

Abstract
The rhesus monkey intrinsic immunity factor TRIM5αrh recognizes incoming capsids from a variety of retroviruses, including human immunodeficiency virus type 1 (HIV-1) and equine infectious anemia virus (EIAV), and inhibits the accumulation of viral reverse transcripts. However, direct interactions between restricting TRIM5α proteins and retroviral capsids have not previously been demonstrated using pure recombinant proteins. To facilitate structural and mechanistic studies of retroviral restriction, we have developed methods for expressing and purifying an active chimeric TRIM5αrh protein containing the RING domain from the related human TRIM21 protein. This recombinant TRIM5-21R protein was expressed in SF-21 insect cells and purified through three chromatographic steps. Two distinct TRIM5-21R species were purified and shown to correspond to monomers and dimers, as analyzed by analytical ultracentrifugation. Chemically cross-linked recombinant TRIM5-21R dimers and mammalian-expressed TRIM5-21R and TRIM5α proteins exhibited similar sodium dodecyl sulfate-polyacrylamide gel electrophoresis mobilities, indicating that mammalian TRIM5α proteins are predominantly dimeric. Purified TRIM5-21R had ubiquitin ligase activity and could autoubquitylate with different E2 ubiquitin conjugating enzymes in vitro. TRIM5-21R bound directly to synthetic capsids composed of recombinant HIV-1 CA-NC proteins and to authentic EIAV core particles. HIV-1 CA-NC assemblies bound dimeric TRIM5-21R better than either monomeric TRIM5-21R or TRIM5-21R constructs that lacked the SPRY domain or its V1 loop. Thus, our studies indicate that TRIM5α proteins are dimeric ubiquitin E3 ligases that recognize retroviral capsids through direct interactions mediated by the SPRY domain and demonstrate that these activities can be recapitulated in vitro using pure recombinant proteins.
Susceptibility to retroviral infections influences species survival and has driven the evolution of cellular restriction factors that inhibit retroviral replication. One important antiretroviral intrinsic immune response is mediated by TRIM5α, which can block early postentry steps in the replication of certain retroviruses in specific primate lineages (3, 37, 55, 58). Under normal restrictive conditions, TRIM5α proteins block accumulation of retroviral reverse transcripts (55) and accelerate the rate at which viral capsids dissociate from high-molecular-weight complexes into lower-molecular-weight subunits (42, 56). The allelic specificity of TRIM5α restriction is illustrated by the fact that rhesus macaque TRIM5α potently inhibits human immunodeficiency virus type 1 (HIV-1) replication, whereas human TRIM5α instead exhibits restriction activity against N-tropic murine leukemia virus but not HIV-1 (23, 29, 43, 55, 67). These differences can be attributed to the differential abilities of TRIM5α proteins to interact with retroviral capsids after viral entry (34, 42, 56).
Like other tripartite (TRIM) family members, TRIM5α contains RING, B-box, coiled-coil, and B30.2/SPRY domains, and each of these domains contributes to restriction activity. The RING domain of TRIM5α has intrinsic E3 ubiquitin ligase activity (63, 64), which is important both for autoubiquitylation and for protein turnover in vivo (12, 26). The E3 ubiquitin ligase activity also contributes to restriction but is not absolutely required (26, 41, 55), and the precise functions of ubiquitylation and the role of the ubiquitin/proteasome system in retroviral restriction are not yet fully understood. Specifically, proteasome activity is not required for TRIM5α antiviral activity per se (12, 41), but proteasome inhibitors do alter the normal progression of TRIM5α restriction, allowing reverse transcripts to accumulate and impairing the ability of TRIM5α to accelerate the dissociation of intact viral capsids (2, 10, 62). Moreover, the restriction of incoming capsids results in proteasome-dependent degradation of TRIM5α (47), again suggesting possible involvement of the proteasome in the normal stepwise progression of restriction.
The B-box domain of TRIM5α plays an essential, but still undefined role in restriction activity, and mutations in this domain can influence protein turnover, intracellular localization, and restriction activity (11). The coiled-coil and ensuing linker 2 (L2) regions of TRIM5α also contribute to efficient capsid binding and restriction, and these elements appear to function primarily in protein oligomerization (27, 36). Cross-linking studies have suggested that TRIM5α may function as a trimer, but trimerization has not been rigorously demonstrated by biophysical studies of pure TRIM5 proteins (36). Finally, the C-terminal B30.2/SPRY domain of TRIM5α is essential for viral capsid recognition and is the key determinant of antiviral specificity (38, 41, 54-56, 68). Several motifs within the SPRY domain have been subject to significant positive selection and exhibit pronounced variability between species (35, 38, 49, 50, 54, 57). In particular, variable region 1 (V1, residues 323 to 350 in rhesus TRIM5α) is critical both for efficient restriction and for capsid binding, and a single amino acid alteration at position 332 of the V1 loop allows human TRIM5α to bind HIV-1 capsids and inhibit HIV-1 replication (34, 54, 68). These and other observations show that the SPRY domain dictates retroviral capsid recognition, although direct binding interactions have not yet been demonstrated and characterized using pure TRIM5α proteins.
TRIM5α proteins appear to recognize the outer shell of the retroviral core particle, which is called the capsid. Lentiviruses such as HIV-1 and equine infectious anemia virus (EIAV) have conical capsids composed of CA protein hexamers that form hexagonal assemblies called fullerene cones (4, 19-21, 28, 32). Intact retroviral core particles can be isolated from membrane-stripped retroviruses and purified by using sucrose-gradient centrifugation, but these assemblies are rather unstable and typically disassemble spontaneously in buffer (1, 17, 30, 46, 60). Structural and biochemical studies of the HIV-1 capsid have therefore frequently used synthetic assemblies created from pure recombinant CA proteins (20). The assemblies formed by pure HIV-1 CA-NC proteins on DNA templates are particularly stable and can be formed under near physiological conditions (7, 21). These CA-NC/DNA assemblies are a mixture of cylindrical and conical structures that recapitulate the hexagonal lattice of CA hexamers found in authentic viral capsids (4, 21, 32). Synthetic HIV-1 CA-NC/DNA assemblies, like membrane-stripped viral capsids, also bind exogenously expressed TRIM5α proteins present in crude cell lysates (51, 56), indicating that the CA-NC/DNA assemblies recapitulate the key elements required for TRIM5α recognition.
In summary, it is now well established that capsid recognition dictates the allelic specificity of TRIM5α restriction. However, direct interactions between pure TRIM5 proteins and retroviral capsids have not yet been demonstrated in vitro, largely owing to the technical challenges of creating suitable capsid targets and of expressing and purifying recombinant TRIM5α proteins. To date, soluble full-length TRIM5α proteins have not been expressed successfully in Escherichia coli, and TRIM5α proteins tend to turnover rapidly and aggregate into cytoplasmic bodies when overexpressed in mammalian systems (6, 12, 53). However, replacing the TRIM5α RING domain with the RING domain from the homologous TRIM21 protein reduces protein turnover without compromising HIV-1 restriction substantially (12, 33). We have taken advantage of this observation to develop systems for expressing and purifying chimeric TRIM5-21R proteins in quantities suitable for biochemical and biophysical characterization.
MATERIALS AND METHODS
Cell cultures.
Human 293T and HeLa cells were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal calf serum. HeLa cells stably expressing rhesus TRIM5α (TRIM5αrh) (55) were maintained with an additional supplement of 1 μg of puromycin/ml.
Expression vectors.
The pLPCX TRIM5-21R expression vector, encoding a hemagglutinin (HA)-tagged TRIM5-21R (12), was used as a PCR template to create a DNA fragment containing the TRIM5-21R gene with KpnI and XhoI restriction sites. This fragment was ligated into the pCAG.OSFT (WISP-08-189) backbone, which itself was modified from the pCAG vector backbone (Addgene) to encode an N-terminal OneStrep and Flag (OSF) epitope tag followed by a TEV protease cleavage site. The resulting vector, pCAG.OSFT-TRIM5-21R (WISP-08-176), was used as a PCR template to amplify a DNA fragment with terminal attB sites to allow for recombination into the pDONR221 Gateway Entry Vector (Invitrogen) to create the pDONR221.OSFT-TRIM5-21R Entry Vector (WISP-08-177). This vector was used as a template for QuikChange site-directed mutagenesis (Stratagene) to create pDONR221.OSFT-TRIM5-21RΔV1 (WISP-08-178) (deletion in the V1 variable loop of the SPRY domain corresponding to TRIM5αrh residues 332 to 344) and pDONR221.OSFT-TRIM5-21RΔSPRY (WISP-08-179) (deletion of the entire SPRY domain and L2 element, TRIM5αrh residues 233 to 497). The pCAG.OSFT-TRIM5αrh and pDONR221.OSFT-TRIM5rh vectors (WISP-08-197 and -08-198) were constructed in an analogous fashion. Recombinant baculoviruses expressing TRIM5-21R, TRIM5-21RΔV1, and TRIM5-21RΔSPRY were prepared in situ by using the BaculoDirect system (Invitrogen). Equivalent mammalian cell expression constructs were generated in the Gateway pcDNA-DEST40 vector (WISP-08-180, -181, -182, and -199, respectively). pET28-based plasmids for bacterial expression of UbcH5 (WISP-08-190), UbcH6 (WISP-08-191), UbcH7 (WISP08-192), and Uba1 (WISP-08-192) were generous gifts from Rachel Klevit (University of Washington) (5, 8), and the Uba1 gene was originally a gift from Richard Vierstra (University of Wisconsin) (22).
Expression and purification of recombinant proteins. (i) TRIM5-21R.
Two liters of SF-21 insect cells were grown to a density of 2 × 106 cells/ml in SF-900 II media (Invitrogen), infected at a multiplicity of infection of 1.0 with recombinant baculoviruses encoding TRIM5-21R and variants, and the expressed proteins were allowed to accumulate for 36 to 44 h (100 rpm shaking, 27°C). All subsequent steps were carried out at 4°C, except where noted. Cells were collected by centrifugation at 500 × g for 8 min, resuspended in 1:25 (vol/vol) of lysis buffer (50 mM NaCl, 50 mM Tris [pH 8.0], 1.5% Triton X-100, 1 mM TCEP, and mammalian protease inhibitor cocktail [Sigma] at 1:150 [vol/vol]), and lysed in a 100-ml Dounce homogenizer (15 strokes). The lysate was clarified by ultracentrifugation (Beckman Ti 50.2 rotor; 45,000 rpm, 184,000 × g, 40 min), filtered (0.45 μm), and loaded at 2 ml/min onto a 10-ml StrepTactin Superflow affinity column (IBA). The bound protein was washed with 50 ml of buffer (50 mM NaCl, 50 mM Tris [pH 8.0], 1 mM TCEP) and eluted with wash buffer supplemented with 2.5 mM d-desthiobiotin. The eluate was loaded directly onto two 5-ml HiTrap Q-Sepharose anion-exchange columns connected in series (GE Healthcare) and eluted with a NaCl gradient from 50 mM to 1 M over 200 ml of buffer (50 mM Tris [pH 8.0], 1 mM TCEP). Purified TRIM5-21R eluted in two peaks (see Fig. 1B). Fractions containing the early eluting peak were pooled, concentrated to 2 ml (Vivaspin concentrator; Sartorius), and loaded onto a 120-ml Superose-6 gel filtration column. The later eluting protein fraction peak tended to aggregate under high-ionic-strength conditions and was therefore loaded directly onto a Superose-6 column without concentration (4-ml fractions). Gel filtration chromatography was performed at 1 ml/min in 25 mM NaCl, 50 mM Tris (pH 8.0), 1 mM TCEP. The gel filtration column was calibrated by using the HMW standard gel filtration calibration kit (GE Healthcare), and the two TRIM5-21R species eluted with apparent molecular masses of 232 kDa (dimer, second anion exchange peak) and 70 kDa (monomer, first anion exchange peak). The purified monomer could be concentrated to 10 mg/ml, whereas the dimer tended to precipitate at concentrations above 3 mg/ml. Concentrated proteins were stored at 4°C for short-term use but tended to precipitate when stored for multiple days. Alternatively, proteins were flash frozen in liquid nitrogen and retained CA-NC binding and autoubiquitylation activities when thawed.
FIG. 1.

TRIM5-21R expression and purification. (A) Western blot (upper, anti-FLAG) and Coomassie blue-stained SDS-PAGE gel (lower) showing the stepwise purification of recombinant TRIM5-21R proteins. Lane 1, soluble lysate from control SF-21 cells; lane 2, lysate from SF-21 cells infected with baculovirus expressing TRIM5-21R; lane 3, StrepTactin affinity-purified TRIM5-21R; lanes 4 and 5, monomeric (M, early eluting peak) and dimeric (D, late eluting peak) TRIM5-21R proteins purified by anion-exchange chromatography; lanes 6 and 7, monomeric (M, late eluting peak) and dimeric (D, early eluting peak) TRIM5-21R proteins purified by gel filtration chromatography. (B) Anion-exchange (upper) and gel filtration (lower) chromatographs showing the elution profiles of monomeric (M) and dimeric (D) TRIM5-21R proteins. Inset (upper panel) shows a Coomassie blue-stained SDS-PAGE analysis of the designated fractions. Elution positions of control molecular mass standards are shown below the gel filtration chromatograph for reference, and estimated molecular masses are given above the peaks of monomeric and dimeric TRIM5-21R.
(ii) Ubiquitin-related proteins.
Recombinant E1 and E2 proteins were expressed in BL21-Codon Plus (DE3)-RIPL bacteria (Stratagene) grown in LB media and purified as described previously (8). Briefly, human UbcH5c, UbcH6, and UbcH7 were purified by cation-exchange chromatography (SP Sepharose) and eluted with a 0 to 0.5 M NaCl gradient in 30 mM MES (morpholineethanesulfonic acid)-1 mM EDTA (pH 6.0). E2-rich fractions were pooled and purified further by size exclusion chromatography in 25 mM sodium phosphate (pH 7.0)-150 mM NaCl. Wheat His6-Uba1 (E1) was purified by Ni2+-affinity chromatography, followed by size exclusion chromatography. Ubiquitin was purified as described previously (44).
Mass spectrometry.
Monomeric and dimeric TRIM5-21R proteins were desalted for electrospray ionization mass spectrometry using a C18 Ziptip (Millipore) and analyzed on a Quattro-II mass spectrometer (Micromass, Inc.). The data were acquired with a cone voltage of 50 eV, a spray voltage of 2.8 kV, and scanning from 800 to 1,400 m/z in 4 s. Spectra were combined, and the multiply charged molecular ions were deconvoluted into a molecular-mass spectrum by using MaxEnt software (Micromass, Inc.).
Liquid chromatography tandem mass spectrometry experiments were used to examine posttranslational modifications of both TRIM5αrh expressed in 293T cells and TRIM5-21R expressed in SF-21 cells. TRIM5αrh contained a C-terminal HA tag (55) and was affinity purified from 293T cells by using α-HA-conjugated Sepharose matrices as described below for cross-linking experiments. Dimeric TRIM5-21R was purified by anion-exchange chromatography, concentrated, and mixed with an equal volume of 2× sodium dodecyl sulfate (SDS) loading buffer. In both cases, samples were boiled, separated by SDS-4 to 20%polyacrylamide gel electrophoresis (PAGE), and visualized using Coomassie blue staining. Major bands corresponding to each TRIM5 protein, as well as minor bands corresponding to higher-molecular-mass (modified) TRIM5 proteins were excised, digested with TPCK (tolylsulfonyl phenylalanyl chloromethyl ketone)-modified trypsin (Promega) and introduced by nanoLC (Eksigent, Inc.) with nano-electrospray ionization (ThermoElectron Corp) into a LTQ-FT hybrid mass spectrometer (ThermoElectron Corp). Peptide molecular masses were measured by Fourier transform ion cyclotron resonance, and peptide sequencing was performed by collision-induced dissociation in the linear ion trap of the instrument. Protein identification and posttranslational modifications (phosphorylation and ubiquitylation) of peptides in tryptic digests were determined by using the MASCOT search engine (Matrix Science) and the NCBInr mammalian taxonomy database using a significance threshold of P < 0.05 and an ion score cutoff value of 20 (Table 1).
TABLE 1.
Posttranslational modifications of TRIM5-21R and TRIM5αrh proteins
| Cell type | Protein | No. of peptides identifieda | % Coverage | MASCOT score (total protein score) |
|---|---|---|---|---|
| SF-21 | TRIM5-21R | 77* | 54 | 7365 |
| Ubiquitin | 7† | 79 | 138 | |
| 293T | TRIM5αrh-HA | 42* | 47 | 2162 |
| Ubiquitin | 2 | 23 | 63 |
*,Two nested TRIM5-21R phosphopeptides were identified: 81LREVKLSPEEGQK93 and 83EVKLSPEEGQK93. †, One ubiquitylated ubiquitin peptide was identified: 43LIFAGKQLEDGR54.
Equilibrium sedimentation analyses.
Equilibrium sedimentation of purified TRIM5-21R proteins was performed using Optima XL-I and XL-A centrifuges (Beckman) at protein subunit concentrations of 6.09, 3.05, and 1.53 μM (monomer) or 6.0, 3.0, and 1.5 μM (dimer) in 50 mM Tris-HCl (pH 8.0)-50 mM NaCl-1 mM β-mercaptoethanol (monomer) or in 25 mM NaCl-50 mM Tris-HCl (pH 8.0)-1 mM TCEP (dimer). Centrifugation (4°C) was performed at two speeds: 14,000 and 18,000 rpm (monomer) and 14,000 and 16,000 rpm (dimer). The resulting six data sets for each oligomeric species were globally fit to single ideal species models with fixed or floating molecular masses using the nonlinear least-squares algorithms in the Heteroanalysis software (9). Solvent density and the partial specific volume of TRIM5-21R were calculated by using the program SEDNTERP (version 1.09) (31).
Autoubiquitylation activity assays.
TRIM5-21R-directed ubiquitylation assays were carried out in 50-μl reaction mixtures containing 1.0 μM TRIM5-21R, 1.0 μM concentrations of the specified E2 enzyme, 20 μM ubiquitin, and 0.5 μM wheat Uba1. Reactions were incubated for 60 min with or without addition of 5 mM ATP-10 mM MgCl2, and reaction products were analyzed by SDS-7.5% PAGE and Western blotting as described below.
Cross-linking reactions. (i) Cross-linking of recombinant TRIM5-21R proteins.
Pure recombinant TRIM5-21R proteins were dialyzed overnight into phosphate-buffered saline (PBS; 1 mM KH2PO4, 5.6 mM Na2HPO4, 154 mM NaCl) plus 1 mM TCEP. Cross-linking reactions were then performed for 10 min at 25°C in 30-μl reaction volumes at final protein concentrations of 0.5 μM, along with 0, 0.1, 0.5, or 1 mM fresh EGS [ethylene glycol-bis(succinimidyl succinate)] cross-linker. Reactions were quenched by addition of 30 μl of 0.1 M Tris-HCl (pH 7.5), and samples were mixed with SDS loading buffer plus 1.2% β-mercaptoethanol, and analyzed by SDS-7.5% PAGE and Western blotting. For reactions performed in the presence of mammalian cell lysate, recombinant TRIM5-21R proteins in PBS plus 1 mM TCEP buffer were incubated in 30-μl reactions containing final concentrations of 50% clarified lysate prepared from 293T cells (see below), 0.25 μM TRIM5-21R, and EGS at 0, 0.1, 0.5, or 1 mM (20 min, 25°C). Reactions were quenched by the addition of 30 μl of 0.1 M Tris-HCl (pH 7.5), diluted to 400 μl with PBS plus 1 mM EDTA, and incubated with 30 μl of StrepTactin Sepharose (IBA) at 4°C for 2 h. The matrix was washed four times with PBS plus 0.5% NP-40 and resuspended in 30 μl of SDS loading buffer plus 1.2% β-mercaptoethanol for Western blot analysis.
(ii) Cross-linking of TRIM proteins expressed in mammalian cells.
For each cross-linking series (four samples), a 10-cm plate of confluent 293T cells expressing OSF-tagged TRIM5-21R or HA-tagged TRIM5αrh was lysed in 300 μl of PBS plus 0.5% NP-40 supplemented 1:150 with protease inhibitor cocktail (Sigma) for 15 min on ice. Lysates were clarified by centrifugation at 16,100 × g for 30 min, and the resulting supernatant was diluted to a final volume of 1.7 ml with PBS plus 1 mM EDTA. Then, 400-μl aliquots of diluted lysate were incubated with 0, 0.1, 0.5, or 1.0 mM EGS (20 min at 25°C). Reactions were quenched by the addition of 400 μl of 0.1 M Tris-HCl (pH 7.5), mixed with 30 μl of pre-equilibrated StrepTactin Sepharose (TRIM21-5R; IBA) or α-HA-conjugated Sepharose (TRIM5αrh; Sigma), and incubated for 2 h at 4°C. Matrices were washed four times with PBS plus 0.5% NP-40 and resuspended in 30 μl of SDS loading buffer plus 1.2% β-mercaptoethanol for Western blot analysis using SDS-7.5% PAGE.
Western blotting.
Proteins were separated by SDS-PAGE, transferred to PVDF-FL membranes (Millipore) in Tris-glycine-10% methanol buffer, blocked in 5% milk-Tris-buffered saline (20 min), and incubated with primary antibodies diluted in 5% nonfat milk-Tris-buffered saline plus 0.1% Tween 20 (16 h, 4°C). Blots were visualized by using an Odyssey infrared imaging system (Li-Cor, Inc.) to detect Alexa 680-nm (Molecular Probes; 1:10,000) or IRDye 800-nm (Rockland; 1:10,000) secondary antibodies. The primary antibodies and dilutions used were as follows: mouse anti-FLAG (Sigma) at 1:3,000 for OSF-TRIM5-21R (Fig. 1, 3 to 5, and 7), mouse anti-HA.11 (Sigma) at 1:1,000 for TRIM5α-HA (Fig. 3), rabbit anti-HIV-1 CA (UT415 made against purified NL4-3 CA protein and affinity purified) at 1:15,000 (Fig. 5), and rabbit anti-EIAV CA (UT418 made against purified EIAV CA protein and affinity-purified) at 1:5,000 (Fig. 7).
FIG. 3.

Recombinant TRIM5-21R dimers and mammalian-expressed TRIM5-21R and TRIM5α proteins exhibit similar cross-linking patterns. (A) Western blot (anti-FLAG) showing purified monomeric (M) and dimeric (D) proteins cross-linked with increasing concentrations of EGS (lanes 3 to 8) or controls without EGS (lanes 1 and 2). (B) Western blots (anti-FLAG) showing purified recombinant TRIM5-21R dimer added to 293T cell lysate and cross-linked in the presence of increasing concentrations of EGS (first panel); TRIM5-21R expressed in 293T cells, cross-linked directly in the lysate, and concentrated by StrepTactin affinity purification (second panel); and TRIM5α expressed in 293T cells, cross-linked directly in the lysate, and concentrated by α-HA affinity purification (third panel). Lanes 1, 5, and 9 show controls in the absence of EGS, and the asterisk denotes contaminating immunoglobulin G heavy chain eluted from the HA-Sepharose beads. Note that all of the samples were run on the same gel, and migration positions can therefore be compared directly.
FIG. 5.

TRIM5-21R binds CA-NC assemblies. (A) Schematic summary (above) and SDS-PAGE/Coomassie blue analyses (below) of purified full-length dimeric TRIM5-21R (lane 3, denoted WT), a dimeric TRIM5-21R construct missing part of the V1 loop (lane 2, denoted ΔV1), and a monomeric TRIM5-21R construct missing the entire SPRY domain (lane 1, denoted ΔSPRY). (B) TRIM5-21R protein binding to CA-NC/DNA assemblies. Monomeric (M) or dimeric (D) TRIM5-21R (WT, lanes 3 and 4 and lanes 7 and 8), TRIM5-21RΔV1 (ΔV1, lanes 2 and 6), or TRIM5-21RΔSPRY (ΔSPRY, lanes 1 and 5) proteins were incubated alone (lanes 1 to 4) or with CA-NC/DNA assemblies (lanes 5 to 8) and then centrifuged through a 70% sucrose cushion to separate CA-NC assemblies and bound proteins (pellet) from unbound and lower-molecular-weight proteins (supernatant). Western blots show the distribution of TRIM (α-FLAG, first, third, and fifth panels) and CA-NC (α-CA, second, fourth, and sixth panels). Note that CA-NC assemblies bound full-length TRIM5-21R constructs better than either TRIM5-21RΔV1 or TRIM5-21RΔSPRY. Ratios of bound CA-NC and TRIM5-21R proteins were obtained by combining known input protein concentrations, quantification of bound CA-NC levels using a standard curve of known CA-NC protein concentrations, and quantification of Western blot band intensities. Estimated bound ratios were as follows: CA-NC/TRIM5-21R dimer = 1.1, CA-NC/TRIM5-21R monomer = 0.8, and CA-NC/TRIM5-21RΔV = 0.18. (C) CA-NC/DNA assemblies bind better to dimeric TRIM5-21R proteins than to monomeric proteins under stringent conditions. Monomeric (lanes 2, 4, and 6) or dimeric (lanes 1, 3, and 5) proteins were incubated alone (lanes 1 and 2) or with CA-NC/DNA assemblies (lanes 3 to 6) and then centrifuged through a 70% sucrose cushion to separate CA-NC assemblies and bound proteins (pellet) from unbound and lower molecular weight proteins (supernatant). Western blots show the distribution of TRIM5-21R (α-FLAG, TRIM panels) and CA-NC (α-CA, CA-NC panels). The concentrations of TRIM5-21R proteins in lanes 5 and 6 were 3.33-fold higher than in lanes 3 and 4, and all binding assays were performed at lower protein concentrations than those shown in part (B) (see Materials and Methods). Note that under these conditions, CA-NC assemblies bound dimeric TRIM5-21R better than monomeric TRIM5-21R. The estimated ratios of bound proteins were as follows: CA-NC/TRIM5-21R dimer (0.15 μM input) = 0.025; CA-NC/TRIM5-21R monomer (0.15 μM input) = 0.01, CA-NC/TRIM5-21R dimer (0.5 μM input) = 0.068; and CA-NC/TRIM5-21R monomer (0.5 μM input) = 0.028.
FIG. 7.

TRIM5-21R binds EIAV cores. (A) Sucrose gradient purification of membrane-stripped cores produced by an EIAV vector. The Western blot (α-CA) shows the distribution of EIAV CA protein across a sucrose density gradient (with fraction densities shown). The CA distribution is quantified below, and the CA protein in fractions 10 to 12 corresponds to core particles. (B) TRIM5-21R protein binding to purified EIAV cores. Dimeric TRIM5-21R (WT, lanes 3 and 6), dimeric TRIM5-21RΔV1 (ΔV1, lanes 2 and 5), or monomeric TRIM5-21RΔSPRY (ΔSPRY, lanes 1 and 4) proteins were incubated alone (lanes 1 to 3) or with purified EIAV cores (lanes 4 to 6) and then centrifuged through a 57% sucrose cushion to separate EIAV cores and bound proteins (pellet) from unbound and lower-molecular-weight proteins (supernatant). Western blots show the distribution of TRIM (α-FLAG, TRIM panels) and EIAV CA (α-CA, CA panels). Note that the pelleted EIAV cores bound full-length TRIM5-21R better than either TRIM5-21RΔV1 or TRIM5-21RΔSPRY.
Restriction assays. (i) HIV-1 restriction assays.
The HIV-1 vector pCMVΔR8.2, which expresses a lacZ reporter, was packaged into vesicular stomatitis virus G-pseudotyped HIV-1 as described previously (48a, 59a). For restriction assays, 3 × 104 HeLa-M cells were seeded in 24-well plates and transfected (Lipofectamine 2000) 24 h later with 1 μg of the control pcDNA-DEST40 vector or with vectors expressing OSFT-TRIM5αrh, OSFT-TRIM5-21R, OSFT-TRIM5-21RΔV1, or OSFT-TRIM5-21RΔSPRY. Cells were incubated for 24 h, washed with media, transduced for 24 h with various quantities of the HIV-1 vector, washed again, and the percentage of infected cells was determined by assaying for lacZ expression at 48 h post infection. Equivalent expression levels of the different TRIM5 constructs were verified by Western blotting.
(ii) EIAV restriction assay.
To assay for restriction, 293T cells were transfected in a 10-cm plate with the CeGFPW EIAV vector system as described below, and infectivity was subsequently assayed as previously described (48). Briefly, dilutions of vector preparations were added to target cells (HeLa or HeLa-TRIM5αrh, 2.0 × 105 cells/well, six-well plate) and incubated for 72 h at 37°C. Transduction efficiencies were determined from the percentage of GFP-positive cells measured by fluorescence-activated cell sorting.
EIAV core isolation.
EIAV cores were prepared by using an adaptation of a method described previously (30). Briefly, EIAV virions were produced by cotransfection of 293T cells (16 × 10-cm plates) with an EIAV vector system. Each 10-cm plate of cells was transfected (CalPhos mammalian transfection kit; Clontech) with 8.1 μg of pEV53 (EIAV structural proteins), 7.5 μg of pSIN6.1CeGFPW (packaged GFP expression vector), and 2.7 μg of phCMV-VSV-G (VSV-G envelope) (39, 69). After 36 h, the supernatants were removed, pooled (four plates/pool), filtered (0.45-μm pore size), and pelleted through a 4-ml 20% sucrose cushion in a Beckman SW-32Ti rotor (134,000 × g, 2 h, 4°C). Each set of pelleted virions was resuspended by gentle pipetting (4 h at 4°C) in 400 μl of ST buffer (10 mM Tris-HCl [pH 7.4], 50 mM NaCl). Four 11.5-ml sucrose gradients (30 and 70% [wt/vol] sucrose in ST buffer) were prepared in 14-by-89-mm tubes using a gradient mixer (Biocomp). The gradients were overlaid with a 0.3-ml cushion of 15% sucrose containing 1% Triton X-100 and then with a 0.3-ml barrier layer of 7.5% sucrose in ST buffer. Each tube of concentrated EIAV particles was carefully layered on top of a gradient and centrifuged in a Beckman SW-41 rotor (210,000 × g, 16 h, 4°C). Twelve 1-ml fractions were collected from the bottom of each tube, and fraction densities were measured by using a digital refractometer (Leica), and EIAV CA was assayed by Western blotting. Three 1-ml fractions of the correct density (1.22 to 1.26 g/ml) contained intact EIAV core particles and were pooled, repelleted by centrifugation in a Beckman SW-41 rotor (2,000 × g, 2 h, 4°C), and resuspended in 40 μl of ST buffer for binding assays.
Capsid binding assays.
Recombinant HIV-1 CA-NC was expressed and purified as described previously (21). Cylindrical and conical assemblies were created by coincubating 10 μM CA-NC and 2 μM d(TG)50 oligonucleotide (15) in 1 ml of assembly buffer (50 mM Tris-HCl [pH 8.0], 500 mM NaCl) for 16 h at 4°C. Assembled complexes were enriched by centrifugation at 10,000 rpm in a microcentrifuge (∼9,000 × g) for 5 min, and pelleted assemblies were resuspended in 200 μl of supernatant. TRIM5-21R proteins were used at final concentrations of 5.0 μM (for the experiments described in Fig. 5B and 6B comparing different TRIM5-21R constructs), 0.5 or 0.15 μM (0.3×) (for the more stringent binding assays described in Fig. 5C), or 0.36 μM (for the EIAV core binding assays described in Fig. 7B). TRIM5-21R proteins were mixed with 20 μl of concentrated CA-NC particles (see the figure legends for quantities) or with purified EIAV cores (∼100 ng, ∼20 nM CA) in reaction volumes of 200 μl, with a final buffer composition of 10 mM Tris, 2 mM KH2PO4, 11 mM Na2HPO4, and 280 mM NaCl (Fig. 5B), 308 mM NaCl (Fig. 5C, 1×), 316 mM NaCl (Fig. 5C, 0.3×), or 250 mM NaCl (Fig. 7B). Reactions were incubated for 45 min at 25°C (CA-NC binding assays) or 4°C (EIAV core assays) with gentle mixing at 10-min intervals. Prior to centrifugation, a 10-μl sample was removed and mixed with SDS loading buffer (input). Binding reactions were layered onto a 4.2-ml cushion of 70% (CA-NC) or 57% (EIAV cores) sucrose (wt/vol; sucrose-PBS plus 1 mM TCEP) in 13-by-51-mm tubes and centrifuged in a Beckman SW-50.1 rotor (110,000 × g, 1 h, 4°C). Samples of the supernatant remaining above the sucrose cushion were saved (supernatant), the sucrose cushions were removed by aspiration, and the pellet was resuspended in 40 μl of 1× SDS loading buffer. Input, supernatant, and pellet samples were analyzed by Western blotting at concentrations in which the supernatant samples corresponded to 1.25% (CA-NC) and 2% (EIAV cores) of the total supernatant, and pellet samples corresponded to 80% of the total pellet. Under these conditions, ∼2.5% of the input EIAV cores pelleted, and ∼1.6% (5.0 μM input TRIM5-21R), 1.6% (0.5 μM input TRIM5-21R), and 2.4% (0.15 μM input TRIM5-21R) of the CA-NC assemblies pelleted.
FIG. 6.

Electron microscopic images of CA-NC assemblies incubated with full-length TRIM5-21R or deletion mutants. Negatively stained, transmission electron microscopic images of CA-NC assemblies incubated with monomeric TRIM5-21RΔSPRY (panels 1 and 2), dimeric TRIM5-21RΔV1 (panels 3 and 4), or full-length dimeric TRIM5-21R (panels 5 to 8). Note that CA-NC/DNA assemblies incubated with TRIM5-21R show enhanced exterior staining and more frequent breaks (arrows) compared to the TRIM5-21RΔV1 or TRIM5-21RΔSPRY controls. TRIM5-21R-induced breaks were quantified by scoring randomly selected electron microscopic images of CA-NC/DNA tubes for the presence of discontinuities (see arrows), with the following results: TRIM5-21RΔSPRY-treated tubes (0% broken, 0 total breaks, n = 26 tubes), TRIM5-21RΔV1-treated tubes (4% broken, 2 total breaks, n = 50), and TRIM5-21R-treated tubes (42% broken, 55 total breaks, n = 50).
Transmission electron microscopy analyses of TRIM-bound CA-NC tubes.
For transmission electron microscopy analyses, binding reactions containing 5-μl suspensions of CA-NC particles (prepared as described above) were incubated with final TRIM5-21R concentrations of 20 to 50 μM (15-μl total volumes, 25°C, 45 min, with gentle mixing every 10 min). Carbon-coated grids were placed on aliquots of each reaction mixture (7 μl, 90 s), washed with 3 to 4 drops of 0.1 M KCl, stained with 3 or 4 drops of 4% uranyl acetate, and air dried. Samples were imaged on Hitachi 7100 and Philips Tecnai 12 transmission electron microscopes at magnifications between ×25,000 and ×100,000. Each electron microscopic analysis was repeated at least three times with proteins from at least three separate purifications.
RESULTS
TRIM5-21R expression and purification.
Wild-type rhesus TRIM5α accumulates at low levels and tends to aggregate when overexpressed in mammalian and insect cells (12; data not shown). These properties have made it difficult to obtain sufficient quantities of pure recombinant TRIM5α proteins for biophysical and structural studies. Substitution of the TRIM5αrh RING domain with the RING domain of the homologous human TRIM21 protein (58% identity), however, generates a fusion protein (designated TRIM5-21R) that has a longer half-life and accumulates to higher levels in mammalian cells than does wild-type TRIM5α (12). Importantly, TRIM5-21R restricts HIV-1 replication, although its activity is slightly reduced relative to wild-type TRIM5α (12; see also Fig. S1A in the supplemental material). TRIM5-21R was therefore an attractive candidate for large-scale expression in insect cells.
TRIM5-21R constructs with N-terminal OSF epitope tags were expressed in SF-21 cells using baculoviral expression systems. TRIM5-21R expression was detectable in Western blots of soluble lysates of infected SF-21 cells but not in control lysates (Fig. 1A, upper panel, compare lanes 1 and 2). The soluble TRIM5-21R protein was affinity purified on a StrepTactin matrix, and elution with d-desthiobiotin yielded TRIM5-21R as essentially the only protein detectable by either Western blotting (upper panel, lane 3) or Coomassie blue staining (lower panel). The protein was further purified by anion-exchange chromatography, where it separated into two distinct species, both of which corresponded to TRIM5-21R (Fig. 1B, upper panel, and Fig. 1A, lanes 4 and 5). The ratios of the two species varied between preparations, but it was common for the late-eluting species to predominate, as shown in Fig. 1B.
Gel filtration chromatography was used as a final step to purify the two distinct TRIM5-21R species. The two species again exhibited different chromatographic behavior, eluting with apparent molecular weights of 232 kDa (late-eluting anion-exchange peak) and 77 kDa (early-eluting anion-exchange peak). The apparent size of the smaller protein suggested that it was likely to be a TRIM5-21R monomer, whereas the apparent size of the larger protein suggested that it was probably either a trimer or a dimer with an extended Stokes radius. Typical yields were 150 μg (larger species) and 200 μg (smaller species) per liter of infected SF-21 cells.
The identities of the two purified recombinant TRIM5-21R proteins were confirmed by electrospray mass spectrometric (ESI/MS) analyses of the intact proteins, and by liquid chromatography tandem mass spectrometric analyses of tryptic digests. The mass of the intact smaller species corresponded to a TRIM5-21R protein that was missing the N-terminal methionine and was acetylated at the N terminus (MWactual = 62,518 g/mol, MWcalculated = 62,520 g/mol), and the mass spectra revealed no evidence for posttranslational modifications. In contrast, the oligomeric species showed a protein of the same mass (MWactual = 62,517 g/mol), as well as a second protein with a mass that corresponded to a singly phosphorylated protein (MWactual = 62,596 g/mol, MWcalc = 62,595; ∼30% of protein). Peptide mapping experiments were therefore performed on TRIM5-21R proteins produced in both SF-21 (insect) and 293T (mammalian) cells. These studies identified a single phosphoserine residue at position 87 within the L1 linker between the RING and B-box domains in TRIM5-21R proteins isolated from both insect and human cells (Table 1, TRIM5αrh numbering). Given its location, this phosphorylation site could, in principle, modulate a postulated interaction between the RING and adjacent L1 elements (24). Ubiquitin-derived peptides were also detected in trypsin digests of purified TRIM5-21R expressed in both insect and mammalian cells, indicating that the protein was also ubiquitylated at low levels. Interestingly, ubiquitin peptides with Lys-48 isopeptide linkages were also detected, indicating that TRIM5-21R could be polyubiquitylated with Lys-48 chains.
In summary, we have developed methods that can be used to express and purify up to milligram quantities of two different TRIM5-21R oligomers, and the larger species can be phosphorylated and ubiquitylated in human and in insect cells.
Recombinant TRIM5-21R proteins are monomers and dimers.
Equilibrium sedimentation centrifugation experiments can provide shape-independent measures of protein solution mass, and this approach was therefore used to determine the oligomeric state of each TRIM5-21R species. Sedimentation data were collected at three different protein concentrations and two rotor speeds, yielding a total of six data sets for each TRIM5-21R species. These six data sets were globally fit to single species models in which the molecular weight was allowed to float and also to monomer, dimer, and trimer models with fixed molecular weights. As illustrated in Fig. 2A, the distribution of the smaller TRIM5-21R species fit a protein with a predicted mass of 59,305 g/mol, which matched the mass expected for a TRIM5-21R monomer (62,520 g/mol, Mobs/Mcalc = 0.95). The data for three initial concentrations of TRIM5-21R centrifuged at 14,000 rpm are shown together with the global fits (Fig. 2A, upper panel), and the small, random residuals indicate that the data were satisfactorily fit by a simple single species model (lower panels). Global fits to the three data sets collected at 18,000 rpm were also satisfactory (data not shown). Similarly, when fixed monomer, dimer, or trimer molecular weights were used during the global fitting procedure, the monomer model was clearly the best fit to the data (see Fig. S2A in the supplemental material).
FIG. 2.

Recombinant TRIM5-21R proteins are monomers and dimers. Equilibrium sedimentation distributions of purified monomeric (A) and dimeric (B) TRIM5-21R proteins (upper panels), and residual differences between the data and the single species models (lower panels). The data are shown for initial subunit protein concentrations of 6.09, 3.05, and 1.53 μM (monomer), and 6.0, 3.0, and 1.5 μM (dimer) at rotor speeds of 14,000 rpm. The data sets were also collected at 18,000 rpm (monomer) and 16,000 (dimer), and all of the data were globally fit to a single species model in which the molecular weight was allowed to float during the refinement. The data points are shown in open symbols, and the best-fit curves are shown as solid lines. Estimated molecular weights were as follows: TRIM5-21R monomer, 59,305 g/mol (MWmonomer = 62,520 g/mol, Mobs/Mcalc = 0.95); and TRIM5-21R dimer, 123,825 g/mol (MWdimer = 125,040 g/mol, Mobs/Mmonomer = 1.98).
In contrast, equilibrium distributions of the larger TRIM5-21R species indicated that this TRIM5-21R protein was a dimer. As shown in Fig. 2B, data fit with a floating molecular weight gave an estimated mass of 123,825 g/mol, which matched the molecular weight expected for a TRIM5-21R dimer (125,040 g/mol, Mobs/Mmonomer = 1.98), and the global fits again exhibited small, random residuals (Fig. 2B and data not shown). The dimer model was also clearly best when the data were globally fit to fixed TRIM5-21R monomer, dimer, or trimer models (see Fig. S2B in the supplemental material). We therefore conclude that the smaller TRIM5-21R species is a monomer and that the larger species is a dimer.
Recombinant TRIM5-21R dimers and mammalian TRIM5-21R and TRIM5α proteins exhibit similar cross-linking patterns.
The observation that recombinant TRIM5-21R formed monomers and dimers was somewhat surprising because previous studies had suggested that TRIM5α proteins might be trimers (36). This conclusion was based upon the SDS-PAGE electrophoretic mobilities of chemically cross-linked, mammalian-expressed TRIM5α proteins, whose mobilities appeared to correspond to trimers (∼150 kDa). To reconcile these observations, we tested whether chemically cross-linked TRIM5 dimers exhibited anomalously slow SDS-PAGE electrophoretic mobilities. Our first set of experiments examined the mobilities of pure recombinant monomeric and dimeric TRIM5-21R proteins cross-linked with EGS. The monomeric protein was used as a control to ensure that protein and cross-linker concentrations were sufficiently low to prevent nonspecific intermolecular cross-linking of nonassociated proteins. As shown in Fig. 3A, TRIM5-21R dimers formed intermolecular cross-links efficiently, whereas TRIM5-21R monomers did not. The major cross-linked species for the TRIM5-21R dimer migrated at an apparent molecular mass of ∼160 kDa on a SDS-7.5% PAGE gel, and a species migrating at ∼125 kDa was also observed at trace levels. Thus, the major cross-linked form of the TRIM5-21R dimer exhibits anomalously slow SDS-PAGE mobility.
In a second set of experiments, we tested whether the oligomeric state or cross-linking properties of TRIM5α proteins could be altered by cross-linking in crude lysates, by expression in human versus insect cells, or by the non-native N-terminal OSF tag or RING domain of the TRIM5-21R construct. As shown in the first panel of Fig. 3B, the cross-linking pattern did not change significantly when the pure recombinant TRIM5-21R dimers were cross-linked either in a buffer solution or in a soluble mammalian cell extract, indicating that TRIM5-21R dimer does not appreciably cross-link with extract proteins under these conditions (compare the first panel of Fig. 3B to Fig. 3A). A very similar cross-linking pattern was also seen when TRIM5-21R proteins were expressed in human 293T cells and cross-linked directly in their soluble extracts (compare Fig. 3B, second and third panels), although in this case the minor, faster-migrating cross-linked species was more evident. This experiment indicates that TRIM5-21R forms similar dimers when expressed in human or insect cells. Finally, we examined the cross-linking of an authentic TRIM5α protein expressed in 293T cells. Although the C-terminally HA-tagged TRIM5α had a slightly lower molecular weight than TRIM5-21R, the cross-linking pattern of the authentic TRIM5α-HA protein expressed in human 293T cells was again very similar to those seen for the various TRIM5-21R proteins. Thus, TRIM5-21R and TRIM5α proteins are predominantly dimeric when expressed in human or insect cells, but the cross-linked dimers exhibit anomalously slow electrophoretic mobilities.
Autoubiquitylation of recombinant TRIM5-21R proteins.
TRIM5α exhibits ubiquitin E3 ligase activity (63, 64), and a recent study showed that a partially purified, recombinant MBP-TRIM5α fusion protein could pair with the UbcH5b E2 enzyme and exhibit ubiquitin E3 ligase activity in vitro (64). We also tested whether our purified TRIM5-21R proteins were active for autoubiquitylation in vitro, using three different pure E2 enzymes. As shown in Fig. 4, both monomeric and dimeric TRIM5-21R proteins paired successfully with UbcH5c and UbcH6, but not with UbcH7 in our in vitro autoubiquitylation assays. Ubiquitylation was particularly efficient in the UbcH5 reactions, where most of the TRIM5-21R dimer was modified with ubiquitin (compare lanes 8 and 9). Control reactions behaved as expected in that ubiquitylated products were not observed in the absence of ATP (compare even and odd lanes).
FIG. 4.
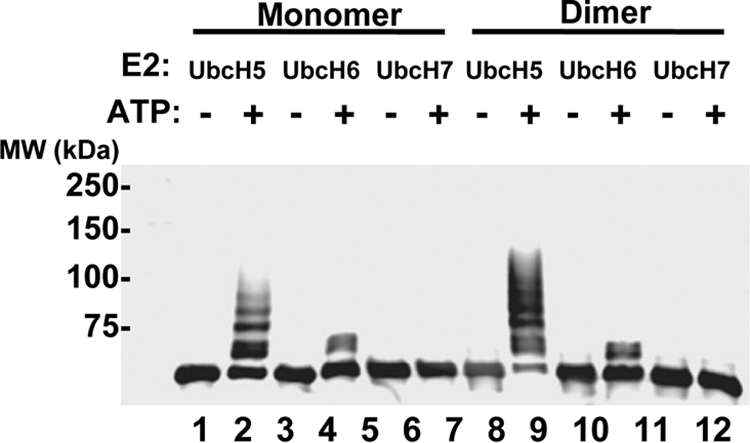
TRIM5-21R proteins can be autoubiquitylated in vitro. Purified monomeric (lanes 1 to 6) and dimeric (lanes 7 to 12) TRIM5-21R proteins were incubated with the wheat Uba1 E1 enzyme, ubiquitin, and one of three ubiquitin E2-conjugating enzymes—UbcH5c, UbcH6, or UbcH7—in the presence or absence of ATP. Reactions were terminated after 60 min, and TRIM5-21R reaction products were analyzed by Western blotting (α-FLAG).
Interestingly, UbcH5 and UbcH6 produced dramatically different product distributions, with UbcH5 catalyzing polyubiquitylation (or possibly multimonoubiquitylation) and UbcH6 catalyzing only monoubiquitylation of TRIM5-21R. These results demonstrate that the purified TRIM5-21R protein is an active ubiquitin E3 ligase and are in good agreement with the report of Yamauchi et al. (64), who found that MBP-TRIM5α was active with UbcH5B but not with UbcH7 (UbcH6 was not tested). Our results are also consistent with the observations that TRIM5α, TRIM21, and TRIM5-21R can self-ubiquitylate to form polyubiquitin chains in vivo, which can contain Lys-48 linkages (64; the present study). Finally, our experiments raise the possibility that TRIM5α and TRIM21 may exhibit different product distributions when paired with different E2 ubiquitin-conjugating enzymes in vivo. Such behavior has been seen previously for other RING and U-box E3 ligases (e.g., BRCA1 and CHIP, respectively) (8, 72) but remains to be confirmed with in vivo studies of nonchimeric TRIM5α and TRIM21 proteins.
Recombinant TRIM5-21R proteins bind HIV-1 CA-NC assemblies.
Although numerous studies have established that the B30.2/SPRY domains of TRIM5α proteins dictate the specificity of retroviral capsid recognition (34, 38, 41, 54-56, 68), direct binding of TRIM5α proteins to retroviral CA protein assemblies has not yet been demonstrated. We therefore tested whether pure recombinant TRIM5-21R proteins could bind the tubular and conical assemblies formed in vitro by pure recombinant HIV-1 CA-NC proteins on DNA templates. Previous studies have shown that these CA-NC/DNA assemblies form the same hexagonal surface lattices as authentic HIV-1 capsids and should therefore present native-like capsid surfaces for TRIM5-21R recognition. Our binding studies took advantage of the observation that CA-NC/DNA assemblies can be separated from free proteins by centrifugation through sucrose cushions (56). TRIM5-21R proteins therefore remained in the supernatants above the cushion, unless they bound and copelleted with the CA-NC/DNA assemblies.
Our studies used both monomeric and dimeric wild-type TRIM5-21R proteins, as well as two mutant TRIM5-21R proteins used as specificity controls (Fig. 5A). The first specificity control (denoted TRIM5-21RΔV1) lacked 13 residues from the middle of the V1 recognition loop of the SPRY domain (10). This loop is required for efficient HIV-1 capsid binding and restriction, although the TRIM5-21RΔV1 protein does retain modest residual HIV-1 restriction activity (10; see Fig. S1A in the supplemental material). The recombinant TRIM5-21RΔV1 protein behaved like wild-type TRIM5-21R in that it formed both monomers and dimers, and the purified TRIM5-21RΔV1 dimer was used in our studies. The second specificity control (denoted TRIM5-21RΔSPRY) lacked the entire SPRY domain and the preceding L2 linker. This control was used because TRIM5 constructs lacking SPRY domains cannot bind HIV-1 capsid in extracts or restrict HIV-1 replication in vivo (56). This construct differed from wild-type TRIM5-21R in that it was exclusively monomeric (data not shown), and the purified monomer was therefore used in our studies.
As shown in Fig. 5B, CA-NC/DNA proteins formed a mixture of assembled tubes and cones, which pelleted to the bottom of the sucrose cushion, and disassembled proteins, which remained in the supernatant (lanes 5 to 8). As expected, all TRIM5-21R proteins remained exclusively in the supernatant in the absence of CA-NC/DNA assemblies (lanes 1 to 4, compare panels 3 and 5). In the presence of CA-NC/DNA assemblies, however, both monomeric and dimeric wild-type TRIM5-21R proteins partitioned between the supernatant and pellet fractions, indicating that both TRIM5-21R proteins could bind CA-NC/DNA assemblies (compare lanes 7 and 8 to lanes 3 and 4 in the sixth panel). The TRIM5-21RΔV1 protein also bound detectably to CA-NC/DNA assemblies (compare lane 6 to lane 3), but at much lower levels than either of the two wild-type TRIM5-21R proteins (compare lane 6 to lanes 7 and 8). A deletion in the SPRY domain V1 loop therefore reduced, but did not entirely eliminate, CA-NC/DNA binding. The TRIM5-21RΔSPRY protein did not bind CA-NC/DNA assemblies detectably in this assay, indicating that the SPRY domain was required for CA-NC/DNA binding (compare lane 5 to lanes 7 and 8). These experiments demonstrate that pure recombinant TRIM5-21R proteins bind directly to CA-NC/DNA assemblies and show that efficient binding requires the SPRY domain and its V1 loop.
Although our binding experiments were not performed under equilibrium conditions, we have nevertheless estimated the concentrations and stoichiometries of the binding reaction components in an initial effort to define the parameters that govern the TRIM5-CA interaction. CA-NC tubes and cones can assemble and disassemble reversibly, and we therefore estimated the concentration of CA-NC protein in the pelletable assemblies by quantifying Western blot band intensities and comparing them to a standard curve of known CA-NC concentrations. Analyzed in this fashion, the pelletable CA-NC protein in each experiment shown in Fig. 5B corresponded to 11 pmol (corresponding to an initial concentration of 55 nM). The initial concentration of TRIM5-21R subunits in each reaction mixture was 5 μM, and the quantity of pelleted TRIM5-21R dimer corresponded to 12 pmol of protein. Thus, in this experiment, the binding reaction was performed with substantial excess of TRIM5-21R dimer over assembled CA-NC protein (∼100-fold), and under these conditions the ratio of bound TRIM5-21 to CA-NC was approximately 1:1. The TRIM5-21RΔV1 protein bound less well in this experiment, at a TRIM5-21ΔV1/CA-NC ratio of 0.18:1. Although these estimated values neglect several potential complications, including possible incomplete CA-NC tube pelleting efficiency and dissociation of CA-NC assemblies during the assay, the data nevertheless imply that TRIM5-21R can bind assembled CA-NC lattices at nearly stoichiometric levels.
CA-NC/DNA assemblies bind TRIM5-21R dimers more efficiently than monomers.
TRIM5α protein oligomerization has been shown to enhance retroviral capsid binding owing to avidity effects (27), and it was therefore surprising that CA-NC/DNA assemblies bound equivalent levels of TRIM5-21R monomers and dimers in the previous assay. We therefore tested whether more stringent binding conditions might reveal differential CA-NC binding of TRIM5-21R monomers and dimers. The stringency was increased by reducing the TRIM5-21R concentration, and under these conditions CA-NC/DNA assemblies bound TRIM5-21R dimers better than monomers. This effect is illustrated in Fig. 5C, which shows that TRIM5-21R dimers bound better at two different initial TRIM5-21R concentrations (0.5 and 0.15 μM, compare lanes 3 and 4 to lanes 5 and 6 in the bottom panel). Under these more stringent conditions, the quantity of pelletable CA-NC protein did not change, but the ratios of pelletable CA-NC to bound TRIM5-21R proteins were substantially lower, demonstrating that the binding conditions were indeed more stringent (see the caption to Fig. 5C). These data suggest that although TRIM5-21R monomers and dimers can both bind CA-NC/DNA assemblies, TRIM5-21R dimerization enhances the efficiency of binding under more stringent conditions, in agreement with the observation that disruption of the coiled-coil oligomerization motif of TRIM5α inhibits restriction (27).
Electron microscopic analyses of TRIM5-21R-decorated CA-NC tubes.
The effects of TRIM5-21R binding to CA-NC tubes were visualized by using transmission electron microscopy of negatively stained CA-NC/DNA assemblies incubated with a 10- to 50-fold excess of TRIM5-21R prior to imaging. As shown in Fig. 6, TRIM5-21R binding did not dramatically alter the appearance of CA-NC/DNA tubes, but two effects were evident. First, CA-NC/DNA tubes incubated with excess TRIM5-21R consistently exhibited increased exterior stain deposition compared to tubes incubated with control TRIM5-21RΔV1 or TRIM5-21RΔSPRY proteins. The simplest explanation for this effect is that it reflects formation of TRIM5-21R coats on the CA-NC/DNA tube exteriors. This effect was sometimes subtle and difficult to discern, but in extreme cases resulted in a thick, dark irregular coat on tube exteriors, as though the TRIM5-21R were aggregating about the tube (Fig. 6, panel 8). Second, CA-NC/DNA tubes decorated with TRIM5-21R frequently appeared more broken and irregular than their counterparts incubated with control TRIM5-21RΔV1 or TRIM5-21RΔSPRY proteins (tube breaks are highlighted by arrows in Fig. 6, panels 5 to 8). This observation suggests that high levels of TRIM5-21R destabilized the tubes and increased their fragmentation, although the effect was modest, and most tubes remained intact. Overall, our electron microscopic data indicate that TRIM5-21R can coat and destabilize CA-NC/DNA assemblies, but both effects were subtle. Higher-resolution cryo-electron microscopic analyses will be required to determine the density and location of TRIM5-21 binding sites on the CA-NC/DNA assemblies.
EIAV core binding assays.
Retroviral cores are the actual targets of TRIM5α restriction, and we therefore tested whether TRIM5-21R could bind authentic, purified core particles. Cores isolated from EIAV vectors were used in these experiments because TRIM5αrh restricts EIAV (10, 23, 71; see Fig. S1B in the supplemental material) and because we are able to purify cores from membrane-stripped EIAV virions more reproducibly and in higher yields than HIV-1 cores. EIAV cores were membrane stripped with a brief Triton X-100 detergent treatment (30) and then purified on a sucrose gradient where they concentrated at the expected density of ∼1.24 g/ml (Fig. 7A).
Purified EIAV cores were incubated with a ∼16-fold excess of pure recombinant TRIM5-21R or control ΔV1 or ΔSPRY variants. As in the CA-NC/DNA binding assay, high-molecular-weight EIAV cores were pelleted by centrifugation through a sucrose cushion (Fig. 7B, lanes 4 to 6 in the sixth panel). Wild-type dimeric TRIM5-21R protein bound and copelleted with the EIAV cores but did not pellet in the absence of cores (compare lanes 3 and 6, fifth panel). In contrast, much lower levels of the control TRIM5-21R ΔV1 protein copelleted with the EIAV cores (compare lanes 5 and 6), and no detectable TRIM5-21R ΔSPRY protein copelleted with the cores (compare lanes 4 and 6). We therefore conclude that TRIM5-21R binds and copellets with authentic EIAV core particles and that this interaction is specific because the wild-type TRIM5-21R protein binds much better than control proteins lacking part of the V1 loop or the entire SPRY domain.
DISCUSSION
We have described the expression, purification, and characterization of TRIM5-21R, which represents the first study of a pure recombinant TRIM5α protein capable of restricting HIV-1 replication. Our experiments provide direct experimental support for the importance of both TRIM5α protein oligomerization and the B30.2/SPRY domain in retroviral capsid recognition. Specifically, we found that deletion of the SPRY domain eliminates binding to both HIV-1 CA-NC assemblies and EIAV cores, that a deletion in the V1 loop reduces binding significantly, and that TRIM5-21R dimerization enhances binding to HIV-1 CA-NC assemblies under stringent binding conditions. These in vitro binding activities correlate well with restriction activities in vivo, indicating that our in vitro experiments accurately recapitulate interactions between capsids and TRIM5 proteins during retroviral restriction.
Our experiments indicate that restricting TRIM5α proteins, including TRIM5α itself, are dimers. Previous studies had established that TRIM5α proteins oligomerize and that the oligomeric state might be trimeric (27, 36). We now show, however, that TRIM5-21R forms dimers but that chemically cross-linked TRIM5-21R dimers exhibit anomalously slow electrophoretic mobility, causing them to appear trimeric. Cross-linked TRIM5α proteins expressed in mammalian cells exhibit very similar mobilities, implying that authentic TRIM5α proteins are also dimeric. It will be of interest to determine whether other members of the TRIM protein family are also dimers. In this regard, we note that others have reported that the related TRIM21 protein forms trimers (45). This conclusion was again based upon the electrophoretic mobility of chemically cross-linked TRIM21 proteins, however, which may be subject to the same uncertainties as cross-linked TRIM5α proteins. Moreover, another group has reported that the TRIM21/SS-A/Ro52 protein may instead be dimeric, based on its gel filtration mobility (59), and two groups have reported that the isolated coiled-coil region of TRIM21 forms dimers rather than trimers (27, 40). We therefore speculate that dimerization may be a common property of many members of the tripartite motif protein family. If so, there are several intriguing similarities between TRIM proteins and immunoglobulin G proteins, since both types of protein can dimerize to display a pair of terminal immunoglobulin-like domains that can recognize antigens, including repeating motifs on viral capsid surfaces.
Once formed, TRIM5-21R dimers are stable and can be purified without appreciable dissociation. Similarly, we saw no evidence of monomer accumulation in equilibrium distributions of TRIM5-21R dimers across a range of micromolar concentrations, again indicating that assembled TRIM5-21R dimers are highly stable (or kinetically inert). Nevertheless, monomeric TRIM5-21R proteins were produced in our baculoviral expression system. These TRIM5-21R monomers did not exhibit a propensity to dimerize, suggesting that they may be kinetically “trapped,” perhaps owing to misfolding of the coiled-coil motif. Not surprisingly, a TRIM5-21R construct missing the SPRY domain and preceding L2 linker was exclusively monomeric, a finding consistent with previous experiments showing that the TRIM5α L2 region is required for oligomerization (40).
SPRY domains of TRIM proteins, presumably including TRIM5α, adopt immunoglobulin-like folds consisting of two antiparallel β-sheets. The ligand binding surface is located at one end of the β-sandwich and is composed of six extended loops and two pockets (25, 61). The loops, which are hypervariable in both amino acid composition and length, share homology with the complementarity-determining region loops of immunoglobulins and account for the species specificity of restriction. In particular, the variable region 1 loop of TRIM5αrh is a critical determinant of capsid binding and antiviral activity, and a deletion within the V1 loop also reduced capsid recognition by TRIM5-21R in our in vitro binding assays. Our results are therefore in good agreement with the prevailing model that retroviral capsid recognition is mediated by the variable loops of the SPRY domain of TRIM5α (34, 54, 68). Furthermore, our experiments demonstrate that this interaction is direct and can be highly efficient. Indeed, under some conditions we observed essentially stoichiometric binding of TRIM5-21R to CA-NC assemblies. Electron microscopic images of CA-NC tubes decorated with TRIM5-21R proteins also suggest that TRIM5-21R can “coat” the CA-NC surface, although higher-resolution studies will be necessary to confirm and extend this finding.
TRIM5α restriction represents a very intriguing problem in molecular recognition because specific TRIM5α alleles are able to recognize a remarkable variety of different retroviral capsids, despite their substantial variation in amino acid sequence and surface topology. This attribute is exemplified by TRIM5αrh, which is able to recognize both EIAV and HIV-1 capsids even though the HIV-1 and EIAV CA proteins share only ∼25% sequence identity. Retroviral capsids do share a common organization, however, since all capsids are assembled on a hexagonal surface lattice composed of CA hexamers. It is therefore likely that both reading heads of the dimeric TRIM5α protein engage similar sites on the capsid surface, and this is most easily accomplished by binding across a local twofold symmetry axis on the capsid lattice (although asymmetric binding is also a formal possibility). Hexagonal HIV-1 CA arrays exhibit two distinct types of local twofold symmetry, one of which is colinear with the local sixfold axes and the other of which connects adjacent hexamers. These two sites have different subunit orientations and spacing, and it is therefore likely that only one of these sites is the preferred TRIM5α binding site.
In solution, HIV-1 CA forms dimers that correspond to the twofold symmetric interactions between adjacent hexamers in the assembled hexagonal lattice. Expression of HIV-1 CA alone does not abrogate restriction, however, indicating that the CA dimer alone probably does not create a high-affinity TRIM5α binding site (14, 16, 51, 52). It is possible that CA alone cannot abrogate restriction because the CA solution dimer is not the preferred TRIM5α binding site or because this CA dimer is weak and/or inherently flexible. However, an interesting alternative is that isolated SPRY domains, and even isolated TRIM5α dimers, may have weak intrinsic capsid binding energies and that high-affinity binding is achieved through the cooperative formation of even higher-order TRIM5α assemblies. This model would be consistent with the observation that TRIM5α proteins are highly susceptible to aggregation or assembly in vitro and in vivo. Indeed, fluorescence images indicate that at least in some cases, large numbers of TRIM5α proteins can assemble on HIV-1 capsids in vivo (6). This model could also help explain how individual TRIM5 alleles can bind such a variety of different retroviral capsid surfaces because direct TRIM5α-CA interfaces would not need to form a large number of favorable interactions but rather could utilize just a few specific interactions and then be stabilized by the cooperative formation of multiple TRIM5α-CA and TRIM5α-TRIM5α interfaces. In contrast, isolated TRIM-Cyp proteins, in which a cyclophilin A domain replaces the SPRY domain, may bind specific retroviral capsids with higher affinities because the cyclophilin A domain can make specific interactions with individual HIV-1 CA subunits (18, 70). This “high-affinity” recognition mechanism could explain why the upstream RING and B-box domains play less important roles in TRIM-Cyp restriction activity (10, 13, 65, 66) but may come at the cost of reducing the ability of TRIM-Cyp restriction factors to recognize as broad a variety of different retroviral capsids as canonical TRIM5α alleles.
Our electron microscopic analyses also indicated that high-level TRIM5-21R binding destabilizes the CA-NC lattice to some degree because TRIM5-21R induced many more breaks in CA-NC/DNA tubes than did nonbinding control TRIM5-21R constructs. Destabilization of the CA-NC lattice could reflect steric hindrance upon saturation binding of TRIM5-21R and/or mismatches in the symmetry or spacing between the CA-NC lattice and putative higher-order TRIM5-21R assemblies. We emphasize, however, that the capsid destabilization induced by TRIM5-21R binding was a relatively minor effect because most CA-NC assemblies remained largely intact, even in the presence of a large excess of TRIM5-21R. Similarly, EIAV cores remained pelletable in the presence of excess TRIM5-21R. Thus, the dramatic destabilization of retroviral capsids observed under restrictive conditions in vivo does not appear to be a direct consequence of TRIM5α binding. Rather, it appears that capsid recognition by TRIM5α and subsequent destabilization/restriction are largely separable events. This idea is consistent with a series of other observations, including the fact that mutations in the B-box region of TRIM5α can abrogate restriction without apparently affecting capsid recognition (11), and that capsid destabilization is abrogated upon inhibition of the proteasome (6, 10).
In summary, our biochemical and biophysical studies have established that purified recombinant TRIM5-21R forms stable dimers that can bind specifically to higher-order HIV-1 CA-NC assemblies and to authentic EIAV core particles. It will be important to ascertain precisely how TRIM5α binds the capsid lattice and to determine the minimal CA assembly required for high-affinity binding, and we anticipate that the availability of recombinant TRIM5-21R protein will facilitate such studies.
Supplementary Material
Acknowledgments
We are grateful to Chad Nelson and the University of Utah Mass Spectrometry facility for ESI and LC mass spectral analyses, Rachel Klevit for the gift of expression vectors for E1 and E2 enzymes, and Chris Hill for critical review of the manuscript.
This research was supported by National Institutes of Health grants AI076121 (to C.A.), AI063978 and AI076094 (to J.G.S.), and AI045405 and GM082545 (to W.I.S.).
Footnotes
Published ahead of print on 17 September 2008.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Accola, M. A., A. Ohagen, and H. G. Gottlinger. 2000. Isolation of human immunodeficiency virus type 1 cores: retention of Vpr in the absence of p6(gag). J. Virol. 746198-6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson, J. L., E. M. Campbell, X. Wu, N. Vandegraaff, A. Engelman, and T. J. Hope. 2006. Proteasome inhibition reveals that a functional preintegration complex intermediate can be generated during restriction by diverse TRIM5 proteins. J. Virol. 809754-9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bieniasz, P. D. 2004. Intrinsic immunity: a front-line defense against viral attack. Nat. Immunol. 51109-1115. [DOI] [PubMed] [Google Scholar]
- 4.Briggs, J. A., T. Wilk, R. Welker, H. G. Krausslich, and S. D. Fuller. 2003. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 221707-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brzovic, P. S., A. Lissounov, D. E. Christensen, D. W. Hoyt, and R. E. Klevit. 2006. A UbcH5/ubiquitin noncovalent complex is required for processive BRCA1-directed ubiquitination. Mol. Cell 21873-880. [DOI] [PubMed] [Google Scholar]
- 6.Campbell, E. M., O. Perez, J. L. Anderson, and T. J. Hope. 2008. Visualization of a proteasome-independent intermediate during restriction of HIV-1 by rhesus TRIM5α. J. Cell Biol. 180549-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell, S., and V. M. Vogt. 1995. Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J. Virol. 696487-6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christensen, D. E., P. S. Brzovic, and R. E. Klevit. 2007. E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat. Struct. Mol. Biol. 14941-948. [DOI] [PubMed] [Google Scholar]
- 9.Cole, J. L. 2004. Analysis of heterogeneous interactions. Methods Enzymol. 384212-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diaz-Griffero, F., A. Kar, M. Lee, M. Stremlau, E. Poeschla, and J. Sodroski. 2007. Comparative requirements for the restriction of retrovirus infection by TRIM5α and TRIMCyp. Virology 369400-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diaz-Griffero, F., A. Kar, M. Perron, S. H. Xiang, H. Javanbakht, X. Li, and J. Sodroski. 2007. Modulation of retroviral restriction and proteasome inhibitor-resistant turnover by changes in the TRIM5α B-box 2 domain. J. Virol. 8110362-10378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diaz-Griffero, F., X. Li, H. Javanbakht, B. Song, S. Welikala, M. Stremlau, and J. Sodroski. 2006. Rapid turnover and polyubiquitylation of the retroviral restriction factor TRIM5. Virology 349300-315. [DOI] [PubMed] [Google Scholar]
- 13.Diaz-Griffero, F., N. Vandegraaff, Y. Li, K. McGee-Estrada, M. Stremlau, S. Welikala, Z. Si, A. Engelman, and J. Sodroski. 2006. Requirements for capsid-binding and an effector function in TRIMCyp-mediated restriction of HIV-1. Virology 351404-419. [DOI] [PubMed] [Google Scholar]
- 14.Dodding, M. P., M. Bock, M. W. Yap, and J. P. Stoye. 2005. Capsid processing requirements for abrogation of fv1 and ref1 restriction. J. Virol. 7910571-10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fisher, R. J., A. Rein, M. Fivash, M. A. Urbaneja, J. R. Casas-Finet, M. Medaglia, and L. E. Henderson. 1998. Sequence-specific binding of human immunodeficiency virus type 1 nucleocapsid protein to short oligonucleotides. J. Virol. 721902-1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forshey, B. M., J. Shi, and C. Aiken. 2005. Structural requirements for recognition of the human immunodeficiency virus type 1 core during host restriction in owl monkey cells. J. Virol. 79869-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forshey, B. M., U. von Schwedler, W. I. Sundquist, and C. Aiken. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 765667-5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gamble, T. R., F. F. Vajdos, S. Yoo, D. K. Worthylake, M. Houseweart, W. I. Sundquist, and C. P. Hill. 1996. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 871285-1294. [DOI] [PubMed] [Google Scholar]
- 19.Ganser-Pornillos, B. K., A. Cheng, and M. Yeager. 2007. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 13170-79. [DOI] [PubMed] [Google Scholar]
- 20.Ganser-Pornillos, B. K., M. Yeager, and W. I. Sundquist. 2008. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 18203-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganser, B. K., S. Li, V. Y. Klishko, J. T. Finch, and W. I. Sundquist. 1999. Assembly and analysis of conical models for the HIV-1 core. Science 28380-83. [DOI] [PubMed] [Google Scholar]
- 22.Hatfield, P. M., J. Callis, and R. D. Vierstra. 1990. Cloning of ubiquitin activating enzyme from wheat and expression of a functional protein in Escherichia coli. J. Biol. Chem. 26515813-15817. [PubMed] [Google Scholar]
- 23.Hatziioannou, T., S. Cowan, S. P. Goff, P. D. Bieniasz, and G. J. Towers. 2003. Restriction of multiple divergent retroviruses by Lv1 and Ref1. EMBO J. 22385-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hennig, J., A. Bresell, M. Sandberg, K. D. Hennig, M. Wahren-Herlenius, B. Persson, and M. Sunnerhagen. 2008. The fellowship of the RING: the RING-B-box linker region interacts with the RING in TRIM21/Ro52, contains a native autoantigenic epitope in Sjogren syndrome, and is an integral and conserved region in TRIM proteins. J. Mol. Biol. 377431-449. [DOI] [PubMed] [Google Scholar]
- 25.James, L. C., A. H. Keeble, Z. Khan, D. A. Rhodes, and J. Trowsdale. 2007. Structural basis for PRYSPRY-mediated tripartite motif (TRIM) protein function. Proc. Natl. Acad. Sci. USA 1046200-6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Javanbakht, H., F. Diaz-Griffero, M. Stremlau, Z. Si, and J. Sodroski. 2005. The contribution of RING and B-box 2 domains to retroviral restriction mediated by monkey TRIM5α. J. Biol. Chem. 28026933-26940. [DOI] [PubMed] [Google Scholar]
- 27.Javanbakht, H., W. Yuan, D. F. Yeung, B. Song, F. Diaz-Griffero, Y. Li, X. Li, M. Stremlau, and J. Sodroski. 2006. Characterization of TRIM5α trimerization and its contribution to human immunodeficiency virus capsid binding. Virology 353234-246. [DOI] [PubMed] [Google Scholar]
- 28.Jin, Z., L. Jin, D. L. Peterson, and C. L. Lawson. 1999. Model for lentivirus capsid core assembly based on crystal dimers of EIAV p26. J. Mol. Biol. 28683-93. [DOI] [PubMed] [Google Scholar]
- 29.Keckesova, Z., L. M. Ylinen, and G. J. Towers. 2004. The human and African green monkey TRIM5α genes encode Ref1 and Lv1 retroviral restriction factor activities. Proc. Natl. Acad. Sci. USA 10110780-10785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kotov, A., J. Zhou, P. Flicker, and C. Aiken. 1999. Association of Nef with the human immunodeficiency virus type 1 core. J. Virol. 738824-8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laue, T., B. Shah, T. Ridgeway, and S. Pelletier. 1992. Computer-aided interpretation of analytical sedimentation data for proteins, p. 90-125. In A. Rowe and J. Horton (ed.), Ultracentrifugation in biochemistry and polymer science. Royal Society of Chemistry, Cambridge, England.
- 32.Li, S., C. P. Hill, W. I. Sundquist, and J. T. Finch. 2000. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407409-413. [DOI] [PubMed] [Google Scholar]
- 33.Li, X., B. Gold, C. O'Huigin, F. Diaz-Griffero, B. Song, Z. Si, Y. Li, W. Yuan, M. Stremlau, C. Mische, H. Javanbakht, M. Scally, C. Winkler, M. Dean, and J. Sodroski. 2006. Unique features of TRIM5α among closely related human TRIM family members. Virology 360419-433. [DOI] [PubMed] [Google Scholar]
- 34.Li, Y., X. Li, M. Stremlau, M. Lee, and J. Sodroski. 2006. Removal of arginine 332 allows human TRIM5α to bind human immunodeficiency virus capsids and to restrict infection. J. Virol. 806738-6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu, H. L., Y. Q. Wang, C. H. Liao, Y. Q. Kuang, Y. T. Zheng, and B. Su. 2005. Adaptive evolution of primate TRIM5α, a gene restricting HIV-1 infection. Gene 362109-116. [DOI] [PubMed] [Google Scholar]
- 36.Mische, C. C., H. Javanbakht, B. Song, F. Diaz-Griffero, M. Stremlau, B. Strack, Z. Si, and J. Sodroski. 2005. Retroviral restriction factor TRIM5α is a trimer. J. Virol. 7914446-14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nisole, S., and A. Saib. 2004. Early steps of retrovirus replicative cycle. Retrovirology 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohkura, S., M. W. Yap, T. Sheldon, and J. P. Stoye. 2006. All three variable regions of the TRIM5α B30.2 domain can contribute to the specificity of retrovirus restriction. J. Virol. 808554-8565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O'Rourke, J. P., J. C. Olsen, and B. A. Bunnell. 2005. Optimization of equine infectious anemia derived vectors for hematopoietic cell lineage gene transfer. Gene Ther. 1222-29. [DOI] [PubMed] [Google Scholar]
- 40.Ottosson, L., J. Hennig, A. Espinosa, S. Brauner, M. Wahren-Herlenius, and M. Sunnerhagen. 2006. Structural, functional, and immunologic characterization of folded subdomains in the Ro52 protein targeted in Sjogren's syndrome. Mol. Immunol. 43588-598. [DOI] [PubMed] [Google Scholar]
- 41.Perez-Caballero, D., T. Hatziioannou, A. Yang, S. Cowan, and P. D. Bieniasz. 2005. Human tripartite motif 5α domains responsible for retrovirus restriction activity and specificity. J. Virol. 798969-8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perron, M. J., M. Stremlau, M. Lee, H. Javanbakht, B. Song, and J. Sodroski. 2007. The human TRIM5α restriction factor mediates accelerated uncoating of the N-tropic murine leukemia virus capsid. J. Virol. 812138-2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perron, M. J., M. Stremlau, B. Song, W. Ulm, R. C. Mulligan, and J. Sodroski. 2004. TRIM5α mediates the postentry block to N-tropic murine leukemia viruses in human cells. Proc. Natl. Acad. Sci. USA 10111827-11832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pickart, C. M., and S. Raasi. 2005. Controlled synthesis of polyubiquitin chains. Methods Enzymol. 39921-36. [DOI] [PubMed] [Google Scholar]
- 45.Rhodes, D. A., and J. Trowsdale. 2007. TRIM21 is a trimeric protein that binds IgG Fc via the B30.2 domain. Mol. Immunol. 442406-2414. [DOI] [PubMed] [Google Scholar]
- 46.Roberts, M. M., and S. Oroszlan. 1989. The preparation and biochemical characterization of intact capsids of equine infectious anemia virus. Biochem. Biophys. Res. Commun. 160486-494. [DOI] [PubMed] [Google Scholar]
- 47.Rold, C. J., and C. Aiken. 2008. Proteasomal degradation of TRIM5α during retrovirus restriction. PLoS Pathog. 4e1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sandrin, V., and F. L. Cosset. 2006. Intracellular versus cell surface assembly of retroviral pseudotypes is determined by the cellular localization of the viral glycoprotein, its capacity to interact with Gag, and the expression of the Nef protein. J. Biol. Chem. 281528-542. [DOI] [PubMed] [Google Scholar]
- 48a.Sanes, J. R., J. L. Rubenstein, and J. F. Nicolas. 1986. Use of recombinant retrovirus to study post-implantation cell lineage in mouse embryos. Embo J. 53133-3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sawyer, S. L., M. Emerman, and H. S. Malik. 2007. Discordant evolution of the adjacent antiretroviral genes TRIM22 and TRIM5 in mammals. PLoS Pathog. 3e197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sawyer, S. L., L. I. Wu, M. Emerman, and H. S. Malik. 2005. Positive selection of primate TRIM5α identifies a critical species-specific retroviral restriction domain. Proc. Natl. Acad. Sci. USA 1022832-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sebastian, S., and J. Luban. 2005. TRIM5alpha selectively binds a restriction-sensitive retroviral capsid. Retrovirology 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi, J., and C. Aiken. 2006. Saturation of TRIM5α-mediated restriction of HIV-1 infection depends on the stability of the incoming viral capsid. Virology 350493-500. [DOI] [PubMed] [Google Scholar]
- 53.Song, B., F. Diaz-Griffero, D. H. Park, T. Rogers, M. Stremlau, and J. Sodroski. 2005. TRIM5α association with cytoplasmic bodies is not required for antiretroviral activity. Virology 343201-211. [DOI] [PubMed] [Google Scholar]
- 54.Song, B., B. Gold, C. O'Huigin, H. Javanbakht, X. Li, M. Stremlau, C. Winkler, M. Dean, and J. Sodroski. 2005. The B30.2(SPRY) domain of the retroviral restriction factor TRIM5α exhibits lineage-specific length and sequence variation in primates. J. Virol. 796111-6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stremlau, M., C. M. Owens, M. J. Perron, M. Kiessling, P. Autissier, and J. Sodroski. 2004. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature 427848-853. [DOI] [PubMed] [Google Scholar]
- 56.Stremlau, M., M. Perron, M. Lee, Y. Li, B. Song, H. Javanbakht, F. Diaz-Griffero, D. J. Anderson, W. I. Sundquist, and J. Sodroski. 2006. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5α restriction factor. Proc. Natl. Acad. Sci. USA 1035514-5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stremlau, M., M. Perron, S. Welikala, and J. Sodroski. 2005. Species-specific variation in the B30.2(SPRY) domain of TRIM5α determines the potency of human immunodeficiency virus restriction. J. Virol. 793139-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Towers, G. J. 2007. The control of viral infection by tripartite motif proteins and cyclophilin A. Retrovirology 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang, D., J. P. Buyon, Z. Yang, F. Di Donato, M. E. Miranda-Carus, and E. K. Chan. 2001. Leucine zipper domain of 52 kDa SS-A/Ro promotes protein dimer formation and inhibits in vitro transcription activity. Biochim. Biophys. Acta 1568155-161. [DOI] [PubMed] [Google Scholar]
- 59a.Ward, D. M., M. B. Vaughn, S. L. Shiflett, P. L. White, A. L. Pollock, J. Hill, R. Schnegelberger, W. I. Sundquist, and J. Kaplan. 2005. The role of LIP5 and CHMP5 in multivesicular body formation and HIV-1 budding in mammalian cells. J. Biol. Chem. 28010548-10555. [DOI] [PubMed] [Google Scholar]
- 60.Welker, R., H. Hohenberg, U. Tessmer, C. Huckhagel, and H. G. Krausslich. 2000. Biochemical and structural analysis of isolated mature cores of human immunodeficiency virus type 1. J. Virol. 741168-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woo, J. S., H. Y. Suh, S. Y. Park, and B. H. Oh. 2006. Structural basis for protein recognition by B30.2/SPRY domains. Mol. Cell 24967-976. [DOI] [PubMed] [Google Scholar]
- 62.Wu, X., J. L. Anderson, E. M. Campbell, A. M. Joseph, and T. J. Hope. 2006. Proteasome inhibitors uncouple rhesus TRIM5α restriction of HIV-1 reverse transcription and infection. Proc. Natl. Acad. Sci. USA 1037465-7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu, L., L. Yang, P. K. Moitra, K. Hashimoto, P. Rallabhandi, S. Kaul, G. Meroni, J. P. Jensen, A. M. Weissman, and P. D'Arpa. 2003. BTBD1 and BTBD2 colocalize to cytoplasmic bodies with the RBCC/tripartite motif protein, TRIM5δ. Exp. Cell Res. 288:84-93. [DOI] [PubMed] [Google Scholar]
- 64.Yamauchi, K., K. Wada, K. Tanji, M. Tanaka, and T. Kamitani. 2008. Ubiquitination of E3 ubiquitin ligase TRIM5α and its potential role. FEBS J. 2751540-1555. [DOI] [PubMed] [Google Scholar]
- 65.Yap, M. W., M. P. Dodding, and J. P. Stoye. 2006. Trim-cyclophilin A fusion proteins can restrict human immunodeficiency virus type 1 infection at two distinct phases in the viral life cycle. J. Virol. 804061-4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yap, M. W., G. B. Mortuza, I. A. Taylor, and J. P. Stoye. 2007. The design of artificial retroviral restriction factors. Virology 365302-314. [DOI] [PubMed] [Google Scholar]
- 67.Yap, M. W., S. Nisole, C. Lynch, and J. P. Stoye. 2004. Trim5α protein restricts both HIV-1 and murine leukemia virus. Proc. Natl. Acad. Sci. USA 10110786-10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yap, M. W., S. Nisole, and J. P. Stoye. 2005. A single amino acid change in the SPRY domain of human Trim5α leads to HIV-1 restriction. Curr. Biol. 1573-78. [DOI] [PubMed] [Google Scholar]
- 69.Yee, J. K., A. Miyanohara, P. LaPorte, K. Bouic, J. C. Burns, and T. Friedmann. 1994. A general method for the generation of high-titer, pantropic retroviral vectors: highly efficient infection of primary hepatocytes. Proc. Natl. Acad. Sci. USA 919564-9568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yoo, S., D. G. Myszka, C. Yeh, M. McMurray, C. P. Hill, and W. I. Sundquist. 1997. Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J. Mol. Biol. 269780-795. [DOI] [PubMed] [Google Scholar]
- 71.Zhang, F., T. Hatziioannou, D. Perez-Caballero, D. Derse, and P. D. Bieniasz. 2006. Antiretroviral potential of human tripartite motif-5 and related proteins. Virology 353396-409. [DOI] [PubMed] [Google Scholar]
- 72.Zhang, M., M. Windheim, S. M. Roe, M. Peggie, P. Cohen, C. Prodromou, and L. H. Pearl. 2005. Chaperoned ubiquitylation: crystal structures of the CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol. Cell 20525-538. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.