

Abstract
Lens epithelium-derived growth factor (LEDGF)/p75 functions as a bimodal tether during lentiviral DNA integration: its C-terminal integrase-binding domain interacts with the viral preintegration complex, whereas the N-terminal PWWP domain can bind to cellular chromatin. The molecular basis for the integrase-LEDGF/p75 interaction is understood, while the mechanism of chromatin binding is unknown. The PWWP domain is homologous to other protein interaction modules that together comprise the Tudor clan. Based on primary amino acid sequence and three-dimensional structural similarities, 24 residues of the LEDGF/p75 PWWP domain were mutagenized to garner essential details of its function during human immunodeficiency virus type 1 (HIV-1) infection. Mutating either Trp-21 or Ala-51, which line the inner wall of a hydrophobic cavity that is common to Tudor clan members, disrupts chromatin binding and virus infectivity. Consistent with a role for chromatin-associated LEDGF/p75 in stimulating integrase activity during infection, recombinant W21A protein is preferentially defective for enhancing integration into chromatinized target DNA in vitro. The A51P mutation corresponds to the S270P change in DNA methyltransferase 3B that causes human immunodeficiency, centromeric instability, and facial anomaly syndrome, revealing a critical role for this amino acid position in the chromatin binding functions of varied PWWP domains. Our results furthermore highlight the requirement for a conserved Glu in the hydrophobic core that mediates interactions between other Tudor clan members and their substrates. This initial systematic mutagenesis of a PWWP domain identifies amino acid residues critical for chromatin binding function and the consequences of their changes on HIV-1 integration and infection.
Integration, catalyzed by the viral integrase protein, is an essential step in the replication cycles of all retroviruses. Integration into cellular chromatin provides an optimal environment for gene expression and ensures that the viral genetic material is inherited by daughter cells upon division. Integration proceeds via the following steps: (i) integrase binding to the cDNA end regions that are synthesized during reverse transcription; (ii) hydrolysis adjacent to invariant CA sequences near both 3′ ends (3′ processing); (iii) transfer of the reactive 3′-OH ends to the 5′-phosphates of a double-stranded cut in cellular chromatin (DNA strand transfer); and (iv) repair of the resulting DNA recombination intermediate, which is likely accomplished by host cell enzymes. (See reference 61 for a detailed overview of retroviral integration.)
Although all retroviruses rely on integrase 3′ processing and DNA strand transfer activities, significant differences exist in the way the various viral genera select their chromosomal integration sites. These differences manifest themselves at the level of local DNA sequence (20, 68) and genetic structure (reviewed in reference 2). Lentiviruses, for example, favor integration into active transcription units, targeting genes fairly equally along their lengths (37, 50). Moloney murine leukemia virus, a γ-retrovirus, more modestly favors genes and transcriptional activity but in stark contrast to lentiviruses displays a marked preference for promoter regions and associated CpG islands (19, 37, 67). Simian foamy virus, a spumaretrovirus, slightly disfavors genes though promoters and CpG islands are targeted significantly over random (43, 60). Other profiled genera, including α-, β-, and δ-retroviruses, display less overall preferences for genes, promoter regions, and CpG islands than their lenti-, γ-, and spumaviral cousins (12, 16, 36, 37, 40). Analyses of Moloney murine leukemia virus/HIV-1 chimera viruses revealed that the cognate integrase protein principally determines local DNA sequence and genetic structure specificities during integration (25). Cell factor(s) that may help guide preintegration complexes (PICs) to promoter regions for integration are unknown. Recent findings by contrast clarify that lens epithelium-derived growth factor (LEDGF)/p75 is a key lentivirus-specific gene targeting factor (9, 21, 34, 55).
LEDGF/p75 behaves as a bifunctional molecular tether during integration (see references 14 and 46 for recent reviews). An α-helical integrase-binding domain (IBD) (8) that encompasses amino acids 347 to 429 of the 530 residue human protein (6, 65) binds directly to integrase, whereas two regions within its N-terminal half, the PWWP domain (residues 1 to 92) and two copies of the AT-hook DNA-binding motif, mediate binding to chromatin (29, 62) (Fig. 1A). Accordingly, the expression of integrase- or chromatin binding-defective mutants in cells depleted for endogenous LEDGF/p75 by RNA interference (27) or gene knockout (55) fails to confer sensitivity to human immunodeficiency virus type 1 (HIV-1) infection. The molecular basis for the interaction between LEDGF/p75 and HIV-1 integrase is well understood (3, 5, 8, 13, 21, 49), whereas the mechanism of LEDGF/p75 chromatin binding function is for the most part unknown. Recent results have revealed that the PWWP domain plays a more important role than the AT-hook motifs during HIV-1 infection (55) and chromatin-dependent integration in vitro (1).
FIG. 1.

LEDGF/p75 schematic and PWWP domain sequence alignments. (A) LEDGF/p75 domain organization and previously described mutants used in the present study. Binding of recombinant protein to DNA in vitro is predominantly mediated by the NLS and dual copy of the AT-hook (ATh) DNA-binding motif, with the PWWP domain (gray shade) contributing a minor role (1, 56, 62). Chromatin binding is largely mediated by the PWWP domain and AT-hooks, with charged regions (CRs) 1 to 3 making secondary contributions (29, 62). MutL1 harbors 12 missense mutations: 6 in the NLS and 3 in each copy of the AT-hook (1, 62). (B) Amino acid alignment of the N-terminal PWWP domains from select human HRP family members. Secondary structural elements elucidated from the solution structure of the HDGF domain (31) are shown above the alignment. Residues targeted for mutagenesis are noted below by triangles; numbers refer to LEDGF/p75 positions. The coloring scheme denotes the following levels of sequence conservation: black, amino acid identity at three or more positions; dark gray, amino acid similarity at all four positions; light gray, chemically similar side chains at three of four positions. (C) Alignment of DNMT3B and LEDGF/p75 PWWP domains. Secondary elements elucidated from the X-ray crystal structure of the murine DNMT3B domain (47) are shown above the corresponding human sequence, whereas HDGF structural elements are drawn below the LEDGF/p75 sequence. The substitution of Pro for Ser-270 (boxed) in DNMT3B is associated with ICF syndrome (53). Black denotes identical amino acids, whereas gray indicates homologous side chains as defined by the following chemical groupings (51): Gly, Ala, Ser, Thr, and Pro; Met, Val, Leu, and Ile; Phe, Tyr, and Trp; Asp, Glu, Asn, and Gln; Lys, Arg, and His; Cys. PR, proline-rich.
The PWWP domain is a ca. 90- to 135-amino acid sequence module present in approximately 60 eukaryotic proteins, many of which, like LEDGF/p75, display an affinity for chromatin (39, 58). Based on amino acid sequence and three-dimensional (3D) structural similarities, the PWWP domain is related to a number of other protein interaction domains that include Tudor, Agenet, malignant brain tumor, and Chromo, which together comprise the Tudor domain “Royal Family” or clan (17, 35). Some of these modules, for example, the heterochromatin protein 1 (HP1) chromodomain (23, 41), interact specifically with posttranslationally modified histone tails. The LEDGF/p75 domain displays affinity for humanized chromatin templates in vitro (1), and the homologous zebra fish Brpf1 domain was recently shown to bind unmodified core histones H2A and H2B (24). To investigate the mechanism of LEDGF/p75 function during integration, we have delineated PWWP domain amino acid residues critical for chromatin binding and HIV-1 infection.
MATERIALS AND METHODS
Plasmids.
Animal cell LEDGF/p75 expression vectors were based in pIRES2-eGFP, and proteins were expressed untagged or as fusions to a C-terminal hemagglutinin (HA) tag as described previously (55). Bacterial expression plasmids were based in pFT-1-LEDGF (63), pFT-1-MutL1 (carrying six missense mutations in the LEDGF/p75 nuclear localization signal [NLS] and three in each AT-hook; Fig. 1A) (62), or pGEX-4T-1-LEDGF1-100 (1). PWWP domain missense mutations were incorporated via PCR-directed mutagenesis as described previously (15, 26), and the resulting coding regions were verified by sequencing. Plasmids pCG-VSV-G (54), pNLX.Luc(R-), and pN/N.Luc(R-) (30) were used to construct single round reporter viruses.
Cells, viruses, and infections.
Human and mouse cell lines were propagated in Dulbecco modified Eagle medium containing 10% fetal bovine serum, 100 IU of penicillin/ml, and 100 μg of streptomycin/ml. Mouse embryo fibroblasts (MEFs) isolated at 13.5 days postconception were transformed via simian virus 40 large T antigen expression as described previously (55). A variety of control and LEDGF/p75 knockout MEFs were used in the course of these studies. The majority of infectivity measurements were conducted with E6(−/−) knockout cells derived from E6(f/f) control MEFs ex vivo via bacteriophage P1 Cre protein expression (55). E1(f/+) and E2(−/−) littermate control and knockout MEFs, respectively, were prepared from embryos following two rounds of mouse mating that began with f/f and Sox2Cre animals as described previously (55). This same mating scheme was used to generate the following control and knockout cell sets from two independent pregnancies: E4(f/+) and E5(−/−); E5(f/+), E3(−/−), and E6(−/−). Heterozygous (+/−) mice lacking Cre were generated by mating Sox2Cre+/− to C57BL/6 animals; littermate matched E17(+/+) and E16(−/−) knockout cells were derived from embryos obtained by intercrossing these heterozygotes.
Single-round HIV-Luc carrying wild-type or D64N/D116N active-site mutant integrase was pseudotyped with vesicular stomatitis virus G (VSV-G) envelope glycoprotein by transient transfection as described previously (54). Viruses whose titers were determined using a 32P-based assay for reverse transcriptase (RT) activity were treated for 1 h at 37°C with 40 U of Turbo DNase (Ambion, Austin, TX)/ml. MEFs (4 × 106) were transfected with 10 μg of pIRES2-eGFP-based vectors expressing untagged LEDGF/p75 using Nucleofector I program T-20 and MEF Nucleofector Solution 1 (Amaxa, Inc., Gaithersburg, MD). Cells sorted by fluorescence-activated cell sorting (FACS) were lysed as described previously (63) for Western blotting or plated in 12-well trays at 4 × 104/well 10 h before infection. Protein concentrations in cell extracts were determined by using a Dc protein assay (Bio-Rad Laboratories, Hercules, CA), and 160 ng was analyzed by Western blotting with anti-LEDGF/p75 A300-847A (Bethyl Laboratories, Inc., Montgomery, TX) or anti-β-actin AC-15 (Sigma-Aldrich, St. Louis, MO) antibodies. MEFs were infected in duplicate with 106 RT cpm of HIV-Luc for 8 h. At 44 h postinfection, cells were processed for duplicate luciferase (Luc) assays. The results (quadruplicate Luc assays of two to four independent infection experiments) are expressed as relative light units (RLU) normalized per μg of total protein in cell extracts (54).
Epifluorescence microscopy.
HeLa TZM-bl cells (66) (4 × 106) were transfected with 10 μg of pIRES2-eGFP variants expressing HA-tagged LEDGF/p75 by using Lipofectamine 2000 (Invitrogen Corp., Carlsbad, CA), and FACS-sorted cells were either lysed as described above for Western blotting or seeded at 5 × 105/well in 12-well plates containing 18-mm coverslips (VWR International, West Chester, PA). The following day, cells washed with phosphate-buffered saline (PBS; Mediatech, Inc., Manassas, VA) were fixed in 4% paraformaldehyde for 10 min. After two washes with PBS, cells were permeabilized with 0.5% (vol/vol) Triton X-100 in PBS for 5 min. Cells blocked in 5% (vol/vol) normal donkey serum (NDS; Chemicon International, Inc., Temecula, CA) for 1 h were incubated for another hour in PBS containing 5% NDS-2% (vol/vol) anti-HA clone 3F10 antibody (Roche Applied Science, Indianapolis, IN). After two washes with PBS-5% NDS, cells were incubated with a 1:1,000 dilution of Alexa Fluor 594-conjugated donkey anti-rat immunoglobulin G (Invitrogen Corp.) for 1 h, followed by several washes with PBS. Samples mounted in Vectashield mounting medium containing DAPI (4′,6′-diamidino-2-phenylindole; Vector Laboratories, Inc., Burlingame, CA) were imaged by using a Zeiss 200M inverted epifluorescence microscope (Carl Zeiss MicroImaging, Inc., Thornwood, NY) equipped with SlideBook software (Intelligent Imaging Innovations, Inc., Denver, CO).
Cellular fractionation.
MEFs were transfected with HA-tagged LEDGF/p75 expression vectors as described above, whereas 293T cells (1.5 × 106/well of a six-well plate) were transfected with 4 μg of DNA by using Lipofectamine 2000. Cells were fractionated 24 h posttransfection by using the method described by Llano et al. (29) with slight modifications. Cells lysed for 15 min on ice in cold CSK I buffer (10 mM PIPES [pH 6.8], 100 mM NaCl, 1 mM EDTA, 300 mM sucrose, 1 mM MgCl2, 1 mM dithiothreitol) supplemented with 0.5% Triton X-100, protease inhibitors (Roche Complete Mini), and 1 mM phenylmethylsulfonyl fluoride were divided into two equal portions and centrifuged at 500 × g for 3 min at 4°C. The supernatants were combined to yield fraction S1, whereas one pellet yielded fraction P1 following solubilization in radioimmunoprecipitation assay (RIPA) buffer (150 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.5% [wt/vol] deoxycholate, 0.1% [wt/vol] sodium dodecyl sulfate, 1% [vol/vol] NP-40). The second pellet, resuspended in CSK II buffer (10 mM PIPES [pH 6.8], 50 mM NaCl, 300 mM sucrose, 6 mM MgCl2, 1 mM dithiothreitol), was treated with 4 U of Turbo DNase for 30 to 60 min followed by extraction with 250 mM NH2SO4 for 10 min at 25°C. After centrifugation at 19,000 × g for 3 min at 4°C, the S2 supernatant was removed from the P2 pellet, the latter of which was solubilized in RIPA buffer. The concentration of total protein in each fraction was determined by using a Dc protein assay kit, and 1 μg was analyzed by Western blotting with anti-HA 3F10 antibodies. The results were quantified by using a FluorChem FC2 imager (Alpha Innotech Corp., San Leandro, CA).
Expression and purification of recombinant proteins.
Wild-type and full-length LEDGF/p75 missense mutant proteins expressed from pFT-1-LEDGF were purified from soluble extracts of Escherichia coli cells as described previously (62, 63). Glutathione S-transferase (GST) fusions to the wild-type or mutated PWWP domains were expressed from pGEX-4T-1-LEDGF1-100 and purified from soluble E. coli extracts essentially as previously described (1). In brief, protein eluted from glutathione-Sepharose beads (GE Healthcare Bio-Sciences Corp., Piscataway, NJ) was further purified by using a Superdex 75 column (GE Healthcare). Two of the mutations, W21A and A51P, negatively impacted protein purity and yield. Fractionation of GST-PWWP/W21A and GST-PWWP/A51P on Hi-Trap heparin columns (GE Healthcare) prior to gel filtration chromatography increased the purity of these final preparations to the same level (>90% as assessed by Coomassie blue staining) as the other GST-PWWP proteins. His-tagged HIV-1 integrase was expressed and purified from a soluble E. coli extract as described previously (1).
In vitro integration and chromatin and DNA binding assays.
LEDGF/p75-dependent integration assays using naked target DNA or matched chromatinized templates were performed as described previously (1). Reaction product formation was quantified by real-time PCR as described therein. Radiolabeled DNA and polynucleosome (PN) binding to wild-type and mutant GST-PWWP domain fusion proteins were performed as described previously (1).
RESULTS
Mutagenesis strategy.
LEDGF/p75, expressed from the PSIP1 gene, is a member of the hepatoma-derived growth factor (HDGF)-related protein (HRP) family that also contains HDGF, HRP1, HRP2, HRP3, and LEDGF/p52 (an alternative mRNA splice variant of PSIP1). The PWWP domain is the most conserved region among these proteins (22) and sequence alignments of protein orthologs (6, 39, 58) and paralogs (22) (Fig. 1B), as well as the 3D structures of the HRP3 (39) and HDGF (31) (Fig. 2) domains, were consulted for mutagenesis. The heart of the PWWP domain is a five-stranded antiparallel β barrel with a 310 helix preceding the fifth strand (Fig. 1B and 2), followed by various numbers of α helices (31, 39, 47, 57). The barrel presents a solvent exposed hydrophobic cavity that is the discerning structural feature of Tudor clan members (35).
FIG. 2.

3D representations of targeted LEDGF/p75 residues. (A) The solution structure of the HDGF PWWP domain (Protein Data Base accession code 2B8A) is shown highlighting the residues that form its hydrophobic cavity (31, 39). (B) Cluster of basic Lys and Arg residues implicated in HDGF DNA binding (31). His-20 of the PHWP motif locates in this general vicinity; the extended N-terminal tail that harbors Arg-3 is not shown. (C) Remaining targeted residues. Ile-11, Pro-19, Val-28, Leu-40, Pro-41, and Ile-42 contribute to the hydrophobicity of the central cavity. Leu-10, Ile-11, Val-28, and Ile-42 are conserved among Tudor clan members (35), whereas Pro-19 is conserved among PWWP domain orthologs (39, 58). Note that Ile-11, Val-28, Leu-40, and Pro-41 are Val, Met, Tyr, and Gln in HDGF, respectively (Fig. 1B); the mutagenesis option in PyMOL (10) was used to model LEDGF/p75 residues at these positions. Residue numbers in panels A to C refer to LEDGF/p75; dark and light side-chain shadings indicate residues more toward the back and front of the page, respectively. Visible secondary structural elements are indicated as in Fig. 1B.
Lys-14, Trp-21, Pro-22, and Phe-45 are the most conserved residues among PWWP domain orthologs (39). Because Pro-22 and Phe-45 are both solvent inaccessible (31, 39), they were not targeted. In addition to Trp-21, LEDGF/p75 residues Met-15 and Tyr-18 from within the β-β arch, Phe-43 from β3, Thr-47 from the β3-β4 loop, and Glu-49 and Ala-51 from β4 (Fig. 1B) line the putative ligand-binding cavity (31, 39) (Fig. 2A). Each of these residues, with the exception of Ala-51, was changed to Ala and/or Glu to test their role in LEDGF/p75 PWWP domain function. The substitution of Ser-270 by Pro within the PWWP domain of DNA methyltransferase (DNMT) 3B is associated with immunodeficiency, centromeric instability, and facial anomaly (ICF) syndrome in humans (53). Since Ser-270 in DNMT3B is analogous to Ala-51 in LEDGF/p75 (boxed in Fig. 1C), Pro was tested in place of Ala-51.
The binding of recombinant LEDGF/p75 protein to DNA in vitro is predominantly mediated by conserved Arg and Lys residues within the NLS and AT-hook motifs (Fig. 1A), with a more minor contribution from the PWWP domain (1, 56, 62). HDGF also binds DNA in vitro (31, 69) but, unlike LEDGF/p75, this protein lacks discernible AT-hook DNA-binding motifs (38). Instead, a positively charged face of the HDGF PWWP domain, comprised of LEDGF/p75 analogous residues Arg-3, Lys-14, Lys-16, Lys-56, Lys-67, Lys-70, Lys-73, Arg-74, and Lys-75 (Fig. 2B), has been implicated in DNA binding (31). These residues were therefore targeted individually or in groups to assess the role of the presumed positively charged LEDGF/p75 face in HIV-1 infection and integration. Other solvent-exposed residues, including Leu-10, Ile-11, Pro-19, His-20, Val-28, Leu-40, Pro-41, and Ile-42, were targeted due to their relative degrees of sequence conservation or proximities to the presumed DNA binding face (Fig. 2B) or hydrophobic cavity (Fig. 2C). Table 1 lists the 24 targeted residues, the reasons for their selection, and resulting 37 novel mutant proteins that were tested for their abilities to support HIV-1 function.
TABLE 1.
Targeted residues and resultant mutant activities
| Residuea | Reason(s) for selectionb | Mutation(s) | HIV-1 infectivity (mean % ± SE)c |
|---|---|---|---|
| Arg-3 | i | R3A | 80.6 ± 16.2 |
| R3A/K14A/K16A | -4.3 ± 5.7 | ||
| Leu-10 | ii | L10A | 50.1 ± 3.9 |
| Ile-11 | ii | I11A | 68.9 ± 14.8 |
| L10A/I11A | 8.0 ± 2.3 | ||
| Lys-14 | i, ii | K14A | 57.9 ± 0.9 |
| K14E | 16.6 ± 1.6 | ||
| Met-15 | ii, iii | M15E | 34.7 ± 0.1 |
| Lys-16 | i | K16A | 91.3 ± 0.2 |
| K16E | 42.0 ± 15.5 | ||
| K14A/K16A | -3.6 ± 6.3 | ||
| Tyr-18 | ii, iii | Y18A | 96.6 ± 1.3 |
| M15E/Y18A | 28.0 ± 0.6 | ||
| Pro-19 | ii | P19A | 107 ± 13.5 |
| P19E | 20.0 ± 6.9 | ||
| His-20 | iv | H20A | 94.8 ± 13.3 |
| H20E | 80.8 ± 9.8 | ||
| Trp-21 | ii, iii | W21A | -4.8 ± 4.7 |
| W21E | -3.4 ± 3.3 | ||
| M15E/Y18A/W21A | -3.1 ± 6.7 | ||
| Val-28 | ii | V28E | 103 ± 12.0 |
| Leu-40 | v | L40E | 42.9 ± 12.1 |
| Pro-41 | v | P41E | 89.0 ± 7.2 |
| L40E/P41E | 6.4 ± 0.3 | ||
| Ile-42 | ii | I42A | 19.4 ± 1.7 |
| Phe-43 | ii, iii | F43A | 41.3 ± 5.4 |
| I42A/F43A | -0.3 ± 3.0 | ||
| Thr-47 | iii | T47E | 8.8 ± 1.6 |
| Glu-49 | iii | E49A | 16.3 ± 0.3 |
| T47E/E49A | 42.3 ± 13.1 | ||
| Ala-51 | iii, vi | A51P | 3.5 ± 3.4 |
| T47E/E49A/A51P | -1.1 ± 4.4 | ||
| Lys-56 | i | K56A | 82.1 ± 9.1 |
| Lys-67 | i | K67A/K70A | |
| Lys-70 | i | K67A/K70A | 69.1 ± 6.0 |
| Lys-73 | i | K73A/K74A/K75A | 61.0 ± 6.3 |
| Arg-74 | i | 4K/R>Ad | 14.2 ± 0.5 |
| Lys-75 | i, ii | 5K/R>Ae | -0.4 ± 2.9 |
Human LEDGF/p75 (National Center for Biotechnology Information accession number NP_150091).
i, role in HDGF DNA binding (31); ii, well conserved among PWWP domain orthologs (39, 58) and/or Tudor domain Royal Family members (35) and surface exposed (31, 39); iii, predictive hydrophobic cavity exposure (31, 39); iv, conserved among HRP family members (6, 22, 58) and exposed on the presumptive DNA binding face (Fig. 2B) (31, 39); v, surface exposed near the hydrophobic cavity (Fig. 2C) (31, 39); vi, analogous DNMT3B residue (Ser-270) implicated in ICF syndrome (53).
That is, the percent HIV-Luc activity from two to four independent experiments relative to E6(-/-) cells transfected with wild-type LEDGF/p75 expression vector. Previously described ΔPWWP and ΔPWWPΔATh deletion mutants functioned at 19.2% ± 2.5% and −3.4% ± 4.2%, respectively, whereas the MutL1 NLS/ATh mutant (Fig. 1A) supported 159% ± 29.7% of wild-type activity.
K67A/K70A/K73A/R74A/K75A.
K56A/K67A/K70A/K73A/R74A/K75A.
Virus infectivity.
LEDGF/p75 plays a critical role in mediating lentiviral integration and hence virus infection (11, 21, 27, 34, 55, 64). We previously generated knockout and matched control LEDGF/p75-expressing mouse cell lines to analyze the role of the host factor in HIV-1 infection. MEFs were infected with VSV-G-pseudotyped single-round HIV-Luc carrying either wild-type or D64N/D116N active-site mutant integrase. At 2 days postinfection, cells were processed for Luc activity, and integrase-dependent levels of HIV-1 infectivity were determined by subtracting the low levels of active site mutant activities from matched wild-type viral infections (55). Accordingly, HIV-Luc infected E6(−/−) knockout cells at 3.4% ± 4.0% (n = 7) of the level of control E6(f/f) cells. These results moreover defined a valuable reverse genetic system, as transiently expressed LEDGF/p75 protein sensitized E6(−/−) cells to HIV-1 infection in an IBD- and PWWP domain-dependent manner (55). Each of the novel PWWP domain mutant proteins was therefore tested for its ability to support E6(−/−) cell infection alongside wild-type LEDGF/p75 and two previously described N-terminal deletion mutants: ΔPWWP, lacking the PWWP domain and ΔPWWPΔATh, which lacked the dual copy of the AT-hook DNA-binding motif in addition to the PWWP domain (Fig. 1A).
As previously established (55), the double ΔPWWPΔATh mutant failed to stimulate the basal level at which E6(−/−) cells became infected, whereas the ΔPWWP deletion mutant supported ca. 19% of the level of wild-type LEDGF/p75 function (Fig. 3A). A perusal of the novel PWWP domain mutant proteins (Fig. 3 and Table 1) revealed a full activity spectrum that spanned from the wild-type level (for example, V28E) to undetectable (for example, K14A/K16A or W21A). Consistent with its high degree of sequence conservation among PWWP domain orthologs (39, 58), Lys-14 appeared the most important of the targeted basic residues. K14A and K14E functioned at ca. 58 and 17% of the level of wild-type LEDGF/p75, respectively, while K14A/K16A was unable to support HIV-1 infection despite efficient expression of the double mutant protein (Fig. 3A). The only other inactive charge-to-alanine mutant, 5K/R>A, harbored six overall amino acid changes (Fig. 3A and Table 1).
FIG. 3.

Mutant protein activities and expression profiles. (A) The indicated untagged mutant proteins were tested for their abilities to sensitize E6(−/−) knockout cells to HIV-1 infection in comparison to wild-type LEDGF/p75 and the empty pIRES2-eGFP expression vector. Protein expression levels are indicated to the right. Lane 1, lysate prepared from cells transfected with empty vector DNA. (B) Additional mutants from the experiment shown in panel A. (C) Same as in panels A and B except that the noted mutant and control proteins were analyzed in a separate Western blotting experiment. Infectivity data minimally compile the results of eight Luc assays (duplicate assays from two to four infections, each conducted in duplicate). In general, only mutants that displayed less than half of the level of wild-type LEDGF/p75 activity are shown (see Table 1 for the complete data set). The levels of endogenous LEDGF/p75 protein in E6(f/f) cells, shown in Fig. 4C below, were below the detection limit of the panel A and C experiments.
Of the residues predicted to comprise the hydrophobic cavity (Fig. 2A), Trp-21 and Ala-51 were the most critical with important roles determined for Met-15, Thr-47, and Glu-49 (Fig. 3B and Table 1). Ile-42 is near the cavity (Fig. 2C), and changing it in concert with Phe-43 also ablated cofactor function (Fig. 3B). Because Y18A conferred the wild-type level of HIV-1 infection, the phenolic side chain was dispensable under these assay conditions. Surprisingly, the activity of the double mutant T47E/E49A protein exceeded the level of either single point mutant (Fig. 3B and Table 1).
The His within the signature PHWP motif appeared unimportant, as H20A and H20E mutants functioned at ca. 95 and 81% of the level of wild-type LEDGF/p75, respectively (Table 1). The first Pro within the motif was also not critical, as P19A functioned at the wild-type level; substituting Glu for Pro-19 though reduced cofactor function approximately fivefold (Fig. 3C and Table 1). The combined L10A/I11A mutant functioned at ca. 8% of wild-type LEDGF/p75, similar to the residual level of activity displayed by the double L40E/P41E mutant protein (Fig. 3C and Table 1).
The observation that a number of point mutant proteins functioned at levels that were significantly less than that observed for the ΔPWWP deletion, which lacked the domain, was unexpected. Because of this, ΔPWWP was tested alongside wild-type LEDGF/p75 and the empty expression vector in expanded sets of knockout and control MEFs. The ΔPWWP mutant functioned at 20.6% ± 1.7% of wild-type in E16(−/−) cells, similar to the level observed in E6(−/−) cells (55) (Fig. 3A), whereas about half this level (11.5% ± 0.6%) was seen using E5(−/−) cells. In contrast, the mutant displayed only residual function (<0.1 to 4.2%) in three other knockout cell lines. A subset of mutant proteins, chosen because they displayed the gamut of activities in E6(−/−) cells, was therefore retested using E2(−/−) cells where the ΔPWWP mutant functioned at 1.7% ± 2.2% (n = 4) of wild-type LEDGF/p75 (Fig. 4A and B). The ΔATh deletion mutant (Fig. 1A), which functioned similar to wild-type LEDGF/p75 in E6(−/−) cells (55), importantly maintained this phenotype in E2(−/−) cells (Fig. 4A and 4B). Levels of K14E, K14A, K16E, and K16A function, which ranged from 17 to 91% of wild-type LEDGF/p75 in E6(−/−) cells (Fig. 3A and Table 1), spanned 8 to 82% when tested in E2(−/−) cells (Fig. 4A). The relative levels of K14E, K14A, K16E, K16A, and K14A/K16A mutant protein function in E6(−/−) cells were furthermore maintained in E2(−/−) cells (compare Fig. 3A and 4A). Due to these results, additional point mutants were not tested in E2(−/−) cells. Of note, the expression level of the ΔPWWP deletion protein in the two knockout cell types did not account for its differential function during infection, since it was expressed just as well if not somewhat better in E2(−/−) versus E6(−/−) cells (Fig. 4C, compare lane 2 to lane 6).
FIG. 4.
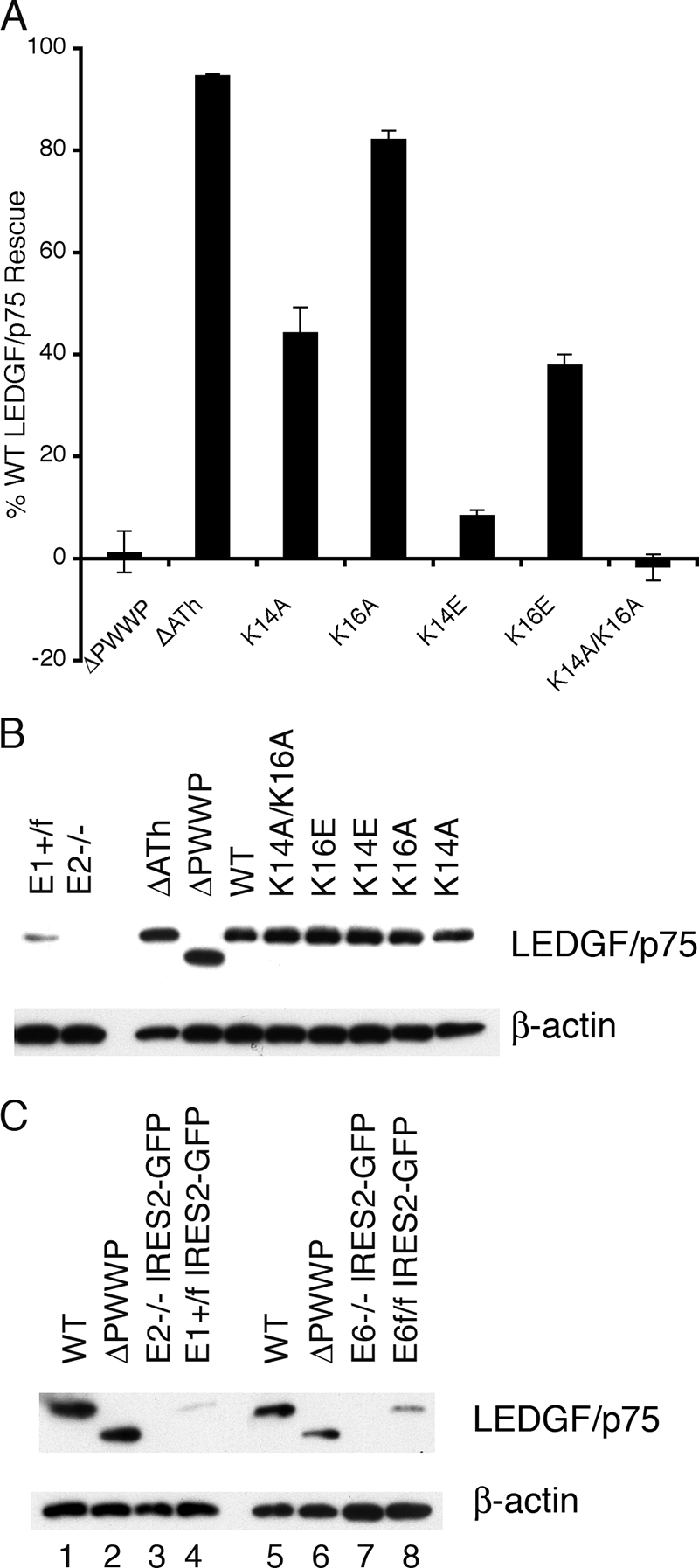
LEDGF/p75 mutant protein activities and expression profiles in E2(−/−) knockout cells. (A) The indicated mutants were tested in comparison to wild-type LEDGF/p75 and empty pIRES2-eGFP vector DNA; data are averages and standard deviations of minimally eight Luc assays from two or more independent transfection and infection experiments. (B) Expression levels of proteins analyzed in panel A. Lanes 1 and 2, endogenous mouse LEDGF/p75 content of parental E1(f/+) and E2(−/−) cells, respectively; HIV-Luc infected these knockout cells at 3.6% ± 0.8% (n = 3) of the level of E1(f/+) cells. (C) Levels of wild-type LEDGF/p75 and ΔPWWP proteins in transiently transfected E2(−/−) (lanes 1 and 2) and E6(−/−) (lanes 5 and 6) cells, respectively. Lanes 3 and 4, endogenous mouse LEDGF/p75 levels in E2(−/−) and E1(f/+) cells, respectively, transfected with empty vector DNA; lanes 7 and 8, endogenous protein levels in transfected E6(−/−) and E6(f/f) cells, respectively.
Association with cellular chromatin.
The PWWP domain, which is present in approximately 60 eukaryotic proteins, plays a central role in LEDGF/p75 chromatin binding (29, 62). We therefore determined the binding properties of a subset of the novel point mutant proteins; K14A/K16A, W21A, I42A/F43A, and A51P were highlighted because each of these failed to sensitize mouse knockout cells to HIV-1 infection despite containing only one or two amino acid changes. LEDGF/p75 interacts intimately with the condensed chromosomes that form during mitosis (7, 29, 33, 42, 44, 62, 65), so the distribution of each mutant in interphase and mitotic cells was compared to the wild type. To maintain consistency with previous mutational studies (29, 33, 62, 65), human cells were used for these analyses.
We previously visualized green fluorescent protein (GFP)-LEDGF/p75 fusion proteins in live HeLa cells by using confocal microscopy (62). To increase the sensitivity of mutant protein detection, we more recently turned to immunodetection of HA-tagged variants in fixed cells using epifluorescence microscopy. To gauge this approach, we first documented the phenotypes of a number of previously analyzed mutant proteins. HeLa TZM-bl cells sorted to enrich for transient transfectants were treated with anti-HA antibodies at 2 days posttransfection, and these results were compared to DAPI-stained images. As determined for the analogous GFP fusion protein (62), deleting the PWWP domain did not significantly alter the localization of HA-tagged LEDGF/p75 during interphase or mitosis (Fig. 5A). Deleting the dual copy of the AT-hook motif marginally affected the chromosome binding capacity of LEDGF/p75, whereas the combined ΔPWWPΔATh deletion mutant, as expected (29, 62), failed to engage chromatin (Fig. 5A, compare rightward merged images). Due to the functional NLS at residues 146 to 152, the double mutant nevertheless was strictly nuclear during interphase (Fig. 5A, left center images). We previously reported GFP-MutL1 (Fig. 1A) as pancellular in interphase and mitotic cells, concluding that the mutant lost chromatin binding (62). In contrast, the MutL1-HA construct retained partial chromatin binding activity: it was predominantly nuclear in interphase cells despite the critical K150A NLS mutation (32) (Fig. 5A, left), indicating that the mutant functionally engaged chromatin during reformation of postmitotic nuclei to acquire its karyophilic phenotype in an NLS-independent manner (65). Accordingly, MutL1-HA displayed affinity for condensed mitotic chromatin, although this appeared somewhat intermediary compared to the wild-type and defective ΔPWWPΔATh-HA mutant phenotypes (Fig. 5A, rightward merged images). Because MutL1 fully sensitized mouse knockout cells to infection, we conclude that direct DNA binding as mediated by the NLS and AT-hooks is dispensable for LEDGF/p75-dependent HIV-1 integration ex vivo (Table 1), as well as in vitro (1).
FIG. 5.

Localization of wild-type and mutant LEDGF/p75 proteins in interphase and mitotic cells. (A) Anti-HA antibody and DAPI stained images of cells transfected with the indicated LEDGF/p75-HA expression constructs. WT, wild type; ΔPΔATh, ΔPWWPΔATh. (B) Same as in panel A except that the indicated PWWP domain point mutants were compared to the wild type. The data are representative of the vast majority of imaged cells; hundreds of interphase cells (left image sets) were observed in each experiment, whereas three to seven mitotic cells (right image sets) were detected. Such results were obtained following a minimum of two independent transfections. (C) Western blotting profiles of the samples analyzed in panels A and B.
Each of the novel PWWP domain point mutant proteins predictably retained the karyophilic behavior of wild-type LEDGF/p75 in interphase cells (Fig. 5B, left sets of images). Significantly, each of these mutants failed to effectively engage condensed mitotic chromatin (Fig. 5B, right image sets). The results of Western blotting revealed that each mutant protein was expressed at a level that equaled or exceeded that of wild-type LEDGF/p75 (Fig. 5C). The interphase and mitotic distributions of the 5K/R>A mutant, which also failed to sensitize mouse knockout cells to HIV-Luc infection (Fig. 3A), was indistinguishable from other defective PWWP domain point mutant proteins (data not shown).
Chromatin association was also assessed following biochemical fractionation of transiently transfected human 293T or mouse knockout cells essentially as previously described (29, 59). Isolated nuclei (fraction P1 in Fig. 6A) were subsequently treated with DNase and then high salt (250 mM NH2SO4) to extract LEDGF/p75 from insoluble cytoskeletal and nuclear matrix materials. Accordingly, the chromatin-bound fraction is defined as the percentage of total LEDGF/p75 protein that partitions to fractions P1 and S2. As expected (29, 59), the vast majority of wild-type LEDGF/p75 (96% ± 1% for n = 4 experiments) was chromatin bound under these conditions (Fig. 6B, lanes 1 to 4). Deleting the PWWP domain marginally impaired association with chromatin (89% bound in Fig. 6B, lanes 5 to 8; 77% ± 7% for n = 4 experiments), whereas approximately half of the ΔPWWPΔATh mutant protein (51% ± 4% for n = 4) was unbound since it partitioned to the initial S1 fraction (Fig. 6B, lanes 9 to 12). The PWWP point mutant proteins behaved similar to the ΔPWWP deletion under these conditions: 76% ± 4% (n = 3) of the W21A mutant partitioned to fractions P1 and S2 (Fig. 6C, lanes 1 to 4), whereas 85% ± 7% (n = 3) of the I42A/F43A mutant remained chromatin bound (Fig. 6C, lanes 5 to 8). Since the ΔATh deletion significantly affected the fractionation phenotype of the ΔPWWP mutant (panel B), the AT-hook motifs were similarly removed from PWWP domain point mutant proteins. In these cases, though, the deletion effected at best a modest influence: 79% ± 5% and 63% ± 2% (n = 2) of the W21A-ΔATh and I42A/F43A-ΔATh mutants, respectively, partitioned to the P1 and S2 fractions (Fig. 6D, lanes 1 to 8). Similar results were obtained using K14A/K16A, A51P, K14A/K16A-ΔATh, and A51P-ΔATh mutant proteins (data not shown).
FIG. 6.

Wild-type and mutant LEDGF/p75 association with chromatin as assessed by biochemical fractionation. (A) Fractionation scheme (adapted from reference 29). (B) Fractionation profiles of 293T cells transfected with plasmids expressing the indicated HA-tagged proteins. Similar results were obtained using E6(−/−) knockout cells. (C and D) Same as in panel B, except with the indicated mutant proteins.
In vitro integration and DNA/chromatin-binding activities.
Purified LEDGF/p75 protein potently stimulates the activities of recombinant lentiviral integrase proteins (4, 6, 7, 62), and the nature of the in vitro reaction conditions can influence the requirements for the different host factor functions. Direct binding to HIV-1 integrase, for example, is crucial under all conditions (6, 8, 45, 48, 62, 71), whereas N-terminal deletion mutants of LEDGF/p75 defective for chromatin binding retained about half the level of wild-type activity when integration was performed with naked target DNA (1, 62). Recombinant ΔPWWP/MutL1 protein by contrast functioned at only ca. 4% of the level of the wild-type when reconstituted PNs were used in place of naked DNA (1). Recombinant K14A/K16A, W21A, I42A/F43A, and A51P proteins, with or without added MutL1 changes, were purified following their expression in E. coli to assess the effects of PWWP domain point mutations on the ability for LEDGF/p75 to stimulate HIV-1 integrase activity in vitro. Three single amino acid changes that effected intermediary phenotypes in the infectivity assay, K14A, I42A, and F43A, were also analyzed.
As previously noted (1), integrase favored PNs ∼10-fold over naked DNA in the absence of added LEDGF/p75 protein (Fig. 7A, inset). Maximal levels of PN-dependent stimulation occurred at 500 nM LEDGF/p75, whereas twice as much protein was required to max out integration into naked DNA (Fig. 7A). Because of this, mutant protein activities were compared to the wild-type at 500 nM and 1 μM LEDGF/p75 when using chromatinized and naked target DNAs, respectively.
FIG. 7.

Activities of select PWWP domain mutant proteins in in vitro integration assays. (A) Effects of LEDGF/p75 protein concentration on levels of integrase (1 μM) DNA strand transfer activity in the presence of PNs (gray line) versus naked DNA (black line). (Inset) Integration (Int) in the absence of added LEDGF/p75 protein. (B) Activities of indicated mutant proteins relative to wild-type LEDGF/p75 (set to 100%) in the presence of 1 μM integrase and naked (black bars) versus chromatinized (gray bars) target DNA. The data are averages ± the standard deviations of a minimum of three independent sets of integration assays. *, P < 0.001 (Student's t test) for the different PN-dependent versus naked DNA integration levels. (C) Percent mutant protein stimulatory activities in the presence of PNs compared to naked DNA. AU, arbitrary units; WT, wild-type.
The mutants functioned at ca. 16% (K14A/K16A/MutL1) to 76% (K14A and I42A) of the level of wild-type LEDGF/p75 for stimulating integration into naked target DNA (Fig. 7B, black bars). Of those that failed to sensitize mouse knockout cells to HIV-1 infection (first four entries in Fig. 7B), W21A supported the most integration (51% of the wild type), whereas K14A/K16A and I42A/F43A were the least active at ca. 23%. The activity of each mutant protein relative to wild-type was moreover reduced when PNs were used in place of naked target DNA (Fig. 7B, compare gray bars to black bars; summarized in panel C as the percent naked DNA stimulatory activity). These changes were most evident for W21A and K14A/K16A. W21A and W21A/MutL1 stimulated PN-dependent integration at ca. 30 and 18% of the levels seen with naked DNA, respectively, whereas the K14A/K16A and K14A/K16A/MutL1 proteins failed to detectably influence integrase activity under these assay conditions (Fig. 7B and C). Titrations revealed that chromatin-binding defective mutants supported more integrase activity at a higher (1.0 or 1.5 μM) LEDGF/p75 concentration (data not shown). These values, however, were invariably less than the suboptimal levels of wild-type activity observed under these conditions (Fig. 7A).
NLS and AT-hook residues primarily mediate the binding of LEDGF/p75 to DNA in vitro (1, 62). The isolated PWWP domain displays affinity for PNs and DNA as well, although this latter activity is comparatively weak due to salt hypersensitivity (1). To ascertain how the different mutations affect PWWP domain-specific chromatin and DNA binding, purified GST fusion proteins harboring the N-terminal 100 amino acid residues of LEDGF/p75 were prebound to glutathione-Sepharose beads and then exposed to radiolabeled DNA or PNs in the presence of low (50 mM) NaCl. After extensive washing, the levels of input substrate pulled down by the various bait proteins were quantified relative to those recovered by the wild-type GST fusion. Accordingly, neither the I42A nor F43A mutation significantly affected DNA or chromatin binding (Fig. 8A; the results of experimental replicates are quantified in panel B). Three of the four mutations that ablated function in the virus infection assay—K14A/K16A, W21A, and I42A/F43A—by contrast dramatically reduced binding to both substrates. GST-PWWP/A51P interestingly recovered ca. 75% of the wild-type level of naked DNA yet only ca. 20% of the input chromatin substrate. In contrast, the K14A mutation significantly reduced binding to both substrates, with DNA capture somewhat more affected than chromatin binding (Fig. 8).
FIG. 8.

PWWP domain DNA and chromatin binding activities. (A) The upper panel shows naked DNA levels recovered via GST pull-down with the indicated proteins. The middle panels show PN pull-downs as assessed by autoradiography (DNA) and Western blotting with anti-histone H3 antibodies. The lower panel shows bound fractions of the different GST proteins. WT, wild-type. (B) Results of four to six independent sets of pull-down assays, expressed as the percent DNA recovery with respect to the wild-type PWWP domain ± the standard deviation. Naked DNA experiments are indicated by black bars, whereas gray bars show the results with PNs. *, P < 0.01 for the differences observed between DNA and chromatin binding using Student's t test.
DISCUSSION
This study confirms and extends the model that LEDGF/p75 acts as a bifunctional molecular tether during HIV-1 integration (27, 55): its C-terminal IBD engages PIC-born integrase, whereas the N-terminal PWWP domain mediates interactions with cellular chromatin. Prior mutagenic (3, 8, 13, 49, 61) and structural biology (5, 8) approaches have significantly clarified the molecular basis of the LEDGF/p75-integrase interaction. Using primary sequence and 3D structural similarities, 24 PWWP domain amino acids were targeted here to elucidate those residues that play important roles during HIV-1 integration (Fig. 1 and 2 and Table 1).
LEDGF/p75 PWWP domain amino acid residues critical for HIV-1 infection.
Nineteen of the twenty-four targeted residues were analyzed via single amino acid substitution and of the 24 resultant mutant proteins, only W21A, W21E, and A51P failed to sensitize E6(−/−) mouse knockout cells to HIV-1 infection (Table 1). These results highlight critical roles for Trp-21 and Ala-51 in mediating lentivirus infectivity. As projected onto the solution structure of the HDGF PWWP domain (31), each of these residues lines the inner surface of the hydrophobic cavity that is the signature structural motif of members of the Tudor clan (Fig. 2A and 9A) (35). Considering that ectopically expressed W21A and A51P proteins failed to engage condensed mitotic chromosomes (Fig. 5B) and bound chromatin less efficiently than wild-type LEDGF/p75 (Fig. 6 and data not shown), our results suggest that the hydrophobic PWWP domain cavity forms an important binding interface for an as of yet-unidentified chromatin binding partner(s). Consistent with this, the W21A change was recently shown to abrogate the transforming activity of an artificial LEDGF/p75 PWWP domain-mixed-lineage leukemia fusion protein (70).
FIG. 9.

Molecular models of crucial Tudor clan member amino acid residues. (A) Amino acids critical for LEDGF/p75 PWWP domain function as defined in the present study. Residues highlighted in text are shown as space fill in the backdrop of the HDGF solution structure (31). The opening in the middle of the structure indicates the hydrophobic cavity; other structural elements are as labeled in Fig. 1B. (B) Crystal structure of the Drosophila HP1 chromodomain bound to a 15-mer histone H3 peptide dimethylated on Lys-9 (Me2K9; PBD code 1KNA). The peptide adopts β strand conformation; the sheet formed by peptide-β1-β2-β3 secondary elements (23) was aligned with β1-β2-β3-β4 from panel A. Tyr-24, Tyr-48, and Trp-45 (behind Me2K9 in this projection) form the hydrophobic binding pocket into which Me2K9 situates. A water molecule (not shown) mediates a hydrogen bond network that bridges Glu-52 backbone and side chain atoms to the dimethyl modification (23). The models were drawn by using PyMOL (10).
The A51P amino acid substitution was modeled after the homologous S270P change in DNMT3B that ablates chromatin-binding function and causes ICF syndrome (18, 53). Ala predominates and Pro is furthermore not found at this position among a collection of 42 PWWP domain sequences (58), suggesting that the loss of chromatin binding could in both cases result from a similar effect on the functionalities of the respective hydrophobic cavities. The isolated LEDGF/p75 domain can bind naked DNA and chromatinized templates in vitro, although the relevance of PWWP domain DNA binding is not entirely clear because it is counteracted by physiological salt concentrations (1). The A51P mutation nonetheless preferentially disrupted the interaction of the PWWP domain with chromatin without gross disruption of in vitro DNA binding (Fig. 8), indicating that the structural integrity of the domain was not overly compromised by this mutation.
By extension, we conjecture that the primary defects of the K14A/K16A and I42A/F43A mutant proteins in supporting HIV-1 infection likewise reside at the chromatin-binding side of the LEDGF/p75 molecular tether. These combination mutations reduced (Fig. 6 and data not shown) or ablated (Fig. 5B) chromatin association in cells, as well as the binding of recombinant GST-PWWP protein to DNA and chromatinized templates in vitro (Fig. 8). Because GST-PWWP/K14A was preferentially defective for binding to naked DNA (Fig. 8), it seems that the K14A/K16A chromatin binding defect could be partially due to a loss of DNA-binding function. This interpretation is consistent with a role for the charged outer face comprised of LEDGF/p75 analogous residues Arg-3, Lys-14, Lys-16, Lys-56, Lys-67, Lys-70, Lys-73, Arg-74, and Lys-75 in the in vitro DNA-binding activity of the HDGF PWWP domain (31). It is tempting to speculate that the hydrophobic cavity supplies the dominant binding interface for an unknown chromatin binding partner, whereas the positively charged outer face (Fig. 2B) could increase the overall affinity of the PWWP domain for chromatin for example, through nonspecific contacts with the DNA phosphodiester backbone. Given sufficient disruption of outer domain face charge-mediated interactions, for example, via the six changes in the 5K/R>A mutant, LEDGF/p75 nevertheless loses chromatin association and hence HIV-1 cofactor function.
The ΔPWWP deletion mutant curiously maintained affinity for mitotic chromatin under conditions where one or two amino acid changes within the domain disrupted function (Fig. 5). These data therefore suggest that the presence of a nonfunctional domain can counteract the abilities for the AT-hook DNA-binding motifs (29, 62) (Fig. 5A) and/or other charged regions within the LEDGF/p75 protein (29) (Fig. 1A) to confer chromatin-binding affinity.
As expected, the K14A/K16A, L10A/I11A, M15E/Y18A, L40E/P41E, and I42A/F43A double-mutant proteins faired less well than their single amino acid constituents at sensitizing mouse knockout cells to HIV-1 infection (Fig. 3 and Table 1). We were therefore somewhat surprised that the T47E/E49A double mutant reproducibly outshone the activities of either T47E or E49A point mutant (Fig. 3B). Thr-47 and Glu-49 participate in hydrophobic cavity formation and moreover abut each other in three dimensions (Fig. 2A and 9A). Our results therefore indicate the requirement for Glu at position 47 or 49, but not both, for effective PWWP domain function during HIV-1 infection. Other Tudor clan members utilize an analogous Glu residue for binding their respective substrates (39). For example, the E134K spinal muscular atrophy mutation in the survival motor neuron (SMN) Tudor domain abrogates binding to spliceosomal Sm protein (52). The co-crystal structure of the Drosophila HP1 chromodomain in complex with a histone H3 N-terminal tail peptide moreover revealed that Glu-52 interacted with the dimethylated Lys-9 side chain required for binding (Fig. 9B) (23). The majority (32 of 42) of PWWP domains harbor Glu or Asp at LEDGF/p75 analogous position 47 or 49 (but never both) (58), indicating that a negatively charged side chain in this general vicinity of the hydrophobic cavity is likely relevant for the function of most PWWP domains.
Conclusions.
We have identified a number of PWWP domain residues, highlighted by Trp-21 and Ala-51, which play critical roles in LEDGF/p75-dependent HIV-1 infection and integration. The results of numerous independent experiments, including sensitization of mouse knockout cells to infection (Fig. 3 and 4), association of ectopically expressed protein with chromatin (Fig. 5 and 6), PN-dependent stimulation of integrase activity in vitro (Fig. 7), and PN pull-down assays (Fig. 8), combine to support a model for the PWWP domain hydrophobic cavity as a crucial chromatin interaction motif (Fig. 9A).
Point mutations in the PWWP (Fig. 3 and Table 1) or IBD (55) can abrogate LEDGF/p75 function during HIV-1 infection. Various PWWP point mutant proteins (for example, W21A or K14A/K16A) were coexpressed with the functionally inactive D366N IBD mutant in knockout cells to test for functional complementation via phenotypic mixing. This approach, however, failed to yield HIV-Luc infectivities beyond those observed with cells expressing sole mutant proteins. Although admittedly negative in nature, these results indicate that LEDGF/p75 may very well function as a monomer during HIV-1 infection. Consistent with this interpretation, purified LEDGF/p75 protein sedimented as a monomer during analytical ultracentrifugation (8).
It is anticipated that the distribution of lentiviral integration is in large part defined by the chromosomal distribution of LEDGF/p75 protein. Lentiviruses may very well utilize a LEDGF/p75 independent pathway to accomplish ca. 2 to 20% of their overall integrations (27, 34, 55), but we speculate that the lion's share occurs through the PIC engaging chromatin-bound LEDGF/p75 via the IBD. Integrase is then encouraged at that moment or soon thereafter to integrate the viral cDNA into the local chromatin vicinity. Concordantly, the W21A and K14A/K16A mutant proteins were preferentially defective for stimulating integration into chromatinized DNA in vitro (Fig. 7B and C). Although anti-LEDGF/p75 antibodies can immunoprecipitate integration-competent lentiviral PICs from cytoplasmic extracts of acutely infected cells (28), the finding that HIV-1 PIC activity did not depend on the host factor (55) suggests that the critical LEDGF/p75 molecule is likely chromatin compared to PIC bound (14). A refined model of LEDGF/p75 function during HIV-1 integration will undoubtedly benefit from identification and verification of salient PWWP domain binding partner(s), a line of investigation that we, and likely others, are pursuing. The mutations described herein will serve as invaluable tools to verify the biological relevance of potential interacting proteins.
Acknowledgments
HeLa TZM-bl cells were obtained through the NIH AIDS Research and Reference Reagent Program from John Kappes, Xiaoyun Wu, and Tranzyme, Inc. We thank Manuel Llano for advice with the fractionation technique used for chromatin binding analyses, Peter Cherepanov and Lavanya Krishnan for their comments on the manuscript, and L. Krishnan for advice with figure preparation.
This study was supported by grants from the National Institutes of Health (NIH) (AI39394 and AI70042 [A.E.], AI45587 [J.L.], and AI60354 [Harvard Medical School Center for AIDS Research]) and the Agence Nationale de Recherche sur le SIDA (2005/004 and 2006/124 [M.L.]). N.Y. was supported by a HMS CFAR Scholar Award, and M.-C.S. was supported by NIH Training Grant AI07245.
Footnotes
Published ahead of print on 17 September 2008.
REFERENCES
- 1.Botbol, Y., N. K. Raghavendra, S. Rahman, A. Engelman, and M. Lavigne. 2008. Chromatinized templates reveal the requirement for the LEDGF/p75 PWWP domain during HIV-1 integration in vitro. Nucleic Acids Res. 361237-1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bushman, F., M. Lewinski, A. Ciuffi, S. Barr, J. Leipzig, S. Hannenhalli, and C. Hoffmann. 2005. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 3848-858. [DOI] [PubMed] [Google Scholar]
- 3.Busschots, K., A. Voet, M. De Maeyer, J. C. Rain, S. Emiliani, R. Benarous, L. Desender, Z. Debyser, and F. Christ. 2007. Identification of the LEDGF/p75 binding site in HIV-1 integrase. J. Mol. Biol. 3651480-1492. [DOI] [PubMed] [Google Scholar]
- 4.Cherepanov, P. 2007. LEDGF/p75 interacts with divergent lentiviral integrases and modulates their enzymatic activity in vitro. Nucleic Acids Res. 35113-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cherepanov, P., A. L. B. Ambrosio, S. Rahman, T. Ellenberger, and A. Engelman. 2005. From the cover: structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl. Acad. Sci. USA 10217308-17313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cherepanov, P., E. Devroe, P. A. Silver, and A. Engelman. 2004. Identification of an evolutionarily-conserved domain in LEDGF/p75 that binds HIV-1 integrase. J. Biol. Chem. 27948883-48892. [DOI] [PubMed] [Google Scholar]
- 7.Cherepanov, P., G. Maertens, P. Proost, B. Devreese, J. Van Beeumen, Y. Engelborghs, E. De Clercq, and Z. Debyser. 2003. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 278372-381. [DOI] [PubMed] [Google Scholar]
- 8.Cherepanov, P., Z.-Y. J. Sun, S. Rahman, G. Maertens, G. Wagner, and A. Engelman. 2005. Solution structure of the HIV-1 integrase-binding domain in LEDGF/p75. Nat. Struct. Mol. Biol. 12526-532. [DOI] [PubMed] [Google Scholar]
- 9.Ciuffi, A., M. Llano, E. Poeschla, C. Hoffmann, J. Leipzig, P. Shinn, J. R. Ecker, and F. Bushman. 2005. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 111287-1289. [DOI] [PubMed] [Google Scholar]
- 10.DeLano, W. L. 2002. The PyMOL molecular graphics system. DeLano Scientific, Palo Alto, CA. http://www.pymol.org.
- 11.De Rijck, J., L. Vandekerckhove, R. Gijsbers, A. Hombrouck, J. Hendrix, J. Vercammen, Y. Engelborghs, F. Christ, and Z. Debyser. 2006. Overexpression of the lens epithelium-derived growth factor/p75 integrase binding domain inhibits human immunodeficiency virus replication. J. Virol. 8011498-11509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Derse, D., B. Crise, Y. Li, G. Princler, N. Lum, C. Stewart, C. F. McGrath, S. H. Hughes, D. J. Munroe, and X. Wu. 2007. HTLV-1 integration target sites in the human genome: comparison with other retroviruses. J. Virol. 816731-6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emiliani, S., A. Mousnier, K. Busschots, M. Maroun, B. Van Maele, D. Tempe, L. Vandekerckhove, F. Moisant, L. Ben-Slama, M. Witvrouw, F. Christ, J. C. Rain, C. Dargemont, Z. Debyser, and R. Benarous. 2005. Integrase mutants defective for interaction with LEDGF/p75 are impaired in chromosome tethering and HIV-1 replication. J. Biol. Chem. 28025517-25523. [DOI] [PubMed] [Google Scholar]
- 14.Engelman, A., and P. Cherepanov. 2008. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog. 4e1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engelman, A., G. Englund, J. M. Orenstein, M. A. Martin, and R. Craigie. 1995. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J. Virol. 692729-2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faschinger, A., F. Rouault, J. Sollner, A. Lukas, B. Salmons, W. H. Gunzburg, and S. Indik. 2008. Mouse mammary tumor virus integration site selection in human and mouse genomes. J. Virol. 821360-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finn, R. D., J. Mistry, B. Schuster-Bockler, S. Griffiths-Jones, V. Hollich, T. Lassmann, S. Moxon, M. Marshall, A. Khanna, R. Durbin, S. R. Eddy, E. L. L. Sonnhammer, and A. Bateman. 2006. Pfam: clans, web tools and services. Nucleic Acids Res. 34D247-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ge, Y.-Z., M.-T. Pu, H. Gowher, H.-P. Wu, J.-P. Ding, A. Jeltsch, and G.-L. Xu. 2004. Chromatin targeting of de novo DNA methyltransferases by the PWWP domain. J. Biol. Chem. 27925447-25454. [DOI] [PubMed] [Google Scholar]
- 19.Hematti, P., B.-K. Hong, C. Ferguson, R. Adler, H. Hanawa, S. Sellers, I. E. Holt, C. E. Eckfeldt, Y. Sharma, M. Schmidt, C. von Kalle, D. A. Persons, E. M. Billings, C. M. Verfaillie, A. W. Nienhuis, T. G. Wolfsberg, C. E. Dunbar, and B. Calmels. 2004. Distinct genomic integration of MLV and SIV vectors in primate hematopoietic stem and progenitor cells. PLoS Biol. 2e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holman, A. G., and J. M. Coffin. 2005. Symmetrical base preferences surrounding HIV-1, avian sarcoma/leukosis virus, and murine leukemia virus integration sites. Proc. Natl. Acad. Sci. USA 1026103-6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hombrouck, A., J. De Rijck, J. Hendrix, L. Vandekerckhove, A. Voet, M. D. Maeyer, M. Witvrouw, Y. Engelborghs, F. Christ, R. Gijsbers, and Z. Debyser. 2007. Virus evolution reveals an exclusive role for LEDGF/p75 in chromosomal tethering of HIV. PLoS Pathog. 3e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Izumoto, Y., T. Kuroda, H. Harada, T. Kishimoto, and H. Nakamura. 1997. Hepatoma-derived growth factor belongs to a gene family in mice showing significant homology in the amino terminus. Biochem. Biophys. Res. Commun. 23826-32. [DOI] [PubMed] [Google Scholar]
- 23.Jacobs, S. A., and S. Khorasanizadeh. 2002. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science 2952080-2083. [DOI] [PubMed] [Google Scholar]
- 24.Laue, K., S. Daujat, J. G. Crump, N. Plaster, H. H. Roehl, C. B. Kimmel, R. Schneider, M. Hammerschmidt, et al. 2008. The multidomain protein Brpf1 binds histones and is required for Hox gene expression and segmental identity. Development 1351935-1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewinski, M. K., M. Yamashita, M. Emerman, A. Ciuffi, H. Marshall, G. Crawford, F. Collins, P. Shinn, J. Leipzig, S. Hannenhalli, C. C. Berry, J. R. Ecker, and F. D. Bushman. 2006. Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS Pathog. 2e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Limón, A., E. Devroe, R. Lu, H. Z. Ghory, P. A. Silver, and A. Engelman. 2002. Nuclear localization of human immunodeficiency virus type 1 preintegration complexes (PICs): V165A and R166A are pleiotropic integrase mutants primarily defective for integration, not PIC nuclear import. J. Virol. 7610598-10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Llano, M., D. T. Saenz, A. Meehan, P. Wongthida, M. Peretz, W. H. Walker, W. Teo, and E. M. Poeschla. 2006. An essential role for LEDGF/p75 in HIV integration. Science 314461-464. [DOI] [PubMed] [Google Scholar]
- 28.Llano, M., M. Vanegas, O. Fregoso, D. Saenz, S. Chung, M. Peretz, and E. M. Poeschla. 2004. LEDGF/p75 determines cellular trafficking of diverse lentiviral but not murine oncoretroviral integrase proteins and is a component of functional lentiviral preintegration complexes. J. Virol. 789524-9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Llano, M., M. Vanegas, N. Hutchins, D. Thompson, S. Delgado, and E. M. Poeschla. 2006. Identification and characterization of the chromatin-binding domains of the HIV-1 integrase interactor LEDGF/p75. J. Mol. Biol. 360760-773. [DOI] [PubMed] [Google Scholar]
- 30.Lu, R., A. Limón, E. Devroe, P. A. Silver, P. Cherepanov, and A. Engelman. 2004. Class II integrase mutants with changes in putative nuclear localization signals are primarily blocked at a post-nuclear entry step of human immunodeficiency virus type 1 replication. J. Virol. 7812735-12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lukasik, S. M., T. Cierpicki, M. Borloz, J. Grembecka, A. Everett, and J. H. Bushweller. 2006. High resolution structure of the HDGF PWWP domain: a potential DNA binding domain. Protein Sci. 15314-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maertens, G., P. Cherepanov, Z. Debyser, Y. Engelborghs, and A. Engelman. 2004. Identification and characterization of a functional nuclear localization signal in the HIV-1 integrase interactor LEDGF/p75. J. Biol. Chem. 27933421-33429. [DOI] [PubMed] [Google Scholar]
- 33.Maertens, G., P. Cherepanov, W. Pluymers, K. Busschots, E. De Clercq, Z. Debyser, and Y. Engelborghs. 2003. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J. Biol. Chem. 27833528-33539. [DOI] [PubMed] [Google Scholar]
- 34.Marshall, H. M., K. Ronen, C. Berry, M. Llano, H. Sutherland, D. Saenz, W. Bickmore, E. Poeschla, and F. D. Bushman. 2007. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS ONE. 2e1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maurer-Stroh, S., N. J. Dickens, L. Hughes-Davies, T. Kouzarides, F. Eisenhaber, and C. P. Ponting. 2003. The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem. Sci. 2869-74. [DOI] [PubMed] [Google Scholar]
- 36.Meekings, K. N., J. Leipzig, F. D. Bushman, G. P. Taylor, and C. R. Bangham. 2008. HTLV-1 integration into transcriptionally active genomic regions is associated with proviral expression and with HAM/TSP. PLoS Pathog. 4e1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitchell, R. S., B. F. Beitzel, A. R. W. Schroder, P. Shinn, H. Chen, C. C. Berry, J. R. Ecker, and F. D. Bushman. 2004. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2e234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakamura, H., Y. Izumoto, H. Kambe, T. Kuroda, T. Mori, K. Kawamura, H. Yamamoto, and T. Kishimoto. 1994. Molecular cloning of complementary DNA for a novel human hepatoma-derived growth factor. Its homology with high mobility group-1 protein. J. Biol. Chem. 26925143-25149. [PubMed] [Google Scholar]
- 39.Nameki, N., N. Tochio, S. Koshiba, M. Inoue, T. Yabuki, M. Aoki, E. Seki, T. Matsuda, Y. Fujikura, M. Saito, M. Ikari, M. Watanabe, T. Terada, M. Shirouzu, M. Yoshida, H. Hirota, A. Tanaka, Y. Hayashizaki, P. Guntert, T. Kigawa, and S. Yokoyama. 2005. Solution structure of the PWWP domain of the hepatoma-derived growth factor family. Protein Sci. 14756-764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Narezkina, A., K. D. Taganov, S. Litwin, R. Stoyanova, J. Hayashi, C. Seeger, A. M. Skalka, and R. A. Katz. 2004. Genome-wide analyses of avian sarcoma virus integration sites. J. Virol. 7811656-11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nielsen, P. R., D. Nietlispach, H. R. Mott, J. Callaghan, A. Bannister, T. Kouzarides, A. G. Murzin, N. V. Murzina, and E. D. Laue. 2002. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature 416103-107. [DOI] [PubMed] [Google Scholar]
- 42.Nishizawa, Y., J. Usukura, D. P. Singh, L. T. J. Chylack, and T. Shinohara. 2001. Spatial and temporal dynamics of two alternatively spliced regulatory factors, lens epithelium-derived growth factor (LEDGF/p75) and p52, in the nucleus. Cell Tissue Res. 305107-114. [DOI] [PubMed] [Google Scholar]
- 43.Nowrouzi, A., M. Dittrich, C. Klanke, M. Heinkelein, M. Rammling, T. Dandekar, C. von Kalle, and A. Rethwilm. 2006. Genome-wide mapping of foamy virus vector integrations into a human cell line. J. Gen. Virol. 871339-1347. [DOI] [PubMed] [Google Scholar]
- 44.Ochs, R. L., Y. Muro, Y. Si, H. Ge, E. K. Chan, and E. M. Tan. 2000. Autoantibodies to DFS 70 kd/transcription coactivator p75 in atopic dermatitis and other conditions. J. Allergy Clin. Immunol. 1051211-1220. [DOI] [PubMed] [Google Scholar]
- 45.Pandey, K. K., S. Sinha, and D. P. Grandgenett. 2007. Transcriptional coactivator LEDGF/p75 modulates human immunodeficiency virus type 1 integrase-mediated concerted integration. J. Virol. 813969-3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Poeschla, E. M. 2008. Integrase, LEDGF/p75 and HIV replication. Cell. Mol. Life Sci. 651403-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiu, C., K. Sawada, X. Zhang, and X. Cheng. 2002. The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat. Struct. Biol. 9217-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raghavendra, N. K., and A. Engelman. 2007. LEDGF/p75 interferes with the formation of synaptic nucleoprotein complexes that catalyze full-site HIV-1 DNA integration in vitro: implications for the mechanism of viral cDNA integration. Virology 3601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rahman, S., R. Lu, N. Vandegraaff, P. Cherepanov, and A. Engelman. 2007. Structure-based mutagenesis of the integrase-LEDGF/p75 interface uncouples a strict correlation between in vitro protein binding and HIV-1 fitness. Virology 35779-90. [DOI] [PubMed] [Google Scholar]
- 50.Schroder, A. R. W., P. Shinn, H. Chen, C. Berry, J. R. Ecker, and F. Bushman. 2002. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110521-529. [DOI] [PubMed] [Google Scholar]
- 51.Schwartz, R. M., and M. O. Dayhoff. 1978. Matrices for detecting distance relationships, vol. 5, suppl. 3. National Biochemical Research Foundation, Washington, DC.
- 52.Selenko, P., R. Sprangers, G. Stier, D. Bühler, U. Fischer, and M. Sattler. 2001. SMN Tudor domain structure and its interaction with the Sm proteins. Nat. Struct. Biol. 827-31. [DOI] [PubMed] [Google Scholar]
- 53.Shirohzu, H., T. Kubota, A. Kumazawa, T. Sado, T. Chijiwa, K. Inagaki, I. Suetake, S. Tajima, K. Wakui, Y. Miki, M. Hayashi, Y. Fukushima, and H. Sasaki. 2002. Three novel DNMT3B mutations in Japanese patients with ICF syndrome. Am. J. Med. Genet. 11231-37. [DOI] [PubMed] [Google Scholar]
- 54.Shun, M.-C., J. E. Daigle, N. Vandegraaff, and A. Engelman. 2007. Wild-type levels of human immunodeficiency virus type 1 infectivity in the absence of cellular emerin protein. J. Virol. 81166-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shun, M.-C., N. K. Raghavendra, N. Vandegraaff, J. E. Daigle, S. Hughes, P. Kellam, P. Cherepanov, and A. Engelman. 2007. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 211767-1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh, D. P., E. Kubo, Y. Takamura, T. Shinohara, A. Kumar, L. T. J. Chylack, and N. Fatma. 2006. DNA binding domains and nuclear localization signal of LEDGF: contribution of two helix-turn-helix (HTH)-like domains and a stretch of 58 amino acids of the N-terminal to the transactivation potential of LEDGF. J. Mol. Biol. 355379-394. [DOI] [PubMed] [Google Scholar]
- 57.Slater, L. M., M. D. Allen, and M. Bycroft. 2003. Structural variation in PWWP domains. J. Mol. Biol. 330571-576. [DOI] [PubMed] [Google Scholar]
- 58.Stec, I., S. B. Nagl, G.-J. B. van Ommen, and J. T. den Dunnen. 2000. The PWWP domain: a potential protein-protein interaction domain in nuclear proteins influencing differentiation? FEBS Lett. 4731-5. [DOI] [PubMed] [Google Scholar]
- 59.Thakar, K., R. Niedenthal, E. Okaz, S. Franken, A. Jakobs, S. Gupta, S. Kelm, and F. Dietz. 2008. SUMOylation of the hepatoma-derived growth factor negatively influences its binding to chromatin. FEBS J. 2751411-1426. [DOI] [PubMed] [Google Scholar]
- 60.Trobridge, G. D., D. G. Miller, M. A. Jacobs, J. M. Allen, H.-P. Kiem, R. Kaul, and D. W. Russell. 2006. Foamy virus vector integration sites in normal human cells. Proc. Natl. Acad. Sci. USA 1031498-1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Turlure, F., E. Devroe, P. A. Silver, and A. Engelman. 2004. Human cell proteins and human immunodeficiency virus DNA integration. Front. Biosci. 93187-3208. [DOI] [PubMed] [Google Scholar]
- 62.Turlure, F., G. Maertens, S. Rahman, P. Cherepanov, and A. Engelman. 2006. A tripartite DNA-binding element, comprised of the nuclear localization signal and two AT-hook motifs, mediates the association of LEDGF/p75 with chromatin in vivo. Nucleic Acids Res. 341663-1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vandegraaff, N., E. Devroe, F. Turlure, P. A. Silver, and A. Engelman. 2006. Biochemical and genetic analyses of integrase-interacting proteins lens epithelium-derived growth factor (LEDGF)/p75 and hepatoma-derived growth factor related protein 2 (HRP2) in preintegration complex function and HIV-1 replication. Virology 346415-426. [DOI] [PubMed] [Google Scholar]
- 64.Vandekerckhove, L., F. Christ, B. Van Maele, J. De Rijck, R. Gijsbers, C. Van den Haute, M. Witvrouw, and Z. Debyser. 2006. Transient and stable knockdown of the integrase cofactor LEDGF/p75 reveals its role in the replication cycle of human immunodeficiency virus. J. Virol. 801886-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vanegas, M., M. Llano, S. Delgado, D. Thompson, M. Peretz, and E. Poeschla. 2005. Identification of the LEDGF/p75 HIV-1 integrase-interaction domain and NLS reveals NLS-independent chromatin tethering. J. Cell Sci. 1181733-1743. [DOI] [PubMed] [Google Scholar]
- 66.Wei, X., J. M. Decker, H. Liu, Z. Zhang, R. B. Arani, J. M. Kilby, M. S. Saag, X. Wu, G. M. Shaw, and J. C. Kappes. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 461896-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu, X., Y. Li, B. Crise, and S. M. Burgess. 2003. Transcription start regions in the human genome are favored targets for MLV integration. Science 3001749-1751. [DOI] [PubMed] [Google Scholar]
- 68.Wu, X., Y. Li, B. Crise, S. M. Burgess, and D. J. Munroe. 2005. Weak palindromic consensus sequences are a common feature found at the integration target sites of many retroviruses. J. Virol. 795211-5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang, J., and A. Everett. 2007. Hepatoma derived growth factor binds DNA through the N-terminal PWWP domain. BMC Mol. Biol. 8101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yokoyama, A., and M. L. Cleary. 2008. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 1436-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu, F., G. S. Jones, M. Hung, A. H. Wagner, H. L. MacArthur, X. Liu, S. Leavitt, M. J. McDermott, and M. Tsiang. 2007. HIV-1 integrase preassembled on donor DNA is refractory to activity stimulation by LEDGF/p75. Biochemistry 462899-2908. [DOI] [PubMed] [Google Scholar]