, the interplay between Sp1 and Sp3 is complex, with Sp3 sometimes acting as a repressor of Sp1 (28, 30). Sp2 lacks one of the N-terminal activation domains and binds to a site different from that of Sp1, Sp3, and Sp4 (33, 42). Sp2 is generally associated with repression of genes activated by the other three members of the major Sp subfamily. Expression of Sp4 has been detected in several cell lines (17, 23). However, in mammals, Sp4 is predominantly expressed in tissues of neuronal origin, while the expression and activity of Sp1 in those tissues are downregulated (27, 31, 57).
Sp1/Sp3 sites have been identified in the replication origins of several DNA viruses, including HPV, SV40, EBV, and HSV-1 (5, 13, 15, 16, 24, 25, 37). The presence of the Sp1/Sp3 sites has been associated with an enhancement of origin-dependent DNA replication. Deletion or mutation of consensus GC boxes within the HPV and SV40 genomes resulted in decreases in DNA replication efficiency. The mechanism of how Sp1 enhances replication in these cases is unknown. However, at least for the case of SV40, the Sp1 site in question does not appear to be involved in transcription of the viral genome (13, 16). Sp1 and possibly other Sp family members also influence origin-dependent EBV DNA replication at the cis-acting element oriLyt (5, 15). This element consists of two essential domains designated the upstream and downstream components. The upstream component contains DNA binding motifs for the EBV transcriptional activator BZLF1. The downstream component is known to be the binding site of cellular transcription factors ZBP-89 and Sp1, which stimulate DNA replication. This stimulation is believed to result from the recruitment of viral DNA replication proteins, since direct interaction of Sp1 and ZBP-89 with the viral DNA polymerase and its processivity factor was demonstrated in protein binding assays (5). The HSV-1 genome contains two oriS core-adjacent regulatory (Oscar) elements, OscarL and OscarR, at the base of the oriS palindrome. Mutation of either element reduced oriS-dependent DNA replication by 60 to 70%, and disruption of both elements reduced replication by 90%. OscarL contains a consensus binding site for the transcription factor Sp1. EMSA and supershift experiments using antibodies directed against the Sp family members of transcription factors demonstrated the presence of Sp1 and Sp3, but not Sp2 or Sp4, in the protein-DNA complexes formed at OscarL (37).
In contrast to our results with GC box 1, we found no evidence of interaction of any of the Sp family members with the second GC-rich sequence (GC box 2), which was also thought to be a possible Sp1 binding site. Instead, the cellular factor YY1 was found to bind to a predicted site overlapping GC box 2. These results represents the first experimentally verified occurrence of YY1 binding to a VZV DNA sequence. YY1 is a 414-amino-acid zinc finger protein that is capable of both repression and activation of cellular genes and transcription in several virus systems, including adeno-associated virus, HPV, parvovirus B19, HSV-1, and murine leukemia virus (26, 46). YY1 has been shown to repress HPV ori-dependent DNA replication in a cell-free replication system. This repression, however, does not require the presence of a YY1 consensus binding site or interaction of YY1 with HPV DNA. Rather, the YY1-mediated repression of HPV DNA replication is believed to be due to direct interaction between YY1 and the HPV E2 protein, a transcription factor that plays important roles in the assembly of the initiation complex at the HPV origin (25).
Our results from DpnI replication assays show that a point mutation of GC box 1 that ablated the formation of the two complexes observed in EMSAs also resulted in a two- to threefold increase in replication efficiency compared to that observed with the wild-type virus. These findings concerning the function of the Sp1/Sp3 binding site within the VZV oriS indicate that interaction of Sp1 and Sp3 with the wild-type GC box 1 sequence results in a suppression of origin-dependent replication. This is in direct contrast to the function of such sites in the EBV and HSV-1 origins and represents a novel finding for VZV. These results also represent the first documentation of a role for Sp3 in the VZV replication cycle. Since these data were obtained using a plasmid containing only the VZV oriS, it will, in the future, be important to correlate these findings with the infectious phenotype of viruses carrying the GC box 1 mutation at the native sites within the VZV genome.
The role or roles played by YY1 in VZV infection are unknown; thus, the identification of a bona fide YY1 binding site within the oriS downstream region site afforded the potential to identify a VZV-specific function for this ubiquitous cellular factor. YY1 has been shown to be capable of direct interaction with Sp1 (24, 45), raising the possibility that bridging could occur between Sp1 and YY1 bound at their respective sites in the downstream region of the VZV oriS. Mutation of the YY1 site with concomitant loss of YY1 binding, however, resulted in no statistically significant effect, eliminating the possibility that a potential Sp1/YY1 bridging interaction might influence replication efficiency under our experimental conditions. This does not eliminate the possibility that YY1 plays an important role in VZV infection. YY1 could influence cellular functions vital for VZV lytic replication or interact functionally with IE62 or other VZV transactivators, such as the IE4 and ORF61 proteins (18, 34, 35) at specific viral promoters. Alternatively, YY1 could influence origin-dependent DNA replication in primary cells or in specific tissues infected by VZV in the human host. As an example, YY1 has been shown to act as a negative regulator of the EBV BRLF1 promoter in both HeLa and Raji cells, but the level of suppression was much greater in HeLa cells (56).
The data gathered on the ability of Sp1 and Sp3 to bind to the GC box 1 site in the downstream region of oriS and the increased oriS-dependent replication efficiency observed with mutations that ablate that binding led to the following model for the role of the GC box 1 sequence analyzed during the course of this work (Fig. 8). At times immediately after infection, endogenous Sp1 and Sp3 bind to the GC box 1 site. Once sufficient IE62 is synthesized, the most active phase of viral gene expression ensues, and at this stage Sp1 and Sp3 exchange off this site and interact physically and functionally with the major VZV transactivator IE62 to efficiently express the viral genome. Previous work by Peng et al. (39) showed that as much as 20 to 25% of total Sp1 is associated with IE62 in VZV-infected cells. Additional support for this possibility comes from experiments showing that ectopic expression of Sp1 and Sp3 but not YY1 enhance IE62-mediated transactivation in transient-transfection assays (data not shown). The resulting decrease of Sp1 and Sp3 at GC box 1 would, based on our data, allow an increase in origin-dependent DNA replication. During the late phase of the VZV life cycle, IE62 is excluded, to a large extent, from the nucleus (21, 22), and Sp1 and Sp3 could potentially rebind to this site and aid in decreasing DNA replication during this phase of infection.
FIG. 8.

Model summarizing a possible mechanism of Sp1/Sp3-dependent modulation of VZV DNA origin-dependent replication during the VZV life cycle. In this model, Sp1 and Sp3 cycle on and off the consensus GGGCGGG site within GC box 1 in response to requirements for their presence for IE62-mediated transcription at promoter sites within the VZV genome. Thus, their presence during the early phase of the virus life cycle would be minimal, resulting in increased DNA replication which also peaks at this phase. IE, immediate early.
This model has broader implications in terms of the multifaceted nature of VZV pathogenesis. Our data show that in MeWo cells, VZV infection reduces expression of the Sp4 protein to a modest but reproducible extent and that interaction of Sp4 with the GC box 1 element is less stable than that observed with Sp1 and Sp3. As indicated above, Sp4 is primarily and dominantly expressed in neuronal tissue and is capable of binding to the consensus Sp1 site, whereas Sp1 expression is downregulated. Therefore, it is possible that, under certain conditions, Sp4 may be able to compete with and displace Sp1 at the VZV GC box 1 site as has been reported in the case of the human ADH5/FDH promoter (23). If Sp4, like Sp1 enhances IE62-mediated transactivation, the combination of normally decreased expression of Sp1 in neurons and reduction of Sp4 levels due to infection could lead to a reduction of VZV gene expression upon initial neuronal infection and a general cessation of viral replication. Alternatively, if Sp4 does not synergize with IE62 but binds to the GC box 1 element, it could, even at reduced levels and possibly in conjunction with neuronal factors, act to suppress VZV DNA replication. Both of these scenarios would likely increase the chances for a switch from lytic to latent infection. Conversely, low levels of Sp1 and decreased levels of Sp4 during reactivation could lead to increased efficiency of viral DNA replication. Thus, the cis-acting GC box 1 element within the downstream region of VZV oriS and the interplay of various levels of Sp1 and Sp4 could potentially be involved in the establishment and/or reactivation of VZV latency.
Acknowledgments
This work was supported by NIH grants AI18449 and AI36884. M.I.K. was supported in part by a scholarship from the Egyptian Ministry of Higher Education.
Footnotes
Published ahead of print on 24 September 2008.
REFERENCES
- 1.Arvin, A. 2001. Varicella-zoster virus, p. 2731-2768. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Arvin, A. M. 1996. Varicella-zoster virus. Clin. Microbiol. Rev. 9361-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balliet, J. W., J. C. Min, M. S. Cabatingan, and P. A. Schaffer. 2005. Site-directed mutagenesis of large DNA palindromes: construction and in vitro characterization of herpes simplex virus type 1 mutants containing point mutations that eliminate the oriL or oriS initiation function. J. Virol. 7912783-12797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balliet, J. W., and P. A. Schaffer. 2006. Point mutations in herpes simplex virus type 1 oriL, but not in oriS, reduce pathogenesis during acute infection of mice and impair reactivation from latency. J. Virol. 80440-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baumann, M., R. Feederle, E. Kremmer, and W. Hammerschmidt. 1999. Cellular transcription factors recruit viral replication proteins to activate the Epstein-Barr virus origin of lytic DNA replication, oriLyt. EMBO J. 186095-6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berarducci, B., M. Sommer, L. Zerboni, J. Rajamani, and A. Arvin. 2007. Cellular and viral factors regulate the varicella-zoster virus gE promoter during viral replication. J. Virol. 8110258-10267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouwman, P., and S. Philipsen. 2002. Regulation of the activity of Sp1-related transcription factors. Mol. Cell. Endocrinol. 19527-38. [DOI] [PubMed] [Google Scholar]
- 8.Chen, D., and P. D. Olivo. 1994. Expression of the variella-zoster virus origin-binding protein and analysis of its site-specific DNA-binding properties. J. Virol. 683841-3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen, J. I., and S. E. Straus. 2001. Varicella-zoster virus and its replication, p. 2707-2730. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 10.Davison, A. J., and J. E. Scott. 1985. DNA sequence of the major inverted repeat in the varicella-zoster virus genome. J. Gen. Virol. 66207-220. [DOI] [PubMed] [Google Scholar]
- 11.Davison, A. J., and J. E. Scott. 1986. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 671759-1816. [DOI] [PubMed] [Google Scholar]
- 12.Deb, S., and M. Doelberg. 1988. A 67-base-pair segment from the Ori-S region of herpes simplex virus type 1 encodes origin function. J. Virol. 622516-2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demeret, C., M. Le Moal, M. Yaniv, and F. Thierry. 1995. Control of HPV 18 DNA replication by cellular and viral transcription factors. Nucleic Acids Res. 234777-4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dynan, W. S., and R. Tjian. 1983. The promoter-specific transcription factor Sp1 binds to upstream sequences in the SV40 early promoter. Cell 3579-87. [DOI] [PubMed] [Google Scholar]
- 15.Gruffat, H., O. Renner, D. Pich, and W. Hammerschmidt. 1995. Cellular proteins bind to the downstream component of the lytic origin of DNA replication of Epstein-Barr virus. J. Virol. 691878-1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo, Z. S., and M. L. DePamphilis. 1992. Specific transcription factors stimulate simian virus 40 and polyomavirus origins of DNA replication. Mol. Cell. Biol. 122514-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hagen, G., S. Muller, M. Beato, and G. Suske. 1992. Cloning by recognition site screening of two novel GT box binding proteins: a family of Sp1 related genes. Nucleic Acids Res. 205519-5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inchauspe, G., S. Nagpal, and J. M. Ostrove. 1989. Mapping of two varicella-zoster virus encoded genes that activate the expression of viral early and late genes. Virology 173700-709. [DOI] [PubMed] [Google Scholar]
- 19.Ito, H., M. H. Sommer, L. Zerboni, H. He, D. Boucaud, J. Hay, W. Ruyechan, and A. M. Arvin. 2003. Promoter sequences of varicella-zoster virus glycoprotein I targeted by cellular transactivating factors Sp1 and USF determine virulence in skin and T cells in SCIDhu mice in vivo. J. Virol. 77489-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kennett, S. B., A. J. Uvadia, and J. M. Horowitz. 1997. Sp3 encodes multiple proteins that differ in their capacity to stimulate or repress transcription. Nucleic Acids Res. 253110-3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinchington, P. R., K. Fite, and S. E. Turse. 2000. Nuclear accumulation of IE62, the varicella-zoster virus (VZV) major transcriptional regulatory protein, is inhibited by phosphorylation mediated by the VZV open reading frame 66 protein kinase. J. Virol. 742265-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinchington, P. R., and S. E. Turse. 1998. Regulated nuclear localization of the varicella-zoster virus major regulatory protein, IE62. J. Infect. Dis. 178S16-S21. [DOI] [PubMed] [Google Scholar]
- 23.Kwon, H.-S., M.-S. Kim, H. J. Edenberg, and M.-W. Hur. 1999. Sp3 and Sp4 can repress transcription by competing with Sp1 for the core cis-elements on the human ADH5/FDH minimal promoter. J. Biol. Chem. 27420-28. [DOI] [PubMed] [Google Scholar]
- 24.Lee, J. S., K. M. Galvin, and Y. Shi. 1993. Evidence for physical interaction between the zinc-finger transcription factors YY1 and Sp1. Proc. Natl. Acad. Sci. USA 906145-6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee, K.-Y., T. R. Broker, and L. T. Chow. 1998. Transcription factor YY1 represses cell-free replication from human papillomavirus origins. J. Virol. 724911-4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee, T.-C., Y. Shi, and R. J. Schwartz. 1992. Displacement of BrdUrd-induced YY1 by serum response factor activates skeletal α-actin transcription in embryonic myoblasts. Proc. Natl. Acad. Sci. USA 899814-9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lerner, L. E., G.-H. Peng, Y. E. Gribanova, S. Chen, and D. B. Farber. 2005. Sp4 is expressed in retinal neurons, activates transcription of photoreceptor-specific genes, and synergizes with Crx. J. Biol. Chem. 28020642-20650. [DOI] [PubMed] [Google Scholar]
- 28.Li, L., S. He, J. M. Sun, and J. R. Davie. 2004. Gene regulation by Sp1 and Sp3. Biochem. Cell Biol. 82460-471. [DOI] [PubMed] [Google Scholar]
- 29.Lynch, J. M., T. K. Kenyon, C. Grose, J. Hay, and W. T. Ruyechan. 2002. Physical and functional interaction between the varicella zoster virus IE63 and IE62 proteins. Virology 30271-82. [DOI] [PubMed] [Google Scholar]
- 30.Majello, B., P. De Luca, G. Hagen, G. Suske, and L. Lania. 1994. Different members of the Sp1 multigene family exert opposite transcriptional regulation of the long terminal repeat of HIV-1. Nucleic Acids Res. 224914-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mao, X., S.-H. Yang, J. W. Simpkins, and S. W. Barger. 2007. Glutamate receptor activation evokes calpain-mediated degradation of Sp3 and Sp4, the prominent Sp-family transcription factors in neurons. J. Neurochem. 1001300-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meier, J. L., and S. E. Straus. 1993. Varicella-zoster virus DNA polymerase and major DNA-binding protein genes have overlapping divergent promoters. J. Virol. 677573-7581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moorefield, K. S., S. J. Fry, and J. M. Horowitz. 2004. Sp2 DNA binding activity and trans-activation are negatively regulated in mammalian cells. J. Biol. Chem. 27913911-13924. [DOI] [PubMed] [Google Scholar]
- 34.Moriuchi, H., M. Moriuchi, S. E. Straus, and J. I. Cohen. 1993. Varicella-zoster virus (VZV) open reading frame 61 protein transactivates VZV gene promoters and enhances the infectivity of VZV DNA. J. Virol. 674290-4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moriuchi, H., M. Moriuchi, S. E. Straus, and J. I. Cohen. 1994. Varicella-zoster virus open reading frame 4 protein is functionally distinct from and does not complement its herpes simplex virus type 1 homolog, ICP27. J. Virol. 681987-1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Narayanan, A., W. T. Ruyechan, and T. M. Kristie. 2007. The coactivator host cell factor-1 mediates Set1 and MLL H3K4 trimethylation at herpesvirus immediate early promoters for initiation of infection. Proc. Natl. Acad. Sci. USA 10410835-10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen-Huynh, A. T., and P. A. Schaffer. 1998. Cellular transcription factors enhance herpes simplex virus type 1 oriS-dependent DNA replication. J. Virol. 723635-3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pajunk, H. S., C. May, H. Pfister, and P. G. Fuchs. 1997. Regulatory interactions of transcription factor YY1 with control sequences of the E6 promoter of human papillomavirus type 8. J. Gen. Virol. 783287-3295. [DOI] [PubMed] [Google Scholar]
- 39.Peng, H., H. He, J. Hay, and W. T. Ruyechan. 2003. Interaction between the varicella zoster virus IE62 major transactivator and cellular transcription factor Sp1. J. Biol. Chem. 27838068-38075. [DOI] [PubMed] [Google Scholar]
- 40.Reference deleted.
- 41.Reference deleted.
- 42.Philipsen, S., and G. Suske. 1999. A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res. 272991-3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rahaus, M., and M. H. Wolff. 1999. Influence of different cellular transcription factors on the regulation of varicella-zoster virus glycoproteins E (gE) and I (gI) UTR's activity. Virus Res. 6277-88. [DOI] [PubMed] [Google Scholar]
- 44.Ruyechan, W. T., H. Peng, M. Yang, and J. Hay. 2003. Cellular factors and IE62 activation of VZV promoters. J. Med. Virol. 70S90-S94. [DOI] [PubMed] [Google Scholar]
- 45.Seto, E., B. Lewis, and T. Shenk. 1993. Interaction between transcription factors Sp1 and YY1. Nature 365462-464. [DOI] [PubMed] [Google Scholar]
- 46.Shi, Y., J.-S. Lee, and K. M. Galvin. 1997. Everything you have ever wanted to know about Yin Yang 1. Biochim. Biophys. Acta 1332F49-F66. [DOI] [PubMed] [Google Scholar]
- 47.Spengler, M. L., W. T. Ruyechan, and J. Hay. 2000. Physical interaction between two varicella zoster virus gene regulatory proteins, IE4 and IE62. Virology 272375-381. [DOI] [PubMed] [Google Scholar]
- 48.Stow, N. D., and A. J. Davison. 1986. Identification of a varicella-zoster virus origin of DNA replication and its activation by herpes simplex virus type 1 gene products. J. Gen. Virol. 671613-1623. [DOI] [PubMed] [Google Scholar]
- 49.Stow, N. D., and E. C. McMonagle. 1983. Characterization of the TRS/IRS origin of DNA replication of herpes simplex virus type 1. Virology 130427-438. [DOI] [PubMed] [Google Scholar]
- 50.Stow, N. D., H. M. Weir, and E. C. Stow. 1990. Analysis of the binding sites for the varicella-zoster virus gene 51 product within the viral origin of DNA replication. Virology 177570-577. [DOI] [PubMed] [Google Scholar]
- 51.Reference deleted.
- 52.Weir, H. M., and N. D. Stow. 1990. Two binding sites for the herpes simplex virus type 1 UL9 protein are required for efficient activity of the oriS replication origin. J. Gen. Virol. 711379-1385. [DOI] [PubMed] [Google Scholar]
- 53.Weller, S. K., A. Spadaro, J. E. Schaffer, A. W. Murray, A. M. Maxam, and P. A. Schaffer. 1985. Cloning, sequencing, and functional analysis of oriL, a herpes simplex virus type 1 origin of DNA synthesis. Mol. Cell. Biol. 5930-942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang, M., J. Hay, and W. T. Ruyechan. 2004. The DNA element controlling expression of the varicella-zoster virus open reading frame 28 and 29 genes consists of two divergent unidirectional promoters which have a common USF site. J. Virol. 7810939-10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang, M., H. Peng, J. Hay, and W. T. Ruyechan. 2006. Promoter activation by the varicella-zoster virus major transactivator IE62 and the cellular transcription factor USF. J. Virol. 807339-7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zalani, S., A. Coppage, E. Holley-Guthrie, and S. Kenney. 1997. The cellular YY1 transcription factor binds a cis-acting, negatively regulating element in the Epstein-Barr virus BRLF1 promoter. J. Virol. 713268-3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou, X., J. M. Long, M. A. Geyer, E. Masliah, J. R. Kelsoe, A. Wynshaw-Boris, and K. R. Chien. 2005. Reduced expression of the Sp4 gene in mice causes deficits in sensorimotor gating and memory associated with hippocampal vacuolization. Mol. Psychiatry 10393-406. [DOI] [PubMed] [Google Scholar]