

Abstract
Signaling mechanisms that regulate astrocyte reactivity and scar formation after spinal cord injury (SCI) are not well defined. We used the Cre recombinase (Cre)-loxP system under regulation of the mouse glial fibrillary acidic protein (GFAP) promoter to conditionally delete the cytokine and growth factor signal transducer, signal transducer and activator of transcription 3 (STAT3), from astrocytes. After SCI in GFAP-Cre reporter mice, >99% of spinal cord cells that exhibited Cre activity as detected by reporter protein expression were GFAP-expressing astrocytes. Conditional deletion (or knock-out) of STAT3 (STAT3-CKO) from astrocytes in GFAP-Cre-loxP mice was confirmed in vivo and in vitro. In uninjured adult STAT3-CKO mice, astrocytes appeared morphologically similar to those in STAT3+/+ mice except for a partially reduced expression of GFAP. In STAT3+/+ mice, phosphorylated STAT3 (pSTAT3) was not detectable in astrocytes in uninjured spinal cord, and pSTAT3 was markedly upregulated after SCI in astrocytes and other cell types near the injury. Mice with STAT3-CKO from astrocytes exhibited attenuated upregulation of GFAP, failure of astrocyte hypertrophy, and pronounced disruption of astroglial scar formation after SCI. These changes were associated with increased spread of inflammation, increased lesion volume and partially attenuated motor recovery over the first 28 d after SCI. These findings indicate that STAT3 signaling is a critical regulator of certain aspects of reactive astrogliosis and provide additional evidence that scar-forming astrocytes restrict the spread of inflammatory cells after SCI.
Keywords: astrocyte, astroglia, cytokine, GFAP, glial fibrillary acidic protein, histochemistry, immunoreactivity, inflammation, spinal cord injury
Introduction
Astrocytes respond to spinal cord injury (SCI) and other CNS insults with a variety of different potential changes, including alterations in many types of molecular expression, cellular hypertrophy, migration, proliferation, and scar formation (Reier and Houle, 1988; Norton et al., 1992; Amat et al., 1996; Silver and Miller, 2004). The intercellular and intracellular signaling mechanisms that regulate different aspects of astrocyte reactivity (astrogliosis) in response to CNS insults in vivo are not well characterized. These signaling mechanisms are important because they determine not only the degree to which reactive astrocytes maintain, modify, or suppress the functions that astrocytes normally execute in uninjured tissue, but also whether or not reactive astrocytes initiate new, injury-induced activities that may be beneficial or harmful. A better understanding of such signaling mechanisms may lead to novel strategies to attenuate negative aspects and augment positive aspects of astrocyte reactivity, and may contribute to combinatorial treatment strategies that address multiple aspects of SCI (Silver and Miller, 2004; Filbin, 2006; Cafferty et al., 2008; Lu and Tuszynski, 2008).
Astrocytes, like other cells, respond to multiple extracellular signaling molecules through a complex assortment of intracellular signal transduction pathways that activate cAMP, MAP (microtubule-associated protein) kinases, nuclear factor κB (NF-κB), Janus kinase-signal transducers and activators of transcription (Jak-STATs), and others, and several of these pathways have been implicated as potential regulators of astrogliosis (Shafit-Zagardo et al., 1988; John et al., 2003; Brambilla et al., 2005; Okada et al., 2006). The STAT3 is a member of the Jak-STAT signaling family that transduces signals for many cytokines and growth factors (Aaronson and Horvath, 2002). STAT3 is activated in many cell types by a number of cytokines implicated in injury responses (Takeda et al., 1997), several of which, including interleukin-6 (IL-6), CNTF, leukemia inhibitory factor (LIF), epidermal growth factor (EGF), and TGFα, have been implicated as triggers of reactive astrogliosis (Balasingam et al., 1994; Winter et al., 1995; Klein et al., 1997; Rabchevsky et al., 1998; Levison et al., 2000; Albrecht et al., 2002). In the CNS, STAT3 is expressed by astrocytes, neurons, and other cell types (Cattaneo et al., 1999), and activation of STAT3 by phosphorylation increases markedly after CNS insults (Acarin et al., 2000; Justicia et al., 2000; Sriram et al., 2004; Yamauchi et al., 2006). Thus, STAT3 is a good candidate to be an activator of certain aspects of astrogliosis.
In this study, we investigated the role of STAT3 signaling in regulating specific aspects of the response of astrocytes to spinal cord injury (SCI) in vivo by using the Cre-loxP system (Sauer, 1994) to achieve a conditional gene deletion or knock-out (CKO) of STAT3 in astrocytes. We cross-bred mice in which Cre recombinase (Cre) was targeted to astrocytes using the mouse glial fibrillary acidic protein (GFAP) promoter (Garcia et al., 2004; Sofroniew, 2005) with mice in which loxP sites were inserted to flank exon 22 of the STAT3 gene, which encodes a phosphorylation site critical for STAT3 activation (Takeda et al., 1998).
Materials and Methods
Animals.
All GFAP-STAT3-CKO and control mice were obtained from the same breeding colony of GFAP-Cre mice of line 73.12 crossed with STAT3-loxP mice on a C57BL/6 background. GFAP-Cre mice of line 73.12 were generated as described (Garcia et al., 2004), using a 15 kb mouse GFAP promoter cassette (clone 445) containing all introns, promoter regulatory elements, exons, and 2 kb of 3′ and 2.5 kb of 5′ flanking regions of the mouse GFAP gene (Johnson et al., 1995). GFAP-Cre mice of line 73.12 were cross-bred with STAT3-loxP mice having loxP sites flanking exon 22 of the STAT3 gene, which encodes a tyrosine residue (tyr705), essential for STAT3 activation, were generated as described (Takeda et al., 1998). Control mice of several genotypes were compared, including mice that (1) carried no transgene or loxP sites (i.e., nontransgenic or wild-type mice), (2) were only heterozygous for GFAP-Cre and carried no STAT3-loxP, or (3) were only heterozygous or homozygous for STAT3-loxP and carry no GFAP-Cre. These three groups exhibited no significant differences (ANOVA) in any parameter evaluated and data from different controls were pooled. GFAP-Cre mice of line 73.12 were also cross-bred with two lines of reporter mice that expressed either β-galactosidase (β-gal) or green fluorescent protein (GFP) downstream of loxP flanked stop signals via either the ROSA promoter for β-gal (Soriano, 1999), or a cytomegalovirus-enhanced actin promoter for GFP (Novak et al., 2000). Mice were housed in a 12 h light/dark cycle in an SPF facility with controlled temperature and humidity and allowed ad libitum access to food and water, and experiments conducted according to protocols approved by the Chancellor's Animal Research Committee of the Office for Protection of Research Subjects at the University of California, Los Angeles.
Cell cultures.
Primary astrocyte cultures were prepared as described previously (Imura et al., 2003; Imura et al., 2006). Briefly, cells were derived from cortices of newborn [postnatal day 0 (P0)–P2] mice and seeded in medium consisting of DMEM/F12 with 10% fetal bovine calf serum. Once confluence was attained, cultures were shaken to remove nonadherent cells and replated (passage 1); when confluence was attained again, this procedure was repeated (passage 2), resulting in cultures of >95% GFAP-positive cells.
Surgical procedures.
All surgical procedures were performed under general anesthesia with isoflurane in oxygen-enriched air using an operating microscope (Zeiss) and rodent stereotaxic apparatus (David Kopf Instruments). Laminectomy of a single vertebra was performed and moderate crush SCI were made at the level of L1/L2 using No. 5 Dumont forceps (Fine Science Tools) ground down to a tip width of 0.4 mm and modified with a spacer so that at maximal closure a 0.4 mm space remained. This closure distance was selected by comparing results achieved with forceps that closed to 1.0 or 0 mm spaces (Faulkner et al., 2004). Forceps were used to compress the cord laterally from both sides for 10 s.
Overall design of in vivo experiments.
The effects of moderate crush SCI were compared in GFAP-STAT3-CKO mice and control mice of different genotypes (see above, Animals). Animals were randomly assigned numbers and evaluated blind to genotype and experimental condition. Mice were evaluated on behavioral tasks to assess hindlimb function and by qualitative and quantitative histological and biochemical procedures as described below.
Behavioral analysis and statistical evaluation.
Mice were evaluated using three behavioral tasks selected to assess hindlimb function before and on days 3, 7, 10, 14 and 28 after SCI (Faulkner et al., 2004). For the open-field locomotor task, the objective of this evaluation was to assess gross voluntary use of the hindlimbs, and did not attempt to define subtle differences in usage that might be correlated with specific neural mechanisms that might underlie dysfunctions. We used a simple six-point scale (Fehlings and Tator, 1995), which focused on gross aspects of hindlimb function, and which we found more appropriate for our goals and our model of upper lumbar SCI. Mice were evaluated in an open field by the same two observers blind to the experimental condition, and received a score for gross voluntary movement of each hindlimb using an operationally defined six-point scale: (0) no voluntary hindlimb movement; (1) little voluntary hindlimb movement; (2) hindlimb movements obvious but did not assist in weight support or stepping; (3) hindlimb assisted in occasional weight support and plantar placement, but not in stepping; (4) hindlimb used for weight support and stepping, but obvious disability; (5) hindlimb function essentially normal (Fehlings and Tator, 1995). Left and right hindlimbs were scored separately and averaged. To assess balance and ability to coordinate stepping, animals were placed on a single lane rotorod (Med Associates) for three trials per session. The rotorod was set for constant acceleration from 3.0 to 30 rpm over 300 s and animals were scored on seconds to fall. Each trial was scored individually and averaged for a final score per session. To assess stepping patterns of forelimbs and hindlimbs after injury, animals were required to run along a paper-lined runway (3 feet long, 3 inches wide) to obtain an edible treat in a darkened box at the end. The plantar surfaces of forelimbs and hindlimbs were brushed with red and black nontoxic paint, respectively, and qualitative analysis was performed on plantar stepping, stride length and width, toe or dorsal foot drags, and overall stepping ability (Ma et al., 2001). All statistical evaluations were performed by ANOVA with post hoc, independent pairwise analysis (Prism; GraphPad). The group sizes used for all statistical comparisons of behavioral evaluations of motor performance were n = 8 or more subjects per experimental group.
Histological procedures.
After terminal anesthesia by barbiturate overdose, mice were perfused transcardially with buffered 4% paraformaldehyde. Spinal cords were removed, postfixed for a further 2 h, and cryoprotected in buffered 30% sucrose overnight. Forty micrometer transverse or 30 μm horizontal frozen sections were prepared using a cryostat microtome (Leica) and processed for various kinds of histological evaluation as described previously (Faulkner et al., 2004). Brightfield immunohistochemistry was performed using biotinylated secondary antibodies (Vector Laboratories), biotin-avidin-peroxidase complex (Vector Laboratories), and diaminobenzidine (Vector Laboratories) as the developing agent. Fluorescence immunohistochemistry was performed using Alexa Fluor-tagged secondary antibodies Alexa 488 (green), Alexa 568 (red), or Alexa 350 (blue) (Invitrogen). Primary antibodies were as follows: rabbit anti-GFAP (1:2000 or 1:20,000; Dako); rat anti-GFAP (1:1000 or 1:5000; Zymed Laboratories); rabbit anti-Cre (1:4000; Covance); mouse anti-Cre (1:100; Covance); rabbit anti-β-gal (1:400 or 1:16,000; MP Biomedicals); rabbit anti-glutathione S-transferase π (GSTπ) (1:150; BD Biosciences); mouse anti-neuronal-specific nuclear protein (anti-NeuN) (1:400 or 1:4000; Millipore); rabbit anti-vimentin (1:6000; Sigma); rabbit anti-glutamine synthetase (1:4000; Sigma); rabbit anti-STAT3 (1:100; Cell Signaling Technology); rabbit anti-phosphorylated STAT3 (pSTAT3) (tyr705; 1:150; Cell Signaling Technology); rat anti-mouse CD45 (1:100 or 1:2000; BD Pharmingen); rabbit anti-fibronectin (1:1000 or 1:5000; Dako). Staining with cresyl violet or luxol fast blue was conducted according to standard procedures. Stained sections were examined and photographed using brightfield and fluorescence microscopy (Zeiss) and scanning confocal laser microscopy (Leica).
Morphometric and statistical evaluation.
Cell counts, cell volume, and lesion volume, were determined using unbiased sampling (Gundersen et al., 1988) and stereological image-analysis software (StereoInvestigator or NeuroLucida; MicroBrightField) operating a computer-driven microscope regulated in the x, y, and z axes (Zeiss) in a manner described in detail previously (Faulkner et al., 2004). For cell density and cell volume measurements in uninjured mice, separate gray and white matter areas were traced in three randomly selected sections per mouse in the L1/L2 region; three sampling frames of 350 × 350 μm were selected per section at random within the traced region by the image-analysis software (StereoInvestigator) and computer-driven stage, and cell numbers were counted and the volume of the counted tissue was calculated. In addition, cell sizes were measured using the nucleator method on >100 cells per animal. For cell volume determinations after SCI, separate gray and white matter areas to be sampled were traced as 200-μm-wide regions immediately adjacent to the wound margin; sampling frames of 35 × 35 μm were selected at random within the traced region by the image-analysis software (StereoInvestigator) and computer-driven stage, and cell sizes were measured using the nucleator method on >100 cells per animal. Lesion volume determinations were made by tracing lesion areas and spinal cord outlines in series of sequentially spaced sections using image-analysis software (NeuroLucida) followed by three-dimensional reconstruction (Faulkner et al., 2004). All statistical evaluations were performed by ANOVA with post hoc, independent pairwise analysis (Prism, GraphPad). The group sizes used for all statistical comparisons of morphometric determinations were n = 4 or more subjects per experimental group.
Western blot analysis.
Total protein was extracted from freshly harvested spinal cord tissue using a lysis buffer: 50 mm Tris-HCl, 150 mm NaCl, 1% Nonidet P-40, 10 mm NaF, 10 mm EDTA, 0.5 mm dithiothreitol, 1 mm Na3VO4, 1 mm phenylmethlsulfonyl fluoride, 4 μg/ml leupeptin, and 4 μg/ml pepstatin A, pH 7.4, and centrifuged at 12,000 × g for 25 min at 4 C. The supernatant was used for measurements of protein as described previously (Qi et al., 2005; Liu and Xu, 2006): Protein concentration was determined using a Bio-Rad DC protein kit. Protein preparations were separated by SDS-PAGE and transferred to a polyvinlyidene difluoride membrane and blocked with 5% nonfat milk Tris-buffered saline–Tween 20 buffer and probed using various antibodies. The Western blots were visualized using GE Healthcare ECL Western Blotting Analysis System and exposed to Kodak x-ray film. Equivalence of protein loading was verified by probing for actin. Antibodies used were as follows: goat anti-GFAP (1:5000; Dako); rabbit anti-pSTAT3 (tyr705; 1:500; Cell Signaling Technology); rabbit anti-pSTAT1 (tyr701; 1:200; Santa Cruz Biotechnology); rabbit anti-actin (1:500; Sigma).
Results
Specificity of Cre targeting to astrocytes
To test the specificity of Cre targeting to astrocytes at the single-cell level in the spinal cord, we cross-bred GFAP-Cre mice (line 73.12) with two strains of reporter mice that encoded the reporter proteins β-gal or GFP. Single- and double-labeling immunohistochemistry showed that both Cre (Fig. 1A,C) and the reporter β-gal (Fig. 1B,D) were expressed throughout the spinal cord by cells with a distribution, density, and morphology typical of astrocytes, and that essentially all cells that expressed detectable levels of Cre (Fig. 1E) or β-gal (Fig. 1F) also expressed GFAP. We quantitatively assessed the efficacy and specificity of Cre enzymatic activity using confocal microscopic analysis of the reporter protein GFP in combination with immunofluorescence of chemical markers for astrocytes, neurons, oligodendrocytes, and microglia/inflammatory cells in the spinal cord after SCI (Figs. 2, 3). In both gray (Fig. 2A) and white (B) matter of spinal cord, >99% of cells that expressed reporter protein also expressed the astrocyte marker, GFAP (Fig. 2E). Conversely, >97% of cells that expressed GFAP also expressed reporter protein (Figs. 2E). We found no cells in the spinal cord that expressed both reporter protein and the neuron marker, NeuN (Figs. 2C,E). In addition, no cells expressed both reporter protein and CD45, a marker for microglia and all white blood cells (Figs. 2D,E). In both gray (Fig. 3A,C,F) and white (B,D,F) matter of the spinal cord, the vast majority of reporter-positive cells did not express the widely used marker of mature oligodendrocytes, GSTπ (Tansey and Cammer, 1991; Arnett et al., 2001; Gregg et al., 2007), and vise versa. A small percentage of cells was colabeled for reporter and GSTπ (Fig. 3E,F), and these cells had the morphological appearance of astrocytes and also expressed GFAP (Fig. 3E,F), suggesting that these cells were a subtype of astrocyte. Our findings are consistent with reports by others that the mouse GFAP promoter is not active in oligodendrocyte progenitor cells in spinal cord (Casper et al., 2007). Together, these findings indicate that in the spinal cord of 73.12 GFAP-Cre reporter mice, essentially all cells that exhibit Cre activity (i.e., reporter protein) are GFAP-expressing astrocytes, and that Cre activity is not exhibited by neurons, oligodendrocytes, or microglia/inflammatory cells.
Figure 1.
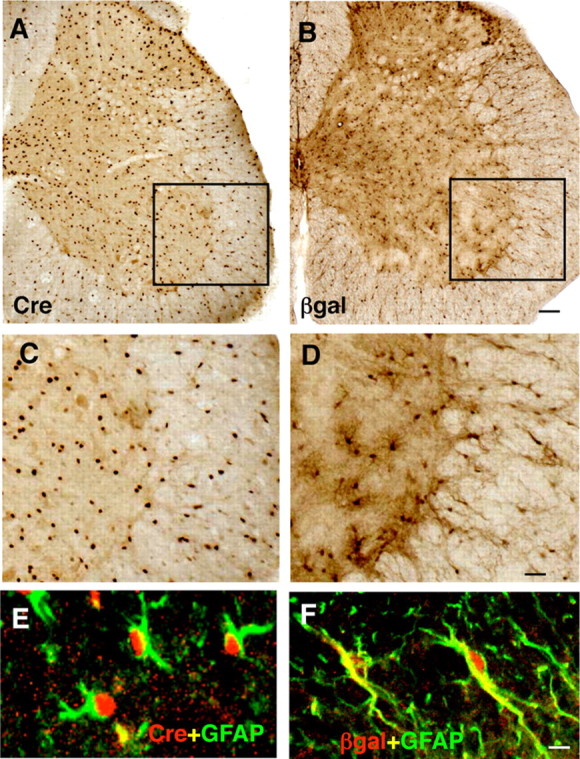
Distribution and appearance of cells expressing Cre and exhibiting Cre-mediated loxP recombination in astrocytes of 73.12 GFAP-Cre reporter mice. A–D, Brightfield immunohistochemistry of Cre (A, C) or the reporter protein β-gal (B, D) in transverse sections of lumbar spinal cord. C, D, Details of boxed areas in A and B, respectively. E, F, Confocal microscopy of two color immunofluorescence showing that Cre (E) and β-gal (F) colocalize with the astrocyte marker GFAP. Scale bars: A, B, 100 μm; C, D, 30 μm; E, F, 5 μm.
Figure 2.
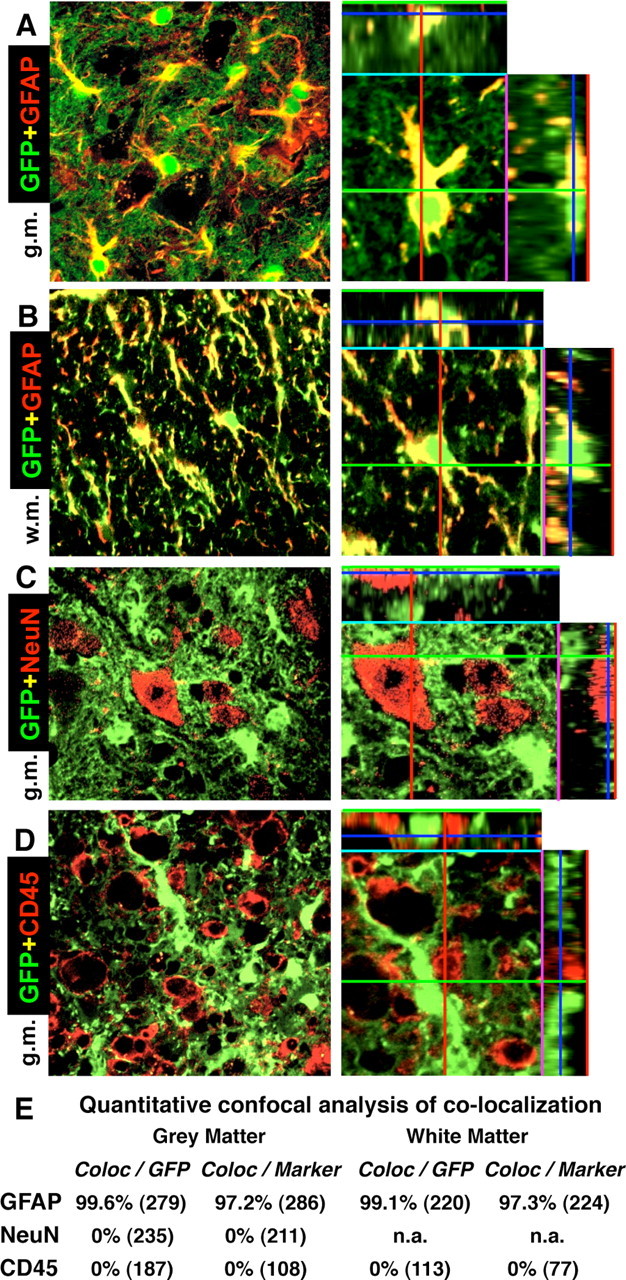
A–D, Specificity of Cre-mediated loxP recombination in astrocytes of 73.12 GFAP-Cre reporter mice at 7 d after SCI in lumbar spinal cord at the immediate margin (A–C) or in the core (D) of the lesion. Survey and detail (ortho) confocal microscopy of the reporter protein GFP combined with immunofluorescence of marker proteins for various cell types is shown. A, B, All cells that express the reporter GFP, also express the astrocyte marker GFAP, and vice versa, in both gray (A) and white (B) matter. Note that the reporter GFP is present in the cell body and throughout many fine distal astrocyte processes, whereas GFAP is present primarily in large proximal processes. C, No cells that express the neuronal marker NeuN also express GFP. D, No cells that express the microglial and white blood cell marker CD45 also express GFP. E, Quantitative confocal analysis of colocalization of reporter GFP and cell type markers. The total numbers of cells evaluated from n = 4 mice are shown in parentheses. Values are expressed as the percentage of cells that exhibit colocalization. g.m., Gray matter; w.m., white matter.
Figure 3.
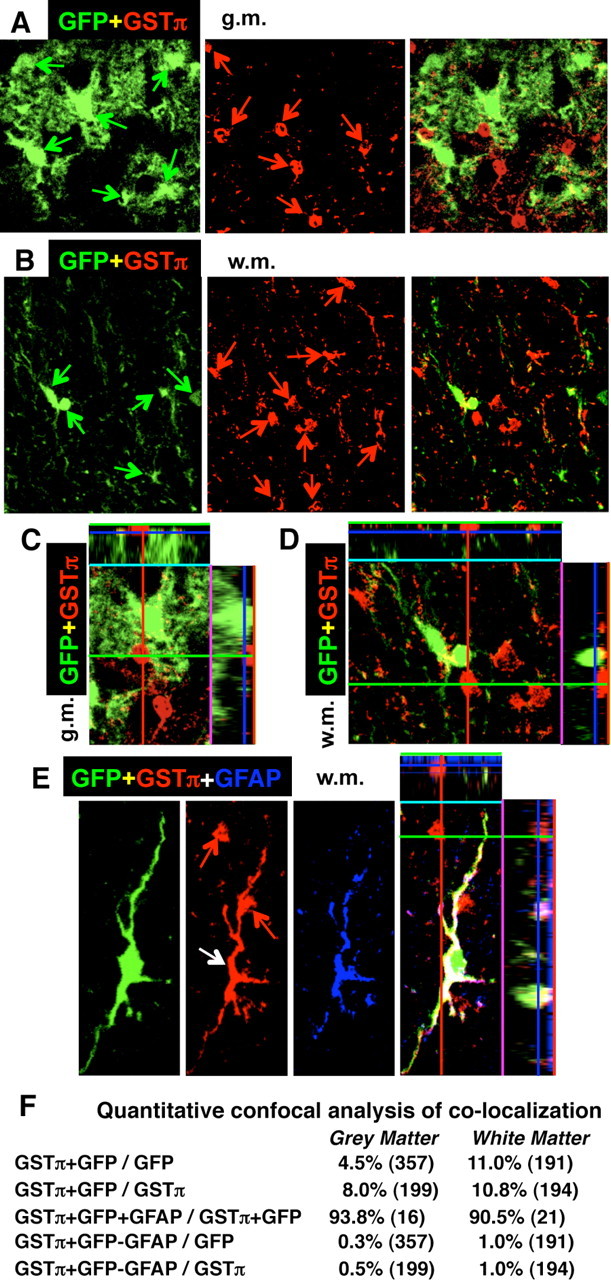
Specificity of Cre-mediated loxP recombination in astrocytes of 73.12 GFAP-Cre reporter mice at 7 d after SCI in lumbar spinal cord at the immediate margin of the lesion. A–E, Confocal microscopy showing single and combined channels for red, green (A–D), and blue (E) of the reporter protein GFP combined with immunofluorescence of marker proteins for various cell types. Survey (A, B) and detail orthoimages (C, D) images of single and combined channels for red and green show that the reporter protein GFP and the mature oligodendrocyte marker GSTπ are expressed in different cells in both gray (A, C) and white (B, D) matter. Red and green arrows indicate cells stained exclusively for one marker or the other. C, D, Details of A and B, respectively. E, Rare cells (white arrow) were observed that expressed both GFP (green) and GSTπ (red), and most of these cells also expressed GFAP (blue) and had the morphological appearance of astrocytes. In E, note also the presence of two presumptive mature oligodendrocytes stained only for GSTπ (red arrows). F, Quantitative confocal analysis of colocalization of reporter GFP and GSTπ alone or in combination with GFAP. The total numbers of cells evaluated from n = 4 mice are shown in parentheses. Values are expressed as the percentage of cells that exhibit colocalization. g.m., Gray matter; w.m., white matter.
As reported previously (Garcia et al., 2004), the forebrain contains anatomically restricted populations of juvenile and adult neural progenitors that express GFAP, and in mice of the line 73.12 used in this study, Cre activity and reporter expression are present in small, specific populations of neurons in olfactory bulb and hippocampus. We also noted reporter expression in a few other scattered populations of neurons such as in a small number of late born neurons in superficial layers (2 and 3) of cerebral cortex (T. Imura and M. U. Sofroniew, unpublished observations). To look for potential reporter expression specifically in afferent neurons that project to the spinal cord and might influence the response to SCI, we conducted single-cell confocal microscopic analysis in GFP reporter mice after injection of retrograde tract tracer into the same region of the lumbar spinal cord that was targeted for SCI and evaluated histologically, and that contains the lumbar hindlimb stepping center at L1/L2 (Kiehn, 2006). In all areas examined, including brainstem reticular formation, vestibular nuclei, red nuclei, motor cortex, and propriospinal pathways, we found that no neurons were colabeled for retrograde tracer and reporter protein (Fig. 4). These findings show that, in mice of the line 73.12 used here, there are no reporter-expressing neurons in spinal cord or any of the spinal cord afferent regions that project to the lumbar spinal cord evaluated after SCI in this study. Together, our various specificity tests were all consistent with the view that our GFAP-Cre mice of line 73.12 comprise a specific and selective model for evaluating the effects on of conditional gene from astrocytes in spinal cord under different experimental conditions in vivo.
Figure 4.

Absence of Cre-mediated loxP recombination in afferent neurons that project to lumbar spinal cord in 73.12 GFAP-Cre reporter mice. A, B, Qualitative (A) and quantitative (B) analysis of confocal microscopy of the reporter protein GFP combined with retrograde tract tracing in various brain and spinal cord regions after injection of fluorescently labeled tracer into lumbar spinal cord. No retrogradely labeled neurons were observed that expressed GFP in any region examined. The total numbers of cells evaluated from n = 4 mice are shown in parentheses. Values are expressed as the percentage of cells that exhibit colocalization. Cx, Cortex; Red n., red nucleus; Ret. F., reticular formation; Vest. n., vestibular nucleus; T, thoracic spinal cord.
Normal appearance of GFAP-STAT3-CKO mice and CNS
To delete STAT3 from GFAP-expressing astrocytes, we cross-bred 73.12 GFAP-Cre mice with mice having loxP sites flanking exon 22 of the STAT3 gene, which contains a phosphorylation site critical for STAT3 activation. CNS tissue from GFAP-STAT3-CKO mice contained a PCR product that was not present in controls and corresponded to recombined-STAT3 (R-STAT3) with exon 22 excised as expected after recombination of these loxP sites (Fig. 5A). To test the efficiency of the conditional gene deletion, we conducted Western blot analysis of primary cell cultures of astrocytes from perinatal mice. Primary astrocytes from both control and GFAP-STAT3-CKO mice grew well in serum under standard conditions. However, whereas control cultures exhibited robust expression of both STAT3 or pSTAT3, both molecules were essentially undetectable in cultures GFAP-STAT3-CKO mice (Fig. 5B), demonstrating effective gene deletion in our Cre-loxP model. GFAP-STAT3-CKO offspring were obtained in expected Mendelian ratios, and developed and thrived in a manner indistinguishable from nontransgenic or partially transgenic littermates. The macroscopic appearance, size, and weight (Fig. 5C), as well as the cytoarchitecture and myelination (Fig. 5D–H) of brains and spinal cords from uninjured GFAP-STAT3-CKO mice, were indistinguishable from those of nontransgenic littermates at all ages.
Figure 5.
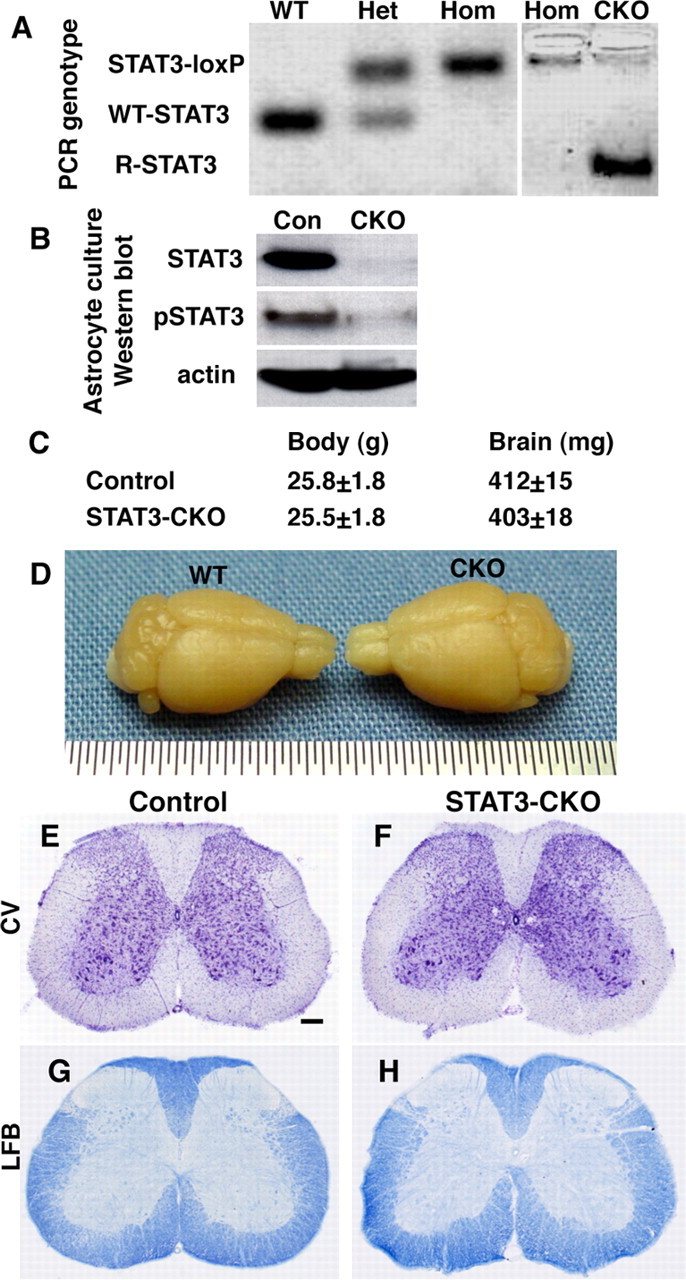
Efficiency of STAT3-CKO from astrocytes, and normal appearance of CNS in GFAP-STAT3-CKO mice. A, PCR of genomic DNA extracted from brain tissue of mice with different genotypes: wild-type (WT), heterozygous (Het), or homozygous (Hom) STAT3-loxP without Cre, and GFAP-Cre-STAT3-CKO (CKO) with Cre+homozygous loxP. CKO brains, but not brains of mice homozygous for loxP but without Cre, exhibit a PCR product of R-STAT3 that is inactivated after recombination of loxP sites to excise exon 22. B, Western blot of primary astrocyte cultures (∼95% astrocytes) grown in serum. Under these culture conditions, astrocytes from control (Con) mice express high levels of STAT3 and pSTAT3, whereas expression of STAT3 and pSTAT3 are almost undetectable in astrocytes from GFAP-STAT3-CKO mice. C, Body and brain weights of GFAP-STAT3-CKO mice are not significantly different from controls (ANOVA, n = 4 per group). D, Macroscopic appearance of GFAP-STAT3-CKO brain is indistinguishable from WT. E–H, Cresyl violet (CV)-stained cytoarchitecture (E, F) and luxol fast blue (LFB)-stained myelin (G, H) exhibit no detectable abnormalities in spinal cords of GFAP-STAT3-CKO (F, H) compared with control (E, G) mice. Scale bar: (in E) E–H, 250 μm.
Appearance of STAT3-CKO astrocytes in the uninjured spinal cord
Jak-STAT signaling through STAT3 and STAT1 has been implicated in astrogliogenesis (Bonni et al., 1997; He et al., 2005). We therefore carefully evaluated the appearance of astroglia in the spinal cord of uninjured GFAP-STAT3-CKO mice in vivo. To do so, we conducted immunohistochemistry for (1) GFAP, the canonical marker for astrocytes, (2) glutamine synthetase, a marker expressed predominantly by astrocytes (Norenberg, 1979; Wilhelmsson et al., 2004) and (3) for the transgenically targeted reporter molecule, β-gal, in uninjured control and GFAP-STAT3-CKO mice. Qualitative analysis of all three markers indicated that astrocytes in gray and white matter were similar in size, shape, and overall appearance in control and STAT3-CKO mice (Fig. 6). Although astrocytes in gray matter exhibited a somewhat reduced staining for GFAP in STAT3-CKO mice (Fig. 6A,B,F,G), the morphology and density of astrocytes stained for either glutamine synthetase (Fig. 6C,H) or β-gal (Fig. 6D,E,I,J) were indistinguishable in control and STAT3-CKO mice. Quantitative analysis of cells stained for glutamine synthetase (Fig. 6K,L) and β-gal (Fig. 6M,N) indicated that both the cell size and the cell density of astrocytes did not differ significantly in spinal cord of control and STAT3-CKO mice.
Figure 6.

Normal distribution and morphology of astrocytes in uninjured GFAP-STAT3-CKO mice. A–J, Brightfield immunohistochemistry of GFAP (A, B, F, G), glutamine synthetase (C, H), and the reporter β-gal (D, E, I, J) in uninjured spinal cord of control (A–E) and STAT3-CKO (F–J) mice. A, B, F, G, Expression of GFAP appears reduced in gray, but not white matter astrocytes of STAT3-CKO mice. C, H, Expression levels glutamine synthetase and distribution of positive cells is indistinguishable in both gray and white matter of control and STAT3-CKO mice. D, E, I, J, Expression levels β-gal and distribution and morphology of β-gal-positive cells is indistinguishable in both gray (D, I) and white (E, J) matter of control and STAT3-CKO mice. K–N, Graphs of morphometric evaluations of astrocytes immunohistochemically stained for glutamine synthetase (Glut. Synth.) (K, L) and β-gal (M, N) in uninjured spinal cords. There was no significant difference (p > 0.05) between control and STAT3-CKO mice in either astrocyte density (K, M) or astrocyte cell size (L, N) in either gray or white matter using either maker to identify astrocytes. In both types of mice, cell density, but not cell size, was significantly greater (p < 0.05) in gray matter compared with white matter. ANOVA with post hoc pairwise comparisons, n = 4 or more per group. Scale bars: (in F) A, F, 150 μm; (in H) B, C, G, H, 70 μm; (in J) D, E, I, J, 15 μm. Error bars indicate SEM.
Activation of pSTAT3 in astrocytes after SCI
Crush injury is a well characterized and validated experimental model of SCI (Inman et al., 2002; Faulkner et al., 2004; Plemel et al., 2008). In the following experiments, we compared the effects of a moderate crush SCI at L1/L2 on various aspects of the response to SCI in GFAP-STAT3-CKO and control mice.
Although STAT3 is expressed by both neurons and glia in adult spinal cord (Cattaneo et al., 1999), levels of active, pSTAT3 are low or undetectable in uninjured adult CNS tissue, but increase markedly after insults such as SCI (Acarin et al., 2000; Justicia et al., 2000; Sriram et al., 2004; Yamauchi et al., 2006). To determine the time course of STAT3 activation in astrocytes after our moderate crush SCI, we used double labeling of immunohistochemistry for pSTAT3 in combination with detection of GFP as an astrocyte marker in GFAP-Cre reporter mice (Fig. 7). In uninjured control mice, pSTAT3 was not immunohistochemically detectable in astrocytes and was occasionally observed in cells with the size and appearance of neurons (Fig. 7A,B). In contrast, at 1 d after SCI in the lesion core (epicenter), essentially all GFP-positive astrocytes in GFAP-Cre reporter mice were strongly positive for pSTAT3 (Fig. 7C). At 1 d after SCI in the tissue surrounding the lesion core, the majority of GFP-positive astrocytes were detectably positive for pSTAT3, and there were many pSTAT3-positive but GFP-negative cells, a portion of which had the appearance of neurons (Fig. 7D). At 2 and 3 d after SCI in the tissue surrounding the lesion, a majority of GFP-positive astrocytes remained detectably positive for pSTAT3, and there were many pSTAT3-positive but GFP-negative cells (Fig. 7E,F). By 7 d after SCI, only a small proportion of GFP-positive astrocytes were positive for pSTAT3 and these were along the immediate peripheral margins of the lesion in the region of the forming glial scar, and there were many pSTAT3-positive but GFP-negative cells (Fig. 7G,H). At 14 d after SCI, GFP-positive astrocytes were rarely positive for pSTAT3, and a number of pSTAT3-positive but GFP-negative cells were present (data not shown). These findings show that a substantial proportion of astrocytes in the immediate vicinity of the lesion are activated by pSTAT3 over a several day period after SCI in wild-type mice. We found no detectable levels of pSTAT3 in astrocytes after SCI in STAT3-CKO mice (data not shown).
Figure 7.
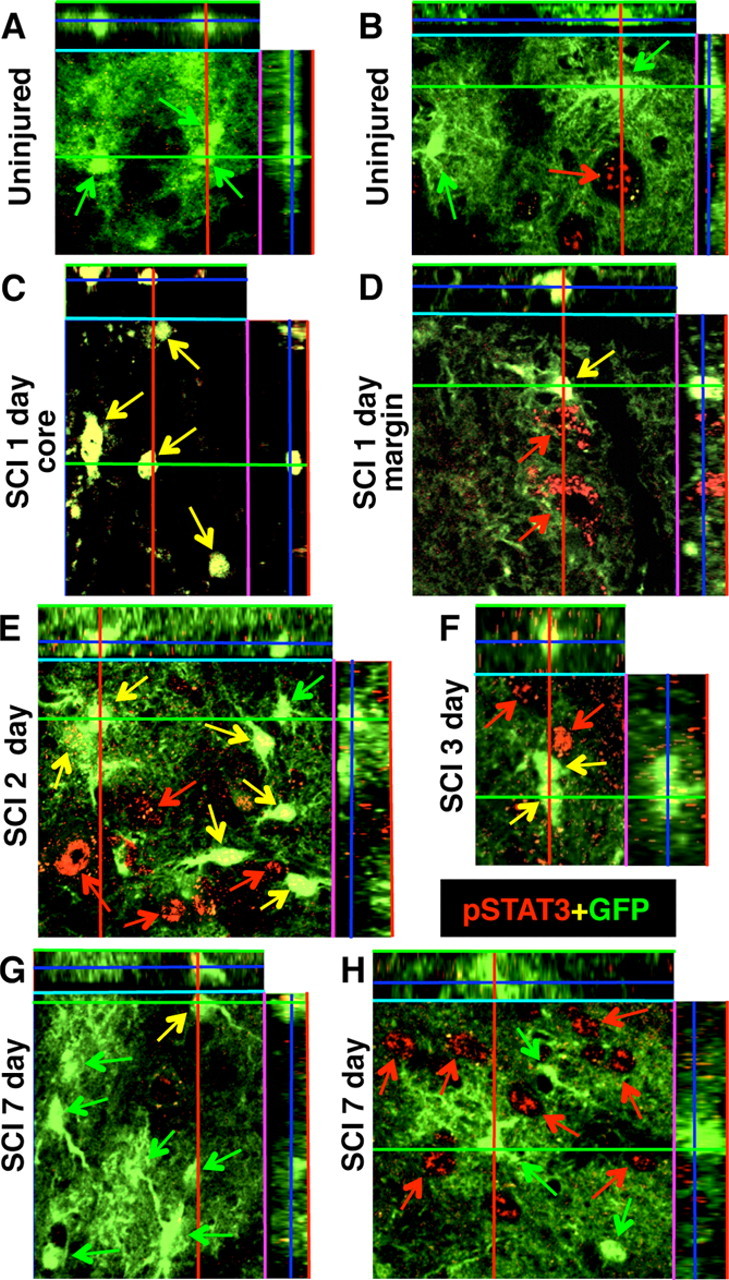
Activation of pSTAT3 in astrocytes and other cell types after SCI. A–H, Confocal microscopy (dual-channel orthoimages) of the reporter protein GFP (green) combined with immunofluorescence of pSTAT3 (red) in lumbar spinal cord of GFAP-Cre-Reporter mice, uninjured and at 1, 2, 3, and 7 d after SCI. A, B, In uninjured mice, no GFP-positive astrocytes exhibit detectable levels of pSTAT3 and there are rare scattered examples of pSTAT3 expression by non-GFP-positive cells that have the size and appearance of neurons. C, At 1 d after SCI in the lesion core or epicenter, all GFP cells are intensely pSTAT3 positive and no other cell types are present that express pSTAT3. D–F, At 1, 2, and 3 d after SCI at the lesion margin where the glial scar subsequently forms, pSTAT3 is expressed by many GFP-positive astrocytes and other cell types that are GFP-negative. G, H, At 7 d after SCI, at the lesion margin only a few GFP-positive astrocytes continue to express pSTAT3, whereas in some regions of the gray matter, pSTAT3 is expressed by many GFP-negative cells. Green arrows indicate GFP-positive cells that do not express pSTAT3; yellow arrows indicate GFP-positive cells that coexpress pSTAT3; red arrows indicate GFP-negative cells that express pSTAT3.
We also examined pSTAT3 and pSTAT1 levels using Western blot analysis. In agreement with previous reports (Yamauchi et al., 2006), both pSTAT3 and pSTAT1 were undetectable in uninjured spinal cord tissue and increased to detectable levels after SCI in control mice (Fig. 8). In our GFAP-STAT3-CKO mice, both pSTAT3 and pSTAT1 were also undetectable in uninjured spinal cord tissue. After SCI in GFAP-STAT3-CKO mice, pSTAT1 increased in a manner that was indistinguishable from that in control mice, whereas pSTAT3 exhibited only a small increase that was markedly attenuated compared with the increase in control mice (Fig. 8).
Figure 8.

Western blots of pSTAT3, pSTAT1, and GFAP in uninjured spinal cord tissue and at 3 d after SCI in control (Con) and GFAP-STAT3-CKO mice. pSTAT3 and pSTAT1 are both undetectable in uninjured control or STAT3-CKO mice. pSTAT3 expression is substantially increased after SCI in control mice and this increase is markedly attenuated but is not absent in STAT3-CKO mice. pSTAT1 expression is moderately increased to similar levels in both control and STAT3-CKO mice. GFAP expression is substantially lower in uninjured spinal cord of GFAP-STAT3-CKO compared with control mice. After SCI, there is a pronounced increase GFAP expression in controls and this increase is markedly attenuated but not entirely absent in GFAP-STAT3-CKO mice.
Response of STAT3-CKO astrocytes to SCI: attenuated astrogliosis
We next determined the effects of astroglial STAT3-CKO on reactive astrogliosis by examining astrocyte activation as indicated by increased expression of intermediate filament proteins and by quantifying the degree of astrocyte hypertrophy. Western blot analysis (Fig. 8) confirmed our immunohistochemical observation (Fig. 6A,B,F,G) of somewhat reduced levels of GFAP expression in the spinal cord of uninjured GFAP-STAT3-CKO mice relative to controls. Western blot analysis also showed that at 3 d after SCI, GFAP-STAT3-CKO mice exhibited an attenuated, but not absent, upregulation of GFAP compared with controls (Fig. 8). Similar observations were made using immunohistochemistry (Fig. 9), which showed a robust upregulation of both GFAP and vimentin at various time points after SCI in control mice (Fig. 9A,C), whereas this upregulation was markedly attenuated in GFAP-STAT3-CKO mice (Fig. 9B,D).
Figure 9.

Attenuated GFAP and vimentin expression, and disrupted scar formation after SCI in GFAP-STAT3-CKO mice. A–D, Horizontal sections of spinal cord stained by brightfield immunohistochemistry for GFAP (A, B) and vimentin (C, D) 14 d after moderate crush SCI in nontransgenic control (A, C) and STAT3-CKO (B, D) mice. In control mice (A, C), SCI induces a marked upregulation of both GFAP and vimentin in astrocytes in both gray and white matter, and this upregulation is markedly attenuated, but not entirely absent, in STAT3-CKO mice (B, D). E–H, Double-labeling immunofluorescence of GFAP (green) and fibronectin (red) examined by conventional (E, F) and confocal (G, H) microscopy. After SCI in control mice, fibronectin-expressing fibromeningeal cells, and GFAP-expressing astrocytes abut in a strictly segregated manner that delineates a sharp border around the entire lesion (E, G). This cellular alignment and border formation is severely disrupted in STAT3-CKO mice (F, H). Scale bars: (in D) A–D, 150 μm; (in F) E, F, 30 μm; (in H) G, H, 10 μm.
To evaluate astrocyte hypertrophy after SCI, we used computer assisted morphometric analysis to measure the cell volume of astrocytes that expressed the reporter protein β-galactosidase in either control mice or GFAP-STAT3-CKO mice, in a precisely defined 200-μm-wide boundary zone around the entire lesion in both gray and white matter. After SCI in control mice, astrocytes exhibited a pronounced and significant hypertrophy of their cell volumes in both gray (Fig. 10A,E,I) and white (C,G,J) matter when compared with uninjured mice. This hypertrophy of astrocytes after SCI did not occur in GFAP-STAT3-CKO mice in either gray (Fig. 10B,F,I) or white (D,H,J) matter, and the astrocytes in GFAP-STAT3-CKO mice were significantly smaller that those in control mice after SCI and were not significantly larger than in uninjured mice.
Figure 10.
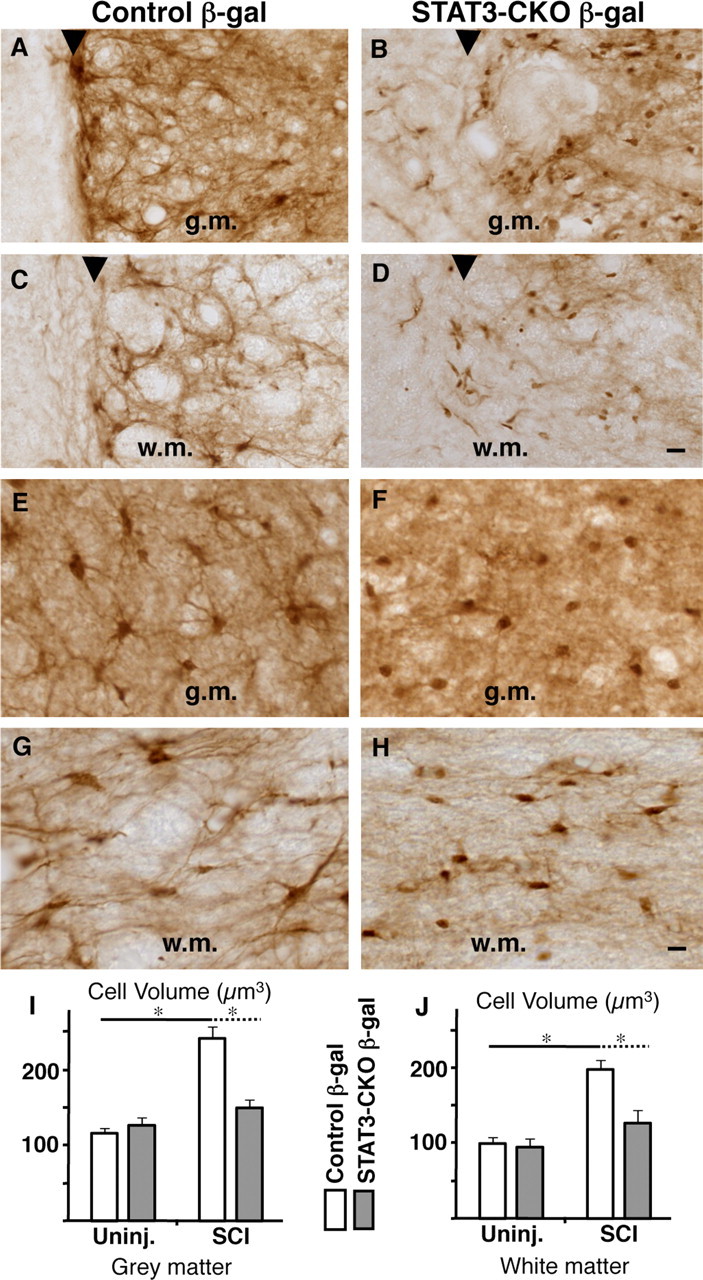
Failure of astrocyte hypertrophy and scar formation after SCI in GFAP-STAT3-CKO mice. A–H, Horizontal sections of spinal cord stained by brightfield immunohistochemistry for the reporter protein β-gal 14 d after moderate crush SCI in nontransgenic control (A, C, E, G) and GFAP-STAT3-CKO (B, D, F, H) mice. A–D show tissue directly along the wound margin. E-H show tissue adjacent to the lesion In control mice, SCI induces a pronounced hypertrophy of β-gal-positive astrocytes with formation of an organized scar that precisely demarcates the lesion boundary (arrowheads) in both gray (A, E) and white (C, G) matter. In contrast, in STAT3-CKO mice, astrocytes fail to hypertrophy after SCI and there is no orderly formation of a glial scar that demarcates the lesion boundary (arrowheads) in either gray (g.m.) (B, F) or white (D, H) matter (w.m.). I, J, Graphs showing the mean cell volume of β-gal stained astrocytes in control and GFAP-STAT3-CKO mice, uninjured (Uninj.), and 14 d after SCI. Stereological measurements of cell volume were performed in a 200 μm boundary zone traced around the entire lesion in both gray and white matter. Astrocytes in both gray (I) and white (J) matter undergo significant hypertrophy after SCI in control mice, but not in STAT3-CKO mice. *p < 0.05 ANOVA with post hoc pairwise analysis; n = 4 or more per group. Scale bars: (in D) A–D, 30 μm; (in H) E–H, 10 μm. Error bars indicate SEM.
Response of STAT3-CKO astrocytes to SCI: disrupted scar formation
Because of the failure of astrocyte hypertrophy in GFAP-STAT3-CKO mice after SCI, we examined in more detail the formation of the glia scar around the SCI lesion, using immunohistochemistry of astrocyte markers (β-galactosidase, GFAP, or vimentin) alone or in combination with markers of fibroblast lineage cells. Fibroblast lineage cells are an important cellular component of the glial scar and interact extensively with astrocytes in an organized manner to form the glial scar around CNS lesions (Bundesen et al., 2003; Silver and Miller, 2004). By 14 d after SCI in control mice, reactive astrocytes within the lesion boundary zone were densely packed and closely abutting with tightly interdigitating processes that formed a continuous unbroken boundary around the central core of the lesion in both gray and white matter (Figs. 9A,C,E,G, 10A,C). In agreement with descriptions by others (Bundesen et al., 2003), we found that fibromeningeal cells were aligned in direct juxtaposition to reactive astrocytes (Fig. 9E,G) in an organized and strictly segregated manner to form a long unbroken palisade like border of glial scar (Figs. 9E,G) that sharply delineated the central necrotic lesion and intact neural tissue, with the fibromeningeal cells on the inside of the lesion and the astrocytes surrounding these cells on the outside (Figs. 9A,C,E,G, 10A,C). In contrast, in GFAP-STAT3-CKO mice glial scar formation was severely disrupted; astrocytes near the lesion were dispersed, their processes did not abut and they did not form a contiguous boundary (Figs. 9B,D,F,H, 10B,D). Moreover, the arrangement of fibromeningeal cells was also disrupted; they aligned along the disorganized astrocytes with the result that there was no clear border of glial scar (Figs. 9F,H) and there was a severe disruption of the boundary between the central necrotic lesion from adjacent intact neural tissue (Figs. 9B,D,F,H, 10B,D). It is interesting to note that this disruption of glial scar formation occurred in GFAP-STAT3-CKO mice after SCI although there was no detectable difference in the density of β-galactosidase-positive astrocytes in control or GFAP-STAT3-CKO mice either directly along the wound margin (Fig. 10A–D) or in the tissue adjacent to the lesion (E–H).
Increased inflammation and lesion size after SCI in GFAP-STAT3-CKO mice
We found previously that transgenically targeted ablation of scar forming reactive astrocytes increased and prolonged inflammatory cell infiltration into neural tissue after CNS injury (Bush et al., 1999; Faulkner et al., 2004; Myer et al., 2006). Because GFAP-STAT3-CKO mice exhibited marked disruption of astroglia scar formation after SCI, we evaluated the extent and severity of inflammation after SCI in these mice using immunohistochemistry for CD45, a marker for macrophages and microglia, alone or in combination with markers for astrocytes. In control mice at 14 d after SCI, inflammatory cells were mostly confined to the central core of the lesion, which contained many large, round, phagocytic macrophages that were clearly separated from surrounding well preserved tissue that exhibited normally appearing microglia but rare or no phagocytic macrophages (Figs. 11A,C). Double staining with GFAP and CD45 showed that inflammatory cells in the central lesion were clearly segregated from the surrounding neural parenchyma by an unbroken palisade of reactive, scar forming astrocytes (Figs. 11E,G). In GFAP-STAT3-CKO mice at 14 d after SCI, the central lesion of large phagocytic inflammatory cells was substantially larger than in control mice and the lesion borders were jagged and disrupted so that there was no clear demarcation of the lesion from surrounding tissue and large phagocytic macrophages spread far into the neural parenchyma (Fig. 11B,D). This later observation was confirmed by double staining with GFAP and CD45, which showed that in GFAP-STAT3-CKO mice, inflammatory cells were not contained or restricted by a glial scar but migrated freely among the attenuated and disorganized astrocytes (Figs. 11F,H).
Figure 11.

Exacerbated inflammation after SCI in GFAP-STAT3-CKO mice. A–D, Horizontal sections of spinal cord stained by brightfield immunohistochemistry for the inflammatory cell marker CD45 14 d after moderate crush SCI in nontransgenic control (A, C) and GFAP-STAT3-CKO (B, D) mice. C, D, Details of boxes in A and B, respectively. E–H, Double-labeling immunofluorescence of GFAP (green) and CD45 (red) examined by conventional (E, F) and confocal (G, H) microscopy. At 14 d after SCI in control mice, many CD45-expressing inflammatory cells remain in the central lesion but rarely extend into or past GFAP-expressing astrocytes that delineate a sharp border between necrotic lesion and intact neural tissue (A, C, E, G). This sharp border formation is severely disrupted in GFAP-STAT3-CKO mice and the severely inflamed area of necrotic lesion is much larger (B) and inflammatory cells penetrate far into adjacent tissue (D, F, H). Scale bars: (in B) A, B, 150 μm; (in D) C, D, 75 μm; (in F) E, F, 60 μm; (in H) G, H, 10 μm.
To look for potential consequences of the increased inflammation after SCI in GFAP-STAT3-CKO mice after moderate crush SCI, we evaluated lesion size and extent of demyelination. In control mice, moderate crush SCI reproducibly caused a small central lesion that was similar in histological appearance and size at 14 and 28 d, with essentially no cavity formation (Fig. 12A,E), as described previously by us and others for C57BL/6 mice (Inman et al., 2002; Faulkner et al., 2004). GFAP-STAT3-CKO mice exhibited significantly greater lesion volume than controls at both 14 and 28 d (Fig. 12B,E), but also exhibited no cavity formation. Control mice regularly exhibited demyelination of white matter in the central lesion, but not of white matter in the lateral columns (Fig. 12C). In contrast, GFAP-STAT3-CKO mice consistently exhibited pronounced demyelination also of white matter in the lateral columns (Fig. 12D).
Figure 12.
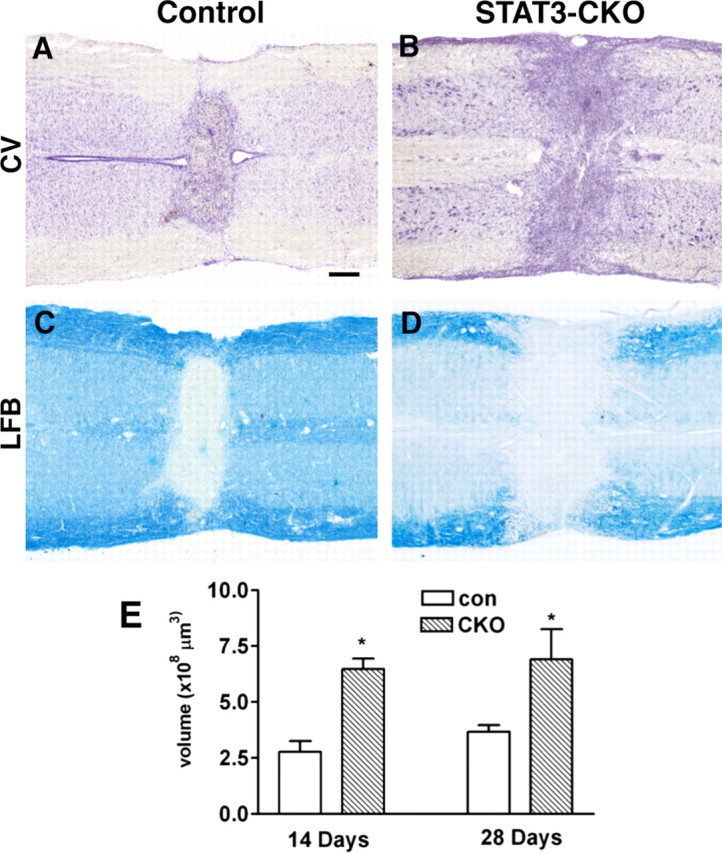
Increased lesion size and exacerbated demyelination in GFAP-STAT3-CKO mice after SCI. A–D, Horizontal sections of spinal cord stained for cresyl violet (CV; A, B) or luxol fast blue (LFB; C, D) at 14 d after moderate crush SCI in control (A, C) and GFAP-STAT3-CKO (B, D) mice showing that the central necrotic lesion is larger and there is more widespread demyelination in GFAP-STAT3-CKO (B, D) compared with control mice (A, C). E, Graph of mean lesion volume in GFAP-STAT3-CKO compared with control (con) mice, stereologically measured on series of sequential horizontal tissue sections through the entire lesion site. Lesion volume was significantly greater in GFAP-STAT3-CKO mice at both 14 and 28 d after SCI (*p < 0.05, ANOVA with post hoc pairwise analysis, n = 8 or more per group per time point). Error bars indicate SEM. Scale bar: (in A) A–D, 150 μm.
Reduced behavioral recovery in GFAP-STAT3-CKO mice after moderate SCI
To evaluate the effects of GFAP-STAT3-CKO on motor behavior after SCI, we assessed open-field and rotorod performance, and conducted footprint analysis. We reported previously that the moderate crush SCI at L1/L2 used here reproducibly causes motor impairments from which nontransgenic mice mostly recover over 14 d (Faulkner et al., 2004). In the present study, in uninjured mice we found no detectable differences between the motor performances of control and GFAP-STAT3-CKO mice in all three of the tests used (Fig. 13). In addition, in a manner similar to our previous studies, we found that control mice exhibited significant impairments in all three motor tests at 3 d after moderate crush SCI, from which they exhibited a gradual and substantial recovery in all three measures of motor function and reached a plateau just below preinjury levels at ∼14 d after SCI (Fig. 13). GFAP-STAT3-CKO mice exhibited somewhat greater levels of impairments in all three motor tests at 3 d after SCI, and also improved with time after SCI. Nevertheless, despite their improvements over time, GFAP-STAT3-CKO mice had moderately but significantly lower scores in all three tests of motor function at all times compared with controls and with preinjury levels (Fig. 13). All mice initially lost bladder function after SCI and required manual bladder expression for at least the first 3 d. Bladder function recovered thereafter at approximately similar rates and to approximately similar degrees in control and GFAP-STAT3-CKO mice, such that bladder function (defined as not requiring manual bladder expression) had returned by 10 d after SCI in 83% of control mice and 70% of GFAP-STAT3-CKO mice and was similar in both groups (>90%) at the time of perfusion at 14 or 28 d.
Figure 13.
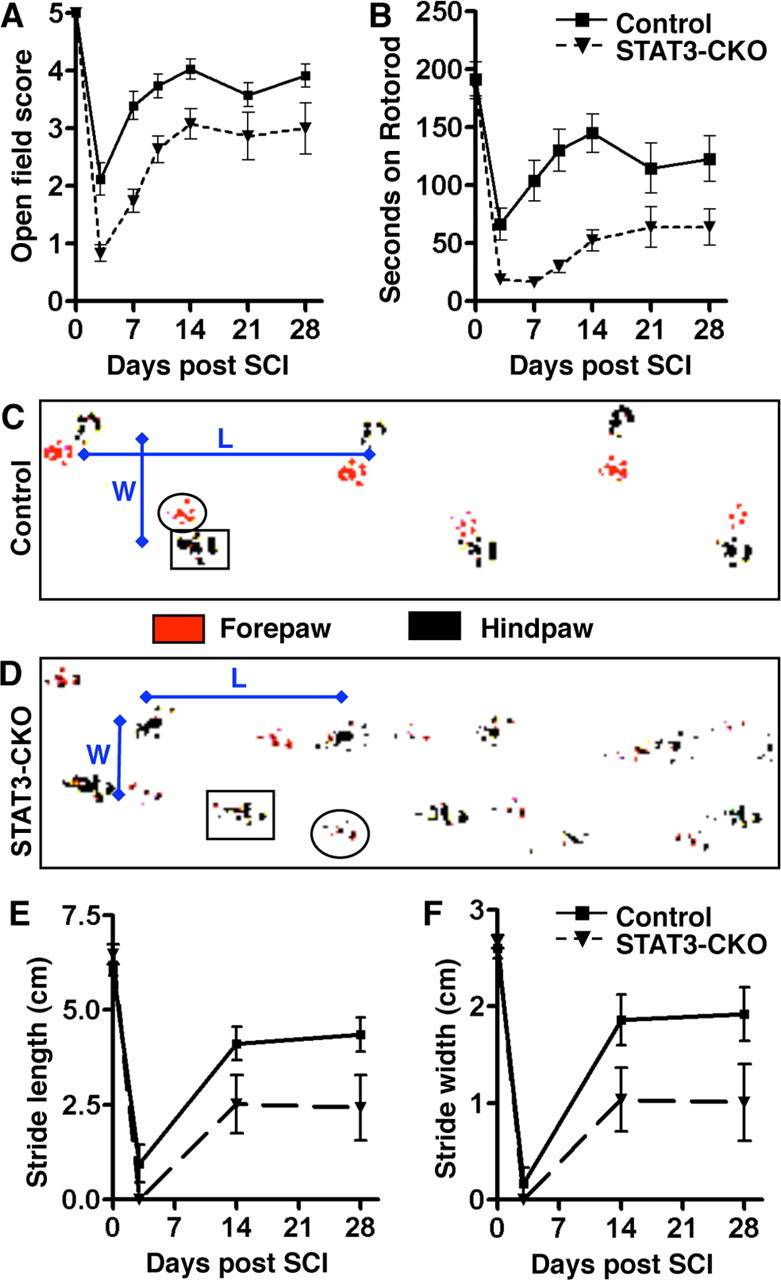
Attenuated motor recovery after moderate crush SCI in GFAP-STAT3-CKO mice. A, B, Graphs show performance of control and GFAP-STAT3-CKO mice in open-field walking (A) and rotorod (B) in uninjured mice (day 0) and over a 28 d period after moderate crush SCI. Open-field walking was scored by two observers blind to the experimental group (scale: 0, hindlimb dragging to 5, normal walking) (for details, see Materials and Methods). Rotorod performance was scored as time in seconds to fall during increasing rotation speed. C, D, Representative footprints of control (C) or GFAP-STAT3-CKO (D) mice at 14 d after SCI showing measurements in stride width (W) and stride length (L). E, F, Graphs of stride length (E) and stride width (F) in uninjured mice (day 0) and over a 28 d period after SCI in control and GFAP-STAT3-CKO mice. On all measures of motor performance, uninjured GFAP-STAT3-CKO mice were indistinguishable from uninjured controls. After moderate crush SCI, both control and GFAP-STAT3-CKO mice exhibited initial severe deficits in motor dysfunction with considerable return of function over the first 14 d; however, in all measures, GFAP-STAT3-CKO mice exhibited significantly lower scores than controls at 14 d and later (n = 11 or more per group per time point; *p < 0.05 ANOVA with post hoc pairwise analysis). Error bars indicate SEM.
Discussion
STAT3 is a signal transducer of various cytokines and growth factors including IL-6, IL-10, EGF, TGFα, LIF, and CNTF, which are important in intercellular signaling after CNS injury. In this study, we found that conditional deletion of STAT3 from GFAP-expressing cells had little detectable effect on the development of astrocytes, but significantly attenuated astrocyte activation after SCI. Our findings show that STAT3 is a critical regulator of certain aspects of reactive astrogliosis, in particular the upregulation of intermediate filaments, astrocyte hypertrophy, and glial scar formation. Our findings also provide evidence that scar-forming astrocytes restrict the spread of inflammatory cells and help to preserve functional tissue after SCI.
Specificity of the transgenic model
We extensively characterized our transgenic mouse model for specificity and efficacy of conditional deletion of STAT3 from astrocytes. Although transgenic targeting is stable within transgenic breeding lines (Feng et al., 2000), every transgenic line exhibits its own specific pattern of targeting and requires detailed characterization at the single-cell level for every population of cells studied. We used transgenic GFAP-Cre mice of line 73.12 generated previously using the entire mouse GFAP promoter and characterized in forebrain (Garcia et al., 2004). Additional characterization of line 73.12 here showed that Cre expression closely mimicked endogenous GFAP in spinal cord. Cre activity, as revealed by reporter gene expression, was confined to molecularly and morphologically identified astrocytes, and Cre activity was selectively targeted to essentially all reactive astrocytes after SCI. Together, these findings indicate that these mice comprise an appropriate targeting model for evaluating effects of conditional gene deletion from astrocytes in SCI. Using the 73.12 GFAP-Cre line, we generated GFAP-STAT3-CKO mice and demonstrated effective STAT3 deletion.
Normal appearance of astrocytes in uninjured STAT3-CKO mice
Jak-STAT signaling through STAT1 and STAT3 has been implicated in astrocyte development (Bonni et al., 1997; He et al., 2005). In vitro studies suggest that commitment of progenitors to astrogliogenesis precedes GFAP expression (Mi and Barres, 1999), in which case Cre expression and STAT3 deletion in our GFAP-STAT3-CKO mice would occur after STAT3 has acted on progenitors to induce astrogliogenesis. We carefully examined CNS architecture and the appearance of astrocytes in GFAP-STAT3-CKO mice. Gross structure and CNS cytoarchitecture revealed by Nissl and myelin stains were indistinguishable in uninjured control and GFAP-STAT3-CKO mice. Quantitative stereological analysis showed that the density and size of astrocytes were identical in uninjured controls and GFAP-STAT3-CKO mice. Reporter- labeled astrocytes were indistinguishable in appearance in uninjured control and GFAP-STAT3-CKO mice. Astrocytes from GFAP-STAT3-CKO mice expressed reduced levels of GFAP, and we cannot rule some level of consequent structural changes that might be revealed by more detailed evaluations, for example with single-cell dye filling (Bushong et al., 2002; Nedergaard et al., 2003). Nevertheless, there were no detectable differences in three motor tests in uninjured controls and GFAP-STAT3-CKO mice. Together, these findings indicate that STAT3-CKO in our model did not detectably disrupt astrocyte development or function in uninjured mice. It is also noteworthy that although STAT3 is present in uninjured spinal cord, we found no detectable activated pSTAT3 in astrocytes in uninjured mice, and that pSTAT3 signaling appears primarily to be activated in response to insults such as traumatic SCI, as seen here and in other studies (Yamauchi et al., 2006). These observations further support the validity of our transgenic strategy.
STAT3 regulation of astrocyte reactivity and scar formation after SCI
The hallmarks of reactive astrogliosis after SCI include a pronounced increase in expression of intermediate filament proteins and cellular hypertrophy that occurs over the first few days, along with scar formation by the end of 2 weeks (Norton et al., 1992; Amat et al., 1996). We found that pSTAT3 is activated specifically in astrocytes over the first several days after SCI and has mostly subsided by 7–14 d after SCI. In addition, many other cells, including neurons, exhibit activated pSTAT3 after SCI and many of these remain activated at 14 d after SCI, highlighting the need to conduct single-cell analyses and to document that conditional deletion is targeted to specific cell types to study to roles of pSTAT3 signaling selectively in those cell types after SCI in vivo. After SCI in our GFAP-STAT3-CKO mice, astrocytes exhibited a markedly attenuated, but not entirely absent, increase in expression of the intermediate filaments, GFAP and vimentin, as well as a failure to undergo cellular hypertrophy. These events may be related. Intermediate filaments are structural proteins (Eng et al., 2000) and astrocytes exhibit attenuated hypertrophy after forebrain injury in mice with global deletion of GFAP and vimentin (Wilhelmsson et al., 2004). It is also noteworthy that signaling pathways other than Jak-STAT are involved in regulating GFAP expression and astrogliosis (Shafit-Zagardo et al., 1988; Gadea et al., 2008), and that signaling through these pathways is likely to be responsible for the small upregulation in GFAP noted after SCI in our GFAP-STAT3-CKO mice.
Contribution to multicellular glial scar formation is a prominent feature of reactive astrocytes after SCI (Reier and Houle, 1988; Silver and Miller, 2004) and, in normal mice, astrocytes and fibroblasts interact to form a clearly delineated and highly aligned border of cells with interlocking processes that surround the lesion and delineate damaged and viable tissue (Bundesen et al., 2003), as noted also in our control mice. In contrast, after SCI in GFAP-STAT3-CKO mice, we found that astrocytes were distributed randomly in the vicinity of the lesion, that the cells did not align and processes did not interlock along the wound margin, that alignment with fibroblasts did not occur, and that there were no clearly delineated borders between necrotic tissue and adjacent neural parenchyma. Although there was no detectable difference in the density of astrocytes in the vicinity of the lesion in control or GFAP-STAT3-CKO mice, the STAT3 deficient astrocytes had failed to hypertrophy and did not exhibit the robust extension of processes noted in control mice. A variety of previous studies have indicated that (1) cytokines that signal through STAT3 are upregulated after SCI (Balasingam et al., 1994; Winter et al., 1995; Klein et al., 1997; Rabchevsky et al., 1998; Levison et al., 2000; Albrecht et al., 2002; Nakamura et al., 2003), (2) tissue levels of pSTAT3 levels increase after SCI (Yamauchi et al., 2006), and (3) broad deletion of STAT3 from neural cells using a nestin-Cre transgenic model increases lesion size and attenuates recovery after SCI (Okada et al., 2006). Our findings are consistent with and extend these observations by providing direct mechanistic evidence in vivo that pSTAT3 signaling acting specifically in astrocytes is essential for certain cellular hallmarks of astrogliosis, including upregulation of intermediate filament proteins, cellular hypertrophy, and formation of a structurally organized glial scar.
Signaling regulation of astrocyte roles after SCI
Astrocytes exert many activities essential to normal CNS function (Kettenmann and Ransom, 2004; Christopherson et al., 2005; Gensert and Ratan, 2006; Iadecola and Nedergaard, 2007; Pellerin et al., 2007). Understanding changes in molecular signaling that occur during reactive astrogliosis may shed light on beneficial as well as detrimental consequences of reactive astrogliosis after CNS insults such as SCI (Sofroniew, 2005), and knowledge of specific functions mediated by specific signaling mechanisms may lead to novel therapeutic strategies. We hypothesized previously that one beneficial role of astrocyte scar formation is to prevent the migration of activated inflammatory cells away from the lesion site into closely adjacent potentially viable neural tissue, based on transgenically targeted astrocyte ablation studies (Bush et al., 1999; Faulkner et al., 2004; Sofroniew, 2005; Myer et al., 2006). Our observations here are consistent with and extend this hypothesis by showing that STAT3 signaling in astrocytes is important in regulating scar formation and that disrupted scar formation exacerbates inflammation after SCI. Similar observations of increased inflammation have been reported recently using related models of transgenically targeted attenuation of reactive astrogliosis after SCI or stroke (Okada et al., 2006; Li et al., 2008). These findings are in line with observations that autoantibodies against an astrocyte protein, AQP4, are associated both with destruction of astrocyte endfeet and autoimmune CNS inflammation in patients with neuromyelitis optica (Lennon et al., 2005). Nevertheless, it is important to note that although reactive astrocyte activities can in some cases be associated with restricting inflammation, limiting lesion size and preserving function (Sofroniew, 2005; Maragakis and Rothstein, 2006; Li et al., 2008), astrocytes also respond to signaling pathways involved in proinflammatory activation (John et al., 2003), such as the NF-κB pathway (Brambilla et al., 2005), and that some reactive astrocyte activities may have proinflammatory effects or cause tissue damage, such as production of reactive oxygen or cytotoxic edema (Murphy et al., 1993; Agulló et al., 1995; Neufeld and Liu, 2003; Antony et al., 2004; Okada et al., 2006). Together, current evidence thus suggests that different potential effects of reactive astrogliosis on overall outcome after CNS insults are likely to be context dependent and regulated by different intracellular signaling pathways. This raises the possibility that different reactive astrocyte activities can be manipulated separately by exogenous interventions. Our study provides direct evidence that pSTAT3 signaling in astrocytes in vivo regulates aspects of astrogliosis that are essential for restricting inflammation and lesion size, thereby preserving functional tissue after SCI. This transgenic strategy will also be useful to dissect the regulation of other roles of reactive astrocytes.
Footnotes
This work was supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke Grants NS042693, NS47386, and NS057624, the Roman Reed Spinal Cord Injury Research Fund of California, and the Adelson Medical Research Foundation.
References
- Aaronson DS, Horvath CM. A road map for those who don't know Jak-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- Acarin L, González B, Castellano B. STAT3 and NFkappaB activation precedes glial reactivity in the excitotoxically injured young cortex but not in the corresponding distal thalamic nuclei. J Neuropathol Exp Neurol. 2000;59:151–163. doi: 10.1093/jnen/59.2.151. [DOI] [PubMed] [Google Scholar]
- Agulló L, Baltrons MA, Garcia A. Calcium-dependent nitric oxide formation in glial cells. Brain Res. 1995;686:160–168. doi: 10.1016/0006-8993(95)00486-a. [DOI] [PubMed] [Google Scholar]
- Albrecht PJ, Dahl JP, Stoltzfus OK, Levenson R, Levison SW. Ciliary neurotrophic factor activates spinal cord astrocytes, stimulating their production and release of fibroblast growth factor-2, to increase motor neuron survival. Exp Neurol. 2002;173:46–62. doi: 10.1006/exnr.2001.7834. [DOI] [PubMed] [Google Scholar]
- Amat JA, Ishiguro H, Nakamura K, Norton WT. Phenotypic diversity and kinetics of proliferating microglia and astrocytes following cortical stab wounds. Glia. 1996;16:368–382. doi: 10.1002/(SICI)1098-1136(199604)16:4<368::AID-GLIA9>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Antony JM, van Marle G, Opii W, Butterfield DA, Mallet F, Yong VW, Wallace JL, Deacon RM, Warren K, Power C. Human endogenous retrovirus glycoprotein-mediated induction of redox reactants causes oligodendrocyte death and demyelination. Nat Neurosci. 2004;7:1088–1095. doi: 10.1038/nn1319. [DOI] [PubMed] [Google Scholar]
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4:1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- Balasingam V, Tejada-Berges T, Wright E, Bouckova R, Yong VW. Reactive astrogliosis in the neonatal mouse brain and its modulation by cytokines. J Neurosci. 1994;14:846–856. doi: 10.1523/JNEUROSCI.14-02-00846.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997;278:477–483. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundesen LQ, Scheel TA, Bregman BS, Kromer LF. Ephrin-B2 and EphB2 regulation of astrocyte-meningeal fibroblast interactions in response to spinal cord lesions in adult rats. J Neurosci. 2003;23:7789–7800. doi: 10.1523/JNEUROSCI.23-21-07789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferty WB, McGee AW, Strittmatter SM. Axonal growth therapeutics: regeneration or sprouting or plasticity? Trends Neurosci. 2008;31:215–220. doi: 10.1016/j.tins.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper KB, Jones K, McCarthy KD. Characterization of astrocyte-specific conditional knockouts. 2007;45:292–299. doi: 10.1002/dvg.20287. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Conti L, De-Fraja C. Signalling through the JAK/STAT pathway in the developing brain. Trends Neurosci. 1999;22:365–369. doi: 10.1016/s0166-2236(98)01378-2. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000) Neurochem Res. 2000;25:1439–1451. doi: 10.1023/a:1007677003387. [DOI] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehlings MG, Tator CH. The relationships among the severity of spinal cord injury, residual neurological function, axon counts, and counts of retrogradely labeled neurons after experimental spinal cord injury. Exp Neurol. 1995;132:220–228. doi: 10.1016/0014-4886(95)90027-6. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple sepctral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Filbin MT. Recapitulate development to promote axonal regeneration: good or bad approach? Philos Trans R Soc Lond B Biol Sci. 2006;361:1565–1574. doi: 10.1098/rstb.2006.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadea A, Schinelli S, Gallo V. Endothelin-1 regulates astrocyte proliferation and reactive gliosis via a JNK/c-Jun signaling pathway. J Neurosci. 2008;28:2394–2408. doi: 10.1523/JNEUROSCI.5652-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principle source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Gensert JM, Ratan RR. The metabolic coupling of arginine metabolism to nitric oxide generation by astrocytes. Antioxid Redox Signal. 2006;8:919–928. doi: 10.1089/ars.2006.8.919. [DOI] [PubMed] [Google Scholar]
- Gregg C, Shikar V, Larsen P, Mak G, Chojnacki A, Yong VW, Weiss S. White matter plasticity and enhanced remyelination in the maternal CNS. J Neurosci. 2007;27:1812–1823. doi: 10.1523/JNEUROSCI.4441-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen HJ, Bendtsen TF, Korbo L, Marcussen N, Moller A, Nielsen K, Nyengaard JR, Pakkenberg B, Sorensen FB, Vesterby A, West MJ. Some new, simple and efficient steriological methods and their use in pathological research and diagnosis. Acta Path Microbiol Immunol Scand. 1988;96:379–394. doi: 10.1111/j.1699-0463.1988.tb05320.x. [DOI] [PubMed] [Google Scholar]
- He F, Ge W, Martinowich K, Becker-Catania S, Coskun V, Zhu W, Wu H, Castro D, Guillemot F, Fan G, de Vellis J, Sun YE. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat Neurosci. 2005:616–625. doi: 10.1038/nn1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- Imura T, Kornblum HI, Sofroniew MV. The predominant neural stem cell isolated from postnatal and adult forebrain but not from early embryonic forebrain expresses GFAP. J Neurosci. 2003;23:2824–2832. doi: 10.1523/JNEUROSCI.23-07-02824.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imura T, Nakano I, Kornblum HI, Sofroniew MV. Phenotypic and functional heterogeneity of GFAP-expressing cells in vitro: Differential expression of LeX/CD15 by GFAP-expressing multipotent neural stem cells and non-neurogenic astrocytes. Glia. 2006;53:277–293. doi: 10.1002/glia.20281. [DOI] [PubMed] [Google Scholar]
- Inman D, Guth L, Steward O. Genetic influences on secondary degeneration and wound healing following spinal cord injury in various strains of mice. J Comp Neurol. 2002;451:225–235. doi: 10.1002/cne.10340. [DOI] [PubMed] [Google Scholar]
- John GR, Lee SC, Brosnan CF. Cytokines: powerful regulators of glial cell activation. Neuroscientist. 2003;9:10–22. doi: 10.1177/1073858402239587. [DOI] [PubMed] [Google Scholar]
- Johnson WB, Ruppe MD, Rockenstein EM, Price J, Sarthy VP, Verderber LC, Mucke L. Indicator expression directed by regulatory sequences of the glial fibrillary acidic protein (GFAP) gene: in vitro comparison of distinct GFAP-lacZ transgenes. Glia. 1995;13:174–184. doi: 10.1002/glia.440130304. [DOI] [PubMed] [Google Scholar]
- Justicia C, Gabriel C, Planas AM. Activation of the JAK/STAT pathway following transient focal cerebral ischemia: signaling through Jak1 and Stat3 in astrocytes. Glia. 2000;30:253–270. doi: 10.1002/(sici)1098-1136(200005)30:3<253::aid-glia5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Ransom BR, editors. Ed 2. New York: Oxford UP; 2004. Neuroglia. [Google Scholar]
- Kiehn O. Locomotor circuits in the mammalian spinal cord. Annu Rev Neurosci. 2006;29:279–306. doi: 10.1146/annurev.neuro.29.051605.112910. [DOI] [PubMed] [Google Scholar]
- Klein MA, Möller JC, Jones LL, Bluethmann H, Kreutzberg GW, Raivich G. Impaired neuroglial activation in interleukin-6 deficient mice. Glia. 1997;19:227–233. doi: 10.1002/(sici)1098-1136(199703)19:3<227::aid-glia5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levison SW, Jiang FJ, Stoltzfus OK, Ducceschi MH. IL-6-type cytokines enhance epidermal growth factor-stimulated astrocyte proliferation. Glia. 2000;32:328–337. doi: 10.1002/1098-1136(200012)32:3<328::aid-glia110>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, Nodin C, Ståhlberg A, Aprico K, Larsson K, Yabe T, Moons L, Fotheringham A, Davies I, Carmeliet P, Schwartz JP, Pekna M, Kubista M, Blomstrand F, Maragakis N, et al. Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab. 2008;28:468–481. doi: 10.1038/sj.jcbfm.9600546. [DOI] [PubMed] [Google Scholar]
- Liu NK, Xu XM. beta-tubulin is a more suitable internal control than beta-actin in Western blot analysis of spinal cord tissues after traumatic injury. J Neurotrauma. 2006;23:1794–1801. doi: 10.1089/neu.2006.23.1794. [DOI] [PubMed] [Google Scholar]
- Lu P, Tuszynski MH. Growth factors and combinatorial therapies for CNS regeneration. Exp Neurol. 2008;209:313–320. doi: 10.1016/j.expneurol.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Basso DM, Walters P, Stokes BT, Jakeman LB. Behavioral and histological outcomes following graded spinal cord contusion injury in C57/Bl/6 mouse. Exp Neurol. 2001;169:239–254. doi: 10.1006/exnr.2001.7679. [DOI] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD. Mechanisms of Disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. 2006;2:679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- Mi H, Barres BA. Purification and characterization of astrocyte precursor cells in the developing rat optic nerve. J Neurosci. 1999;19:1049–1061. doi: 10.1523/JNEUROSCI.19-03-01049.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy S, Simmons ML, Agulló L, Garcia A, Feinstein DL, Galea E, Reis DJ, Minc-Golomb D, Schwartz JP. Synthesis of nitric oxide in CNS glial cells. Trends Neurosci. 1993;16:323–328. doi: 10.1016/0166-2236(93)90109-y. [DOI] [PubMed] [Google Scholar]
- Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Houghtling RA, MacArthur L, Bayer BM, Bregman BS. Differences in cytokine gene expression profile between acute and secondary injury in adult rat spinal cord. Exp Neurol. 2003;184:313–325. doi: 10.1016/s0014-4886(03)00361-3. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci. 2003;26:523–530. doi: 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Neufeld AH, Liu B. Glaucomatous optic neuropathy: when glia misbehave. Neuroscientist. 2003;9:485–495. doi: 10.1177/1073858403253460. [DOI] [PubMed] [Google Scholar]
- Norenberg MD. Distribution of glutamine synthetase in the rat central nervous system. J Histochem Cytochem. 1979;27:756–762. doi: 10.1177/27.3.39099. [DOI] [PubMed] [Google Scholar]
- Norton WT, Aquino DA, Hozumi I, Chiu FC, Brosnan CF. Quantitative aspects of reactive gliosis: a review. Neurochem Res. 1992;17:877–885. doi: 10.1007/BF00993263. [DOI] [PubMed] [Google Scholar]
- Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–155. [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55:1251–1262. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- Plemel JR, Duncan G, Chen KW, Shannon C, Park S, Sparling JS, Tetzlaff W. A graded forceps crush spinal cord injury model in mice. J Neurotrauma. 2008;25:350–370. doi: 10.1089/neu.2007.0426. [DOI] [PubMed] [Google Scholar]
- Qi JW, Nakamura K, Hosokawa S, Okada Y, Horiuchi R, Yamamoto K. Time-dependent induction of midazolam-1-hydroxylation enzymes in rats treated with St. John's wort. Biol Pharm Bull. 2005;28:1467–1471. doi: 10.1248/bpb.28.1467. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Weinitz JM, Coulpier M, Fages C, Tinel M, Junier MP. A role for transforming growth factor alpha as an inducer of astrogliosis. J Neurosci. 1998;18:10541–10552. doi: 10.1523/JNEUROSCI.18-24-10541.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reier PJ, Houle JD. The glial scar: its bearing on axonal elongation and transplantation approaches to CNS repair. Adv Neurol. 1988;47:87–138. [PubMed] [Google Scholar]
- Sauer B. Site-specific recombination: developments and applications. Curr Opin Biotech. 1994;5:521–527. doi: 10.1016/0958-1669(94)90068-x. [DOI] [PubMed] [Google Scholar]
- Shafit-Zagardo B, Kume-Iwaki A, Goldman JE. Astrocytes regulate GFAP mRNA levels by cyclic AMP and protein kinase C-dependent mechanisms. Glia. 1988;1:346–354. doi: 10.1002/glia.440010507. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;5:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- Sriram K, Benkovic SA, Hebert MA, Miller DB, O'Callaghan JP. Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration: key signaling pathway for astrogliosis in vivo? J Biol Chem. 2004;279:19936–19947. doi: 10.1074/jbc.M309304200. [DOI] [PubMed] [Google Scholar]
- Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A. 1997;94:3801–3804. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161:4652–4660. [PubMed] [Google Scholar]
- Tansey FA, Cammer W. A pi form of glutathione-S-transferase is a myelin- and oligodendrocyte-associated enzyme in mouse brain. J Neurochem. 1991;57:95–102. doi: 10.1111/j.1471-4159.1991.tb02104.x. [DOI] [PubMed] [Google Scholar]
- Wilhelmsson U, Li L, Pekna M, Berthold CH, Blom S, Eliasson C, Renner O, Bushong E, Ellisman M, Morgan TE, Pekny M. Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J Neurosci. 2004;24:5016–5021. doi: 10.1523/JNEUROSCI.0820-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter CG, Saotome Y, Levison SW, Hirsh D. A role for ciliary neurotrophic factor as an inducer of reactive gliosis, the glial response to central nervous system injury. Proc Natl Acad Sci USA. 1995;92:5865–5869. doi: 10.1073/pnas.92.13.5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi K, Osuka K, Takayasu M, Usuda N, Nakazawa A, Nakahara N, Yoshida M, Aoshima C, Hara M, Yoshida J. Activation of JAK/STAT signalling in neurons following spinal cord injury in mice. J Neurochem. 2006;96:1060–1070. doi: 10.1111/j.1471-4159.2005.03559.x. [DOI] [PubMed] [Google Scholar]