

Abstract
Purpose:
Eradication of post-treatment residual myeloma cells is needed to prevent relapses and immunostimulatory monoclonal antibodies (mAbs) such as anti-CD137, CTLA-4, CD40, etc, that enhance the immune response against malignancies represent a means of achieving this purpose. This study explores anti-CD137 mAbs for mutiple myeloma (MM) treatment in preclinical models of the disease because they safely augment tumor immunity and are in clinical trials for other cancers.
Experimental design:
The anti-tumor effect of anti-CD137 mAb on mouse plasmacytomas derived from HOPC and NS0 cell lines was studied and compared with that of anti-CTLA-4, anti-CD40 and anti-ICAM-2 mAbs. The anti-tumor effect of anti-CD137 mAb was also examined in a mouse syngeneic disseminated myeloma (5TGM1) model, which more closely resembles human MM. Depletions of specific cell populations and gene-targeted mice were used to unravel the requirements for tumor rejection.
Results:
Agonistic mAb against CD137 and blocking anti-CTLA-4 mAb showed activity against intra-peritoneal HOPC tumors, resulting in extended survival of mice that also became immune to re-challenge. Anti-CD137 mAbs induced complete eradications of established subcutaneous NS0-derived tumors that were dependent on IFN-γ, NK cells and CD8+ T lymphocytes. NK cells accumulated in tumor draining lymph nodes (TDLNs) and showed increased IFN-γ production. Anti-tumor efficacy of anti-CD137 mAb was preserved in CD28-deficient mice, despite the fact that CD28 signaling increases the expression of CD137 on CD8+ T cells. Importantly, anti-CD137 mAb treatment significantly decreased systemic tumor burden in the disseminated 5TGM1 model.
Conclusions:
Anti-CD137 mAb's immune-mediated anti-tumor activity in mouse models holds promise for myeloma treatment in humans.
Keywords: CD137 (4-1BB), myeloma, NK cells, immunotherapy, Interferon-γ
INTRODUCTION
Multiple myeloma (MM) is a fatal neoplasm characterized by the uncontrolled proliferation of monoclonal plasma cells (1). Currently, the two most efficacious treatment options for patients with MM are tandem high-dose chemotherapy followed by autologous stem cell infusion, or allogeneic haematopoietic stem cell transplantation after myeloablative therapy or reduced-intensity conditioning (1, 2). New drugs have recently been incorporated in our armamentarium including the proteasome inhibitor bortezomib (Velcade) (3) and thalidomide derivatives that act as immunomodulators (4). Nevertheless, cure is very rarely achieved, due to persistence of residual disease. Therefore, new therapeutic approaches to control or even eradicate residual tumor cells are definitely needed, opening an opportunity for immunotherapy (5).
Over the last few years, cancer immunotherapy has emerged as a novel experimental treatment modality in multiple myeloma (6). This approach harnesses the potential of the host immune system to recognize and eradicate neoplastic tissue. Therefore, the success of cancer immunotherapy depends on the efficient induction and maintenance of endogenous anti-tumor immune responses mediated by innate and adaptive immune cells, that in the case of myeloma are counterbalanced by immunosuppressive factors produced by the tumor (6).
Immunostimulatory monoclonal antibodies (mAbs) represent a new and exciting strategy in cancer immunotherapy to potentiate the immune responses of the host against the malignancy (7). Such agonistic or antagonistic mAbs bind to key receptors in cells of the immune system acting to enhance antigen presentation (e.g. anti-CD40), provide co-stimulation (e.g anti-CD137), or to counteract immunoregulation (e.g. anti-CTLA-4). The aim is to boost weak, ineffectual, endogenous anti-tumor immunity to therapeutic levels. This potential has been demonstrated in animal models with a number of these mAbs showing impressive therapeutic activity in preclinical settings (7, 8). Anti-CTLA-4 mAbs are in advanced clinical trials for melanoma and other indications (8, 9). However, a possible obstacle to the clinical development of some of the immunostimulatory mAbs is their associated toxicity, most commonly reversible autoimmunity and/or systemic inflammatory reactions (7). In this regard, anti-CD137 is one of the most interesting immunostimulatory mAbs tested for cancer therapy (10-12), since the very same anti-CD137 mAbs that potently enhance tumor rejection are capable of reducing the incidence and severity of experimental autoimmune diseases (12-16).
CD137 (also called 4-1BB) is a T-cell co-stimulatory receptor induced upon TCR activation (11, 17). In addition to its expression on activated CD4+ and CD8+ T cells, CD137 is also expressed on CD4+CD25+ regulatory T cells (Tregs), NK and NK-T cells, monocytes, neutrophils and dendritic cells (DCs). Its natural ligand, CD137L has been described on APCs including B cells, monocyte/macrophages and DCs (17). Upon interaction with its ligand, CD137 leads to increased TCR-induced T cell proliferation, cytokine production, functional maturation, and prolonged CD8+ T cell survival (11, 17). Moreover, ligation of CD137 increases the proliferation and IFN-γ secretion of NK cells in response to IL-2 (18). Consistent with the co-stimulatory function of CD137, agonistic mAbs against this receptor have been shown to provoke powerful tumor-specific T cell responses capable of eradicating tumor cells in a variety of murine syngeneic tumor models leaving the animal immune to re-challenge (10, 19). Depletion and functional experiments indicate that CD8+ T and NK cells are the most consistent protagonists of the immune rejection process (10, 11, 19-21).
However, little is known about the potential therapeutic effect of this and other immunostimulatory mAbs in MM. In this study, we examined and compared the anti-myeloma effect of various immunostimulatory mAbs including anti-CD137 in two distinct mouse plasmacytoma models and investigated the requirements for the anti-tumor response generated by anti-CD137 mAb in these models. Finally, we have corroborated the anti-myeloma effect of anti-CD137 mAb in a disseminated myeloma model transplantable to immunocompetent mice that more closely resembles many features of the corresponding human disease.
MATERIALS AND METHODS
Mice and cell lines
Female BALB/c wild-type mice (5-6 weeks old) were purchased from Harlan Laboratories (Barcelona, Spain). Female C57BL/KaLwRijHsd mice (6-8 weeks old) were from Harlan (The Netherlands). IFN-γ−/− (C.129S7 (B6)-Ifngtm1Ts/J) and CD28−/− mice (C.129S2 (B6)-Cd28tm1Mak/J) and respective wild type (WT) littermates, both on BALB/c background were obtained from the Jackson Laboratory (Bar Harbor, ME) and were bred in our animal facility under specific pathogen-free conditions. Rag-1−/− BALB/c mice were also purchased form Jackson. All animal procedures were conducted under institutional guidelines (study approval number 003/02) that comply with national laws and policies.
The HOPC, NS0, P815 and YAC-1 cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA). CT26 cells were received from Dr. MP. Colombo (Milano. Italy). Cell lines were maintained in complete RPMI medium (RPMI 1640 with Glutamax [Gibco, Invitrogen, CA] containing 10% heat-inactivated FBS [SIGMA-ALDRICH, UK], 100 IU/ml penicillin and 100 μg/ml streptomycin [Biowhittaker, Walkersville, MD] and 5×10−5 mol/L 2-mercaptoethanol [Gibco]). Mouse 5TGM1 myeloma cells expressing enhanced GFP (5TGM1-GFP) (22), were generated from the parental 5TGM1 cell line (23), which was in turn established from the transplantable mouse 5T33 myeloma (24).
Antibodies and reagents
The hybridoma cell lines anti-CD40 (FGK-45), anti-CD4 (GK 1.5), anti-CD8β (H3S-17-2) and anti-IFN-γ (HB170) were obtained from ATCC; the anti-4-1BB (2A) and anti-CTLA-4 (9H10) hybridomas were kind gifts respectively from Dr. L. Chen (Johns Hopkins. Baltimore, MD) (25) and from Dr. J. Allison (Memorial Sloan Kettering, New York, NY) (26). Anti-ICAM 2 (4G8) mAb was produced in our laboratory as described before (27, 28). The monoclonal antibodies produced by these hybridomas were purified from respective culture supernatant by affinity chromatography in sepharose protein G columns according to manufacturer's instructions (GE Healthcare Bio-sciences AB, Uppsala, Sweden). IgG from rat serum was used as control antibody and was obtained from SIGMA (SIGMA-ALDRICH, UK). Anti-Asialo GM1 antiserum was used for in vivo NK cell depletion and was purchase from Wako (Wako, Neuss, Germany). PolyI:C was purchased from Pharmacia (Uppsala, Sweden).
In vivo tumor growth and depletion of lymphocyte subsets
For the intra-peritoneal (i.p.) myeloma models, BALB/c mice received an i.p. injection of either HOPC or NS0 cells (5×105 per mouse) on day 0, and on days 4 and 7 were treated intravenously (i.v.) with the corresponding mAb at 100μg per injection. These mice were examined weekly for palpable abdominal tumors or ascites.
For the subcutaneous (s.c.) myeloma model, BALB/c mice received an s.c. injection of 5×105 NS0 cells on day 0, and on days 9, 11, 13 and 15 were treated i.p. with either anti-CD137 mAb or the control rat IgG at 100μg per injection. Tumor diameters were measured using a electronic caliper every 2-4 days, and tumor size was determined by multiplying perpendicular diameters.
For in vivo leukocyte subset depletion, mice bearing NS0 s.c. tumors were injected with either depleting CD4 or CD8β-specific mAbs (200μg per dose), or anti-Asialo GM1 antiserum (50 μl per dose) prior to anti-CD137 treatment. Both depleting mAbs and anti-Asialo GM1 antiserum were administered daily for 5 consecutive days beginning 3 days before treatment onset and then every 5th day for the remainder of the experiment.
For in vivo IFN-γ blockade, mice bearing s.c. NS0 tumors were given 200 μg of neutralizing anti-IFN- γ one day after treatment onset and every 4 day thereafter for the next 2 weeks.
Experiments with the 5TGM1 MM model
5TGM1-GFP cells (106 per mouse) were intravenously inoculated, via tail vein, into 6-8 weeks old female naïve C57BL/KaLwRijHsd mice. Immediately following tumor cell inoculation, mice were randomly assigned to one of four different groups (n ≥ 8/group) and treated thereafter for 28 days by i.p. injection according to the following protocol: Group I: Vehicle (PBS); Group II: Bortezomib (1mg/kg body weight, three times a week); Group III: Rat IgG (control IgG) (100μg on days 4, 8, 14, 18 post-tumor cell inoculation). Group IV: anti-CD137 mAb (100μg on days 4, 8, 14, 18 post-tumor cell inoculation). Body weights were obtained at base-line and at weekly intervals thereafter. At the end of the experiment, mice were sacrificed and whole skeletons and visceral organs (spleens, livers, kidneys, ovaries, brains, lungs, hearts) were harvested and immediately imaged for fluorescent tumor foci as previously described (22). Briefly, selective excitation of EGFP was achieved using an Illumatools fiberoptic fluorescence lighting system (Epi model LT-9500; Lightools Research Inc., Encinitas, CA) with a 470/40 nm band-pass filter and a dichroic mirror. Emitted fluorescence was collected through a long-pass. Filter at 515nm with a MagnaFire® SP cooled color charge-coupled device (CCD) camera (Optronics, Goleta, CA) with an 11-48mm zoom lens, with same exposure times. High-resolution images (1,300 × 1030 pixels) were captured directly on a Macintosh laptop computer and are presented here unadjusted for contrast or brightness. Tumor burden was also assessed by assaying 5TGM1-specific monoclonal paraprotein (IgG2bκ) in sera prepared from whole blood obtained by retro-orbital sinus bleed of tumor-bearing mice just prior to sacrifice, under light methoxyflurane-induced anesthesia. Mouse IgG2bκ levels were assayed using a specific in-house sandwich ELISA as previously described (23), with a rat IgG1κ that binds mouse IgG2 γ heavy chain (CLONE LO-MG2b-2; Research Diagnostics Inc., Concord, MA) as the capture antibody and a horseradish peroxidase-conjugated rat IgG1κ that binds mouse IgG κ light chain (CLONE LO-MK-2; Biodesign) as detection antibody. In this ELISA, there is no species cross-reactivity or cross-reactivity to other mouse immunoglobulins.
Isolation of mononuclear cells from lymph nodes and tumors
At the indicated time points, tumor draining lymph nodes (TDLNs) or minced tumor nodules were harvested from tumor-bearing mice that had been treated with either anti-CD137 mAb or the control Ab. Both axillary and inguinal TDLNs from individual mice were pooled. Then, the LNs were incubated in Collagenase and DNase (Roche Basel, Switzerland) for 15 minutes at 37°C, and pressed between 2 semi-frosted microscopic slides. Finally the dissociated cells were passed through a 70 μm nylon mesh filter (BD Falcon, BD Bioscience, San Jose, CA) and washed before further use.
FACS Analysis and cytotoxicity experiments
Cells resuspended in PBS with 5% FBS and pre-treated with Fc-Block (anti-CD16/32, eBioscience, San Diego, CA) to reduce the non-specific staining. Afterwards, cells were incubated with the staining Abs. Monoclonal antibodies to the following mouse antigens conjugated to fluorescein isothiocyanate (FITC), phycoerythrin (PE), allophycocyanin (APC) or biotin were used: CD40, CD137, ICAM 2, CD80 , CD86, IAd , H2Kd, CD49b, CD3, CD4, CD8, IFN-γ, and CD69. These specific mAbs and their corresponding isotype controls were obtained from BD Pharmingen. Biotinylated antibodies were visualized with streptavidin-FITC (Sav-FITC) (BD Pharmingen). A FACS Calibur (BD) was used for cell acquisition and data analysis was carried out using Cell Quest Pro (BD) or FlowJo (Tree Star, Inc.) software. Five hour 51Cr release assays were performed to measure NK and CTL activity in spleen and LN cell suspensions as previously described (20, 27). For CTL activity five day restimulation cocultures with irradiated (120 Gy) NSO cells were performed as described (27, 29).
Intracellular cytokine staining
Lymphocytes from TDLNs were resuspended in complete RPMI medium, set up in 96-well plates at a concentration of 106 cells/well, and incubated with PMA (5ng/ml) and ionomycin (500ng/ml) for 5 hours. After 1 hour of incubation, Brefeldin-A was added at 10μg/ml and monensin at 5μg/ml. Finally, the cells were surface-stained using NK markers, processed with the Fix & Perm kit (BD Biosciences), and stained for the intracellular cytokine IFN-γ. In case of T cells restimulated with irradiated NS0 cells a similar protocol was used but surface staining with FITC-tagged anti-CD107a mAb (BD) was performed prior to intracellular staining.
In vitro antibody stimulation
Naïve CD8+ lymphocytes were prepared from spleen cells of naïve BALB/c mice by generating a single-cell suspension which was enriched in CD8+ T cells by negative selection using immuno-magnetic beads according to the manufacturer's protocol (Miltenyi Biotec). For stimulation with anti-CD3 96-well plates were coated with 0.1 ml/well of anti-CD3 mAb (145.2C11) at 1 μg/ml for 3 hours at 37°C. When anti-CD28 (BD Biosciences) was used it was added at 5 μg/ml. After 4-day incubation, CD137 expression was analyzed on CD3+CD8+ population by FACS.
Statistical analysis
Kaplan-Meier plots were used to analyze survival. Prism software (Graph Pad Software, Inc.) was used to analyze tumor growth and the percentages of NK cells and to determine statistical significance of difference between groups by applying an unpaired Student's t-test. Comparison of survival curves was made by the log-rank test. P values of <0.05 were considered significant. For tumor burdens, comparison of means was performed by ANOVA and posthoc analysis by Fisher's PLSD test using Statview Software (SAS Institute, Cary, NC).
RESULTS
Anti-CD137 mAb increases overall survival in aggressive plasmacytoma models, inducing long-lasting tumor-specific immunity
Anti-CTLA-4, anti-CD40, and anti-CD137 are some of the most efficacious immunostimulatory mAbs (7). In numerous animal models, they have been shown to promote powerful tumor-specific T cell responses capable of clearing established tumors. In addition, we have previously observed that ICAM-2 specific mAb exhibited anti-tumor activity in the CT26 mouse colon cancer model that is mediated by inhibition of activation induced cell death (AICD) in T lymphocytes (27, 28).
In this study, we examined and compared the therapeutic efficacy of these four immunostimulatory mAbs in the highly tumorigenic HOPC myeloma model. To this end, HOPC cells (5×105 cells per mouse) were inoculated in the peritoneal cavity of BALB/c mice on day 0, and on days 4 and 7 the corresponding mAb was i.v. injected. In this experimental system, we found that both anti-CD137 and anti-CTLA-4 mAbs showed a clearly defined anti-myeloma effect, with 40-50% of animals surviving long-term (>120 days), while anti-ICAM-2 and anti-CD40 mAbs at these dose regimes showed little and no therapeutic activity, respectively (Fig. 1A). It is noteworthy that while CD137, CTLA-4 and CD40 are absent from the plasma membrane of tumor cells, ICAM-2 is readily expressed (Fig1B).
Fig.1. Mice treated with anti-CD137 mAb showed increased survival to HOPC plasmacytoma and acquire long-lasting and tumor-specific immunity. The therapeutic effect of anti-CD137 treatment was independent from direct targeting of HOPC myeloma cells.

(A) To compare the relative efficacy of the different immunostimulatory mAbs on the treatment of HOPC tumors, BALB/c mice (16 per group) were i.p. injected with 5×105 HOPC viable cells on day 0, and on days 4 and 7 were i.v. treated with control rat IgG or the indicated mAbs at the dose of 100μg. These mice were examined weekly for palpable abdominal tumors or ascites. Mice survival was plotted using the Kaplan-Meier method and analyzed for significance using the log-rank test. Pooled data from two identical experiments are shown. (B) HOPC tumor cells were evaluated for MHC I, MHC II, CD80, CD86, ICAM-2, CD40, CD137 and CTLA-4 expression by FACS. The grey area represents the fluorochrome-tagged isotype control antibody and the white area represents the relevant antibody. (C) To examine whether a long-lasting and tumor-specific immunity could be generated, mice that had been cured of HOPC tumors with either anti-CTLA-4 or anti-CD137 mAbs were re-challenge 4 month later with HOPC and CT26 cells. Tumor cells (5×105 per mouse) were s.c. inoculated in the opposite flanks of long-term surviving mice from experiment (A) as represented in the scheme. Mice were monitored for tumor growth and compared to naïve age-matched mice. Tumor sizes were assessed by measuring (in millimeters) perpendicular diameters of tumors and the results are expressed as tumor area. These experiments were repeated at least twice yielding similar results. Representative data are shown.
To determine whether treatment with anti-CD137 or anti-CTLA-4 mAbs concomitantly elicits anti-tumor immunity that is long-lasting, mice that had been cured of the HOPC tumor by treatment with these two immunostimulatory mAbs were re-challenged s.c. with HOPC cells. We found that while cured mice did not develop palpable HOPC tumors, all naive mice showed progressive tumor growth (Fig. 1C). In parallel, we observed that the long-lasting immune memory developed in mice cured by anti-CD137 or anti-CTLA-4 mAbs was specific for HOPC, since these mice did not reject the syngeneic CT26 tumor cell line, inoculated on their opposite flank (Fig. 1C). These findings indicate that both anti-CD137 and anti-CTLA-4 mAbs may be useful for myeloma therapy.
Although, there was previous published information about anti-CTLA-4 mAbs in plasmacytoma models (30), the potential of anti-CD137 mAbs which have a safer preclinical profile (31, 32) remains unexplored. Therefore, we next confirmed the anti-tumor effect of anti-CD137 mAb in another experimental plasmacytoma model. With this aim, NS0 myeloma cells (5×105) were inoculated i.p. in BALB/c mice on day 0, and on days 4 and 7 post-tumor cell inoculation, anti-CD137 mAb or the control rat IgG were i.v. injected. In this experimental system, we found that anti-CD137 mAb showed a potent therapeutic effect, with 70-80% of animals surviving long-term (>120 days) (Fig. 2A).
Fig.2. Potent therapeutic effects of the agonistic anti-CD137 mAb in mice bearing NS0 non-immunoglobulin secreting plasmacytomas accompanied by CTL induction;

(A) To evaluate the relative efficacy of anti-CD137 mAb on prolonging survival in of NS0 tumor-bearing mice, NS0 cell (5×105) were i.p. injected into syngeneic BALB/c mice (12 per group) and antibodies (100 μg/mouse) were given i.v. on days 4 and 7 after tumor injection. Survival was plotted using the Kaplan-Meier method and analyzed for significance using the log-rank test. (B) NS0 tumor cells were evaluated for expression of the indicated surface markers by flow cytometry. The grey area represents the isotype control antibody and the white area represents the relevant antibody. (C) Evaluation of therapeutic effect of anti-CD137 mAb assessed on established subcutaneous tumors by sequential measures of tumor areas (fraction of surviving tumor free mice is provided in each graph). BALB/c mice (6 per group) received a s.c. injection of 5×105 NS0 cells on day 0, and on days 9, 11, 13 and 15 mice were treated i.p with mAb 2A or a control IgG at 100μg per injection. Statistical analyses were performed by the T test. This experiment was repeated at least three times yielding similar results. Representative data are shown and compiled data for statistical analysis are presented in a separate graph. (D) Cell suspensions from spleens and tumor draining lymph nodes of mice cured from NS0 sc tumors by anti-CD137 mAb or naïve mice were restimulated in cocultulture with iradiated NS0 cells (1: 25) for 5 days and tested in 51Cr-release assays (mean±SEM from six independent mice) against CT26 and NSO cells (left) and for upregulation of intracellular IFNγ and surface CD107a in gated CD3+CD8+ splenocytes by FACS (right). Percentage of double positive cells expressed as the mean±SEM from 5-day cocultures prepared from 6 different mice are shown inside the corresponding dot plots.
Interestingly, neither HOPC cells nor NS0 cells expressed the CD137 molecule on their plasma membranes, indicating that the therapeutic effect of the agonistic mAb is not mediated via direct targeting of the malignant plasma cells (Fig 1B and 2B).
We also evaluated the therapeutic effect of anti-CD137 mAb in established subcutaneous NS0 tumors that were clearly palpable before commencement of treatment. To this end, mice received a s.c. injection of 5×105 NS0 cells on day 0, and on days 9, 11, 13 and 15 post-tumor cell inoculation were treated i.p. with either anti-CD137 mAb or control rat IgG. Consistent with our previous results, anti-CD137 mAb treatment resulted in profound inhibition of tumor growth and more than 60% of mice bearing NS0 tumors were completely cured (Fig. 2C). This robust therapy model was thereafter chosen to study the mechanistic requirements behind the therapeutic effects of anti-CD137 mAb.
NSO tumor rejections are accompanied by CTL induction
The spleen and tumor draining lymph nodes from the mice that had been cured from NS0 subcutaneous tumors by anti-CD137 mAb treatment contained cells that upon 5 day restimulation in culture with irradiated NSO cells showed tumor specific cytolytic activity against NS0 cells in 51Cr-release assays (Fig 2D right graphs). Moreover, these cocultures contained CD8+ T cells that upregulated intracellular IFN-γ and showed degranulation (measured as surface CD107a) specifically upon rexposure to NSO cells, but not CT26 cells (Fig 2D dot plots on the left). This indicates that CD8 T cells can recognize tumor antigens on NS0 cells.
Both NK and CD8+ T cells are required for tumor rejection
To identify the cell types responsible for the anti-tumor activity of anti-CD137 mAb, we carried out in vivo leukocyte subset depletion prior to anti-CD137 treatment. As shown in Fig. 3A, depletion of either NK cells or CD8+ T cells significantly impaired the therapeutic effect of the treatment. In this regard, we found that NSO cells are almost as susceptible to NK-mediated lysis as the sensitive YAC-1 cells (inset to figure 3A in the NK depletion graph) despite of the fact that NSO cells intensely express surface MHC class I (Figure 2B). In vitro NK cytotoxicity was observed with NK cells obtained from poly I:C-preinjected Rag−/− syngeneic mice. These NK cells unsuccessfully killed NK-resistant P815 cells in the same assays (inset to figure 3A NK depletion graph). In contrast, CD4+ T cell depletion had no significant effect on tumor rejection. These results indicate that both NK and CD8+ T cells, but not CD4+ T cells, are required for tumor rejection. It is noteworthy that depletion of CD8+ subset was performed with an anti-CD8β depleting antibody to ensure that only peripheral CD8+ T cells but not CD8α+ DCs were affected.
Fig.3. Absolute requirements of IFN-γ, NK cells, and CD8+ T lymphocytes for eradication of NS0 plasmacytomas after anti-CD137 mAb treatment.

(A) Involvement of CD4+, CD8+ T cells and NK cells in the eradication of tumors after anti-CD137 treatment as in Fig. 2C was assessed. BALB/c mice, in groups of 6, bearing s.c. NS0 tumors were injected i.p. with either anti-CD4 or anti-CD8β mAbs or i.v with anti-Asialo GM1 antiserum. A total of 200μg per dose of each mAb were injected into recipient mice for depleting CD4+ and CD8+ T cells and 50 μl per dose of anti-Asialo GM1 were administered for depleting NK cell. Both CD4 and CD8β specific mAbs and anti-Asialo GM1 antiserum were administered as described in Materials and Methods. Fraction of surviving tumor free mice is provided in each graph. In the graph corresponding to NK cell depletion an inset is provided that shows specific lysis (mean±SEM) in 51Cr release assays demonstrating the sensitivity of NS0 cells to killing by activated DX5+ NK cells isolated from the spleens of Rag1−/− mice that had been pretreated 18h earlier with 50 μg of poly I:C ip. % of lysis were compared to those achieved against YAC-1 cells and P815 targets. (B) To determine whether IFN-γ was required for eradication of NS0-derived tumors after anti-CD137 mAb treatment, WT or IFN-γ−/− (IFN-γ K.O.) BALB/c mice (6 per group) were inoculated s.c. with 5×105 NS0 viable cells and then treated with either anti-CD137 mAb or control antibody. Alternatively, tumor-bearing mice (n = 6) were treated with anti-CD137 mAb, and were subsequently given 200 μg of neutralizing anti-IFN-γ as described in Materials and Methods. Statistical analyses were performed using the t test. These experiments were performed at least twice, yielding similar results. Representative data are shown and compiled data for statistical analysis are presented in separate graphs for A and B.
Normal function of IFN-γ is an absolute requirement for tumor rejection
IFN-γ production is critical for the cell-mediated anti-tumor immune response. Here, we examined whether IFN-γ was required for the anti-myeloma effect of anti-CD137 treatment as described for other tumor models (33). To this end, both WT and IFN-γ-deficient mice were inoculated with NS0 cells and treated with anti-CD137 mAb or the control rat IgG as described in Materials and Methods. Whereas tumors in the WT BALB/c mice regressed after treatment, all of IFN-γ-deficient mice developed progressively growing tumors (Fig. 3B). Similarly, tumor regression was significantly impaired in mice that received a neutralizing anti-IFN-γ mAb (Fig. 3B). The fact that the anti-IFN-γ mAb did not completely abolish the therapeutic effect of anti-CD137 mAb may simply be a result of incomplete blockade. Therefore, tumor eradication after treatment with agonistic anti-CD137 mAb is dependent on IFN-γ.
Anti-CD137 mAb induced NK cell augment in TDLNs and IFN-γ; production by NK cells
The abolishment of efficacy upon NK cell depletion was remarkable (Fig 3A). At the time when tumor rejections were first observed (8-10 days after treatment onset), both inguinal and axillary TDLNs had increased relative and absolute numbers of NK cells (CD3−DX5+cells) when the mice had been treated with anti-CD137 mAb (Fig. 4A). We wondered if such NK cells under anti-CD137 treatment would also show a higher degree of activation and we found that DLN NK cells showed similar levels of CD69 expression regardless of the antibody used for treatment. However, anti-CD137 treatment up-regulated the capability of NK cells in DLN to produce IFN-γ as assessed by intracellular staining (Fig. 4B). These effects on NK biology at the DLN are likely involved in the absolute requirement of NK cells for the antitumor effects.
Fig.4. NK cells are increased and activated in tumor draining lymph nodes (TDLNs) while CD8 T cells predominate in the tumor rejecting infiltrates.

(A) BALB/c mice (6 per group) received a s.c. injection of 5×105 NS0 viable cells on day 0, and on days 9, 11, 13 and 15 were treated i.p. with either anti-CD137 mAb or a control IgG at 100μg per injection. The average percentages and total cell numbers of NK cells (DX5+ CD3− cells) in TDLN were determined by flow cytometry. Pooled data from 2 experiments are shown. Statistical analyses were performed by the T test. (B) NK cells were evaluated for CD69 expression by flow cytometry. Histograms show CD69 expression on this cell subset. The percentages ± SEM of positive cells are indicated. Data are representative of 2 independent experiments. (C) Intracellular expression of IFN-γ by NK cells recovered from TDLNs of either control IgG or anti-CD137-treated-tumor-bearing mice was examined. The mononuclear cells from TDLNs were stimulated with PMA/Ionomycin in vitro for 5 hours and subsequently stained for intracellular IFN-γ. The percentages ± SEM of NK cells producing IFN-γ are indicated. In panels (B) and (C) shaded histograms represent isotype control antibody and white histograms represent the relevant antibody. (D) percentage± SEM of CD4 (CD3+CD4+), CD8 (CD3+CD8+) and NK cells (CD3−DX5+) in lymphoid cell suspensions obtained from disaggregated NS0 tumor nodules. The lesions were excised on day 19 from mice that had been treated with anti-CD137 mAb or control antibody on days 9,11,13,15 after tumor cell injection. Absolute numbers also showed clear increases in intratumoral CD8 T cell counts and decreases in CD4 T cell counts (data not shown).
The tumor infiltrate of NSO tumors that are responding to anti-CD137 treatment are enriched in CD8 T cells
Cell suspensions from NSO tumors taken from mice which have been treated with anti-CD137 and showed signs of growth delay or shrinkage were obtained to study their lymphocyte content. It was found that there was an increase of CD8 T cells while NK and CD4 cells were not increased in the tumor rejecting infiltrate (Figure 4C). These data indicate that the main role at the final execution of tumor rejection corresponds to CD8 T cells that abundantly populate the tumor lesion. This does not preclude that NK cells could be playing a role at earlier stages or that they cooperate with CTLs in an orchestrated fashion (29). However percentage and absolute numbers (not shown) of intratumour leukocytes provide evidence indicating that CTLs are the main players when rejections become clinically meaningful.
The anti-tumor effect of anti-CD137 is independent of CD28
To determine whether or not co-stimulation through CD28 is a pre-requisite for CD137 signaling, we first investigated whether triggering the CD28 pathway contributes to the up-regulation of CD137 on naïve T cells. To this end purified CD8+ T cells were stimulated in vitro with plate bound anti-CD3 mAb in the presence or absence of soluble anti-CD28 mAb. We observed that TCR triggering by anti-CD3 mAbs in combination with CD28 co-stimulation was much more efficient at inducing surface expression of CD137 on CD8+ T cells than TCR-CD3 triggering alone (Fig. 5A).
Fig. 5. CD28 signals up-regulate CD137 expression on CD8+ T cells but anti-CD137 mAb treatment of NS0 tumors is independent from CD28 function.

(A) Dot plot analysis of CD137 expression on CD8+ splenocytes after 96 h in vitro activation with anti-CD3 mAb or anti-CD3 + anti-CD28 as indicated. (B) Comparison of subcutaneous growth of individual tumors derived from NSO treated either with rat IgG or anti-CD137 mAb (100μg on days 9, 11, 13, 15) in Balb/c WT mice or in CD28−/− mice as indicated in the figure the fraction of tumor-free surviving mice is provided. Representative results from two similarly performed are shown.
Based on these results we hypothesized that the in vivo blockade of the CD28 pathway would abrogate the anti-myeloma effect of anti-CD137 mAb. To test this hypothesis both WT and CD28-deficient mice were inoculated with NS0 cells and treated with anti-CD137 mAb or the control rat IgG as described before. Surprisingly, the anti-myeloma effect of anti-CD137 mAb was preserved in CD28-deficient mice. Thus, although CD28 signaling increases the expression of CD137 on CD8+ T cells in vitro, the therapeutic effect of anti-CD137 is independent of CD28 (Fig 5B).
Anti-CD137 decreased tumor burden in 5TGM1 myeloma-bearing mice
To extend our observations about the anti-plasmacytoma effect of anti-CD137 to a MM model considered more relevant to the human disease, we used the 5TGM1 cell line transfected with the EGFP gene to accurately monitor tumor progression (22). This cell line does not express detectable levels of CD137 on its plasma membrane upon FACS analysis (data not shown). Intravenous injection of these tumor cells in naïve syngeneic C57BL/KaLwRijHsd mice gives rise to a disseminated tumor model in which fluorescent tumor cells can be visualized as tumoral foci in multiple organs in a distribution that very closely resembles severe cases of MM in humans. This is true particularly with regard to skeletal involvement (Fig. 6A and B). An Anti-CD137 mAb treatment course started four days after tumor cell inoculation clearly reduced multi-organ tumor burden in a significant proportion of mice (Fig. 6 A and B), although it did not attain the efficacy achieved by a course of bortezomib in a similar experimental schedule (Fig. 6A). Anti-CD137 mAb-mediated reduction of tumor burden was evident in the skeleton of 7 out of 10 mice and although tumor disappearance was complete in only 3 out of 7 mice, the tumor fluorescent foci in the remaining four mice was nonetheless significantly reduced and comparable to those seen in bortezomib-treated mice. The efficacy of anti-CD137 mAb treatment was even more dramatic in visceral organs with spleen and liver involvement in only one out of 10 mice inoculated with tumor cells compared to ≥ 7 out of 10 in tumor-inoculated mice treated either with vehicle or a control IgG (Fig. 6A, B). Consistent with these observations, weight loss assessed on day 30 post-tumor cell inoculation when the mice were sacrificed (data not shown), was also less pronounced in anti-CD137 mAb-treated mice compared to the other two groups. Moreover, the serum monoclonal paraprotein concentration, a marker of overall myeloma tumor load, was significantly decreased by the course of anti-CD137 mAb treatment compared to the vehicle or rat control Ab treatment (Fig. 6C), although in this respect anti-CD137 mAb was again surpassed by the efficacy of bortezomib. Overall these data indicate that anti-CD137 mAb is clearly efficacious in this MM model even as monotherapy and further suggest the clinical potential of the approach.
Figure 6. Anti-CD137 mAb treatment significantly decreases tumor burden in a disseminated multiple myeloma model.
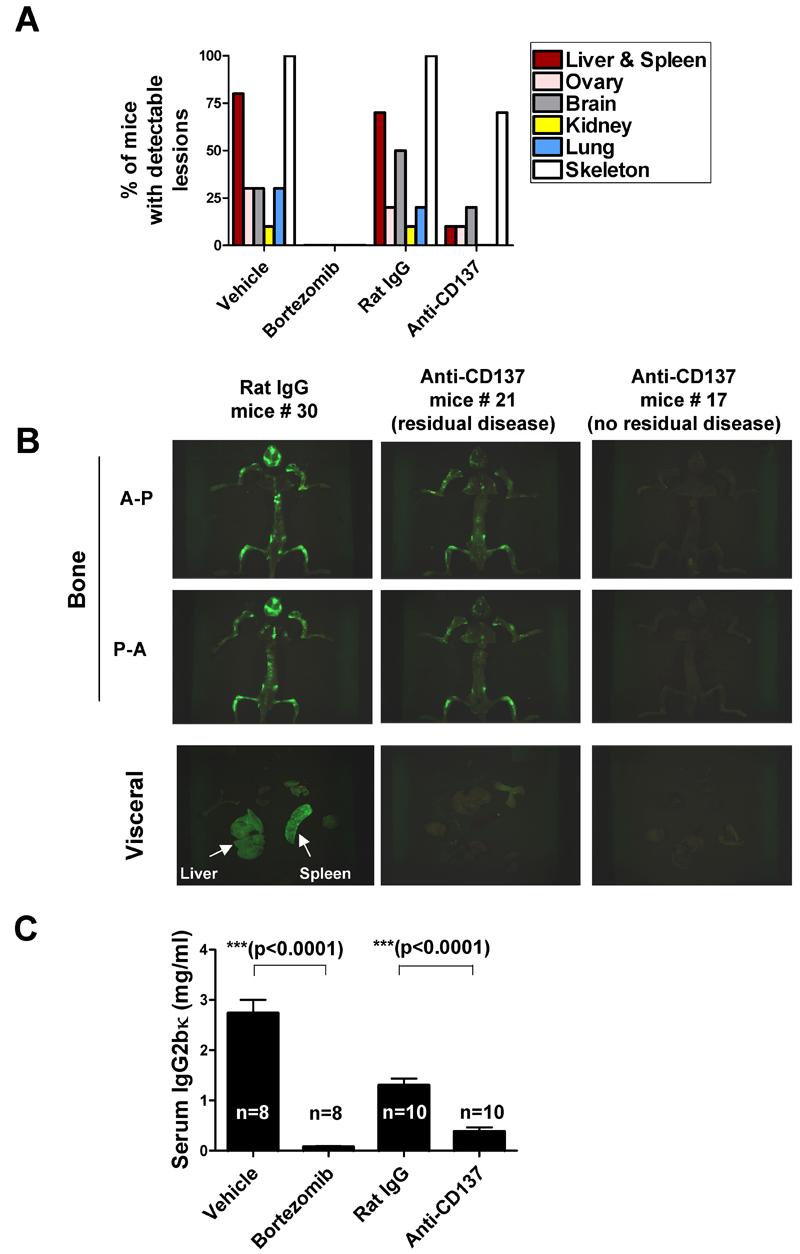
To investigate the anti-tumor efficacy of anti-CD137 mAb in a model of myeloma with widespread skeletal involvement, 5TGM1-GFP myeloma cells (106) were i.v. injected into syngeneic C57BL/KaLwRijHsd mice (≥ 8 per group) and then randomly assigned to four groups which received either vehicle, bortezomib, control rat IgG or anti-CD137 mAb for 4 weeks (see Materials and Methods for dosing protocol).
(A) Fraction of mice with detectable EGFP+ foci in the indicated organs of mice immediately after sacrifice on day 30. Tissues were optically imaged, as described under Materials and Methods, in antero-posterior (A-P) and postero-anterior (P-A) orientations and then scored. Data represent the % of mice in each group with ≥ 2 fluorescent foci in both orientations in ≥3 bones. (B) Representative pictures of green fluorescence emitted (A-P and P-A views) of mice from (A) that had been eviscerated during necropsy and the corresponding pictures form the explanted organs. In the case of anti-CD137 mAb, the mice photos shown are from representative animals displaying either fluorescence under the threshold of detection (representing completely eradicated tumor/no residual disease) or reduced but detectable fluorescence (representing some residual disease) as indicated in the figure (c) Serum concentrations (mean ± SEM) of the 5TGM1 monoclonal paraprotein (IgG2bκ) from mice in the different treatment groups measured 30 days after tumor cell inoculation.
DISCUSSION
This study demonstrates that pharmacological agonistic manipulation of CD137 with a specific mAb shows benefit in various murine myeloma models and provides important clues on the involvement of CD8+ T lymphocytes and NK cells in the therapeutic immune response. The effects of anti-CD137 mAb are not directed against the malignant plasma cells but indirectly mediated by immune regulation, as suggested by the absence of CD137 from the cell membrane of the different plasmacytomas used.
It has been postulated that the antibody engages CD137 expressed on activated T cells and maybe on some activated NK cells in the tumor bearing mice (7). CD137 induction on CD8+ T lymphocytes must be the result of tumor antigen presentation either by tumor cells or professional APCs. The advantageous effects of CD137 ligation on CTL differentiation and memory have been extensively reported (34-37). However, the direct or indirect pathway that leads in vivo to the activation of NK cells, as we have observed in DLN, is still unclear (38). We are investigating whether ligation of the CD137 molecules expressed on NK cells plays a role in tumor rejection. In this regard, we have recently reported that tumor rejection is often the result of the concerted action of multiple leukocyte partners including NK cells, which play a pivotal role and can be found to be producing IFN-γ in vivo (29). The finding of the sensitivity of NSO cells to autologous NK cells despite high surface MHC class I indicates the expression of ligands for NK activatory receptors (38).
NK-cell production of IFN-γ at sentinel LN have been reported to be important in the initiation of T cell immunity, and tumoricidal NK cells have been detected in tumor rejecting infiltrates (38). In our case, NK cells at tumor DLNs show activation features and numeric increases that could be dependent on either recruitment or local proliferation of NK cells. In spite of the absolute need of NK cells to achieve efficacy, it is clear that at the time when the tumors start to be clinically controlled by treatment, the rejecting infiltrate is dominated by CTLs. More investigation is needed to precisely define the train of events that leads to tumor rejection through the cooperative crosstalk between NK and CTLs. Our results further highlight an important role for NK cells operating at the draining LN (38).
IFN-γ is a pleiotropic cytokine and it has been reported to induce changes in tumor endothelial cells thereby facilitating homing of effector T cells into solid tumors and to inhibit angiogenesis. In addition, IFN-γ may damage tumor cells by means of anti-proliferative and pro-apoptotic signaling pathways (39), while up-regulating antigen presentation, as well as CTL and macrophage activity (40). In our hands, recombinant IFN-γ had no effect on the proliferation and survival of NS0 plasma cells in culture (data not shown), strongly suggesting that it plays an indirect role in the efficacy of anti-CD137 mAb treatments. The critical requirement of IFN-γ for tumor rejection suggests its potential as a biomarker to correlate with efficacy in eventual clinical trials for myeloma.
The injected therapeutic antibody can bind CD137 on other leukocyte and endothelial cell lineages (17). The role of these interactions at inducing tumor immunity remains unexplored. We can conclude from selective depletions that intervention of CD4+ T cells is dispensable at least in the NS0 model. In this sense, it has been recently reported that in mouse melanoma CD4 depletion is even beneficial for antitumor effects (41).
We confirm that anti-CTLA-4 mAb can also have a role in the treatment of myeloma (30) in a fashion that looks comparable to the effects anti-CD137 mAb as observed in an i.p. plasmacytoma model in which suboptimal efficacy is attained by both antibodies. The mechanism of action of anti-CTLA-4 has been described by the group of J. Allison (42) and in essence involves blockade of anti-CTLA-4 negative signals, in such a way that the co-stimulatory activity of CD28 is liberated. Therefore the mechanism of action of anti-CTLA4 mAb is different from that of anti-CD137 mAb, although both converge in the generation of a tumor-rejecting lymphocyte infiltrate. It is noteworthy that tumor cells used in this work express CD80 that may bind CTLA-4 and CD28 on activated T cells. Thus, in these cases, CTLA-4 mAb could block this inhibitory interaction upon in vivo lymphocyte-tumor cell encounter(43).
Over the last few years, there has been intense interest in whether or not the CD80/86-CD28 co-stimulatory pathway is required for CD137 mAb activity. In 1997, TH. Watts et al reported that ligation of CD137 can co-stimulate human and mouse CD28−/− T cells (44), suggesting that CD137 triggering can take place independently of the CD28 pathway (45). This group also demonstrated that enforced co-stimulation through CD137 by CD137L could promote anti-tumor responses in the presence or in the absence of CD28 (46). However, R Offringa and colleagues (47) recently reported that blockade of the costimulatory pathway abrogated the capacity of agonistic anti-CD137 mAb to trigger CTL immunity in response to the Ad5E1A peptide vaccine. This was probably because under weaker TCR signals, CD137 expression on naive T cells is not attained, and therefore the susceptibility of these cells to anti-CD137 mAbs required both stimulation of the TCR and CD28-dependent co-stimulation. However, in our hands, CD28 function is dispensable for CD137 activity, albeit it is easy to find CD8+ T cell activation conditions in which CD28 co-stimulation results in much higher CD137 expression by CD8+ T cells in vitro, as previously suggested by experiments with the natural ligand (48).
This study describes the therapeutic effects of anti-CD137 mAb as monotherapy in myeloma but it is clear that CD137 targeted therapy would be most efficacious in combination immunotherapy. Combinations of tumor vaccines, adoptive T cell therapy, chemo and/or radiotherapy are known to display additive and synergistic effects in cancer, and combination therapies of anti-CD137 mAb and other immunostimulatory mAbs have shown remarkable activity in other tumor models (7, 32, 49, 50). However for practical reasons, work on myeloma models should first define anti-CD137 combination potential with standard treatments for MM such as bone-marrow transplantation and chemotherapy.
The significant reduction of tumor burden in the experiments performed with the 5TGM1 model is remarkable due to the nature of the systemic dissemination of this experimental disease. The comparison of anti-CD137 mAb with bortezomib seems to favor the latter agent but should be considered with caution. On the one hand, bortezomib has direct cytotoxic effects on myeloma cells and likely acts immediately on the tumor cells in vivo, whereas the full effect of the anti-CD137 mAb would need a latency period until the immune response is sufficiently up-regulated. On the other hand, the full potential of anti-CD137 mAb has yet to be exploited, in particular with regard to combination strategies either with a tumor vaccine (25) or an intervention to increase tumor immunogenicity. There is also the possibility of combining sub-optimal doses of bortezomib with anti-CD137 mAb in order to reduce some of the well known side effects of bortezomib such as neuropathies. The use of the 5TGM1 myeloma model for such combination therapy studies in the future has obvious advantages since it utilizes immunocompetent mice and accumulation of the monoclonal paraprotein in serum accurately reflects tumor burden. In addition, transfection of 5TGM1 cells with EGFP permits image assessment (22) of tumor load in real time without increasing immunogenicity, since CTLs do not recognize EGFP as foreign in the H-2b class I molecules (29).
There are a number of rationale for proposing clinical trials with agonist anti-CD137 mAb in myeloma: (i) the results of the present study on three different myeloma mouse models, including the disseminated 5TGM1 model that shares many characteristic features with the human disease; (ii) a safe clinical leading profile (ASCO 2008, abstract 3007) and (31)); (iii) the abundance of NK cells and memory T cells in the bone marrow which is the most common primary site for malignant plasma cells in myeloma (1); (iv) an agent of this kind (BMS663513) is already undergoing phase I and II trials for melanoma, renal cell carcinoma, lung and ovarian cancer. The effects of the anti-CD137 mAb on the disseminated 5TGM1 model reported herein provide a base-line to further optimize combination strategies and improve preclinical efficacy to guide future clinical trial design.
Acknowledgements
We are grateful to Dr. L. Chen both for providing an agonist anti-CD137 producing hybridoma as well as for helpful criticism and discussion. Dr. James Allison kindly provided anti-CTLA-4 mAb producing hybridoma cells. Drs. Jesús Prieto, Ascensión Lopez-Diaz de Cerio, Mercedes Rodriguez-Calvillo, Maurizio Bendandi, and Juan José Lasarte are acknowledged for support and scientific discussion. Elena Ciordia, Javier Guillén, Juan Percaz and Eneko Elizalde are acknowledged for excellent animal care.
Grant suport: Research grants were from: Ministerio de Educación y Ciencia (MEC-SAF2005-03131), Departamento de Educación del Gobierno de Navarra, Departamento de Salud del Gobierno de Navarra (Beca Ortiz de Landázuri). Redes temáticas de investigación cooperativa RETIC (RD06/0020/0065), European commission VII famework program (ENCITE) and “UTE for project FIMA”. OM and AA were recipients of scholarships from Ministerio de Educación (MEC) and Fondo de investigación sanitaria (BEFI) respectively. S H-S is sponsored by the AECC (Asociación Española contra el Cancer). BOO is supported by a NIH/NCI Career Development Award (KO1 CA104180). The use of the flow cytometry facilities at UTHSC at San Antonio is supported by a Cancer Center Support Grant from the NCI (P30 CA054174) to the Cancer Therapy & Research center (CTRC@UTHSCSA).
Footnotes
STATEMENT OF CLINICAL RELEVANCE.
Multiple myeloma treatment is fast improving. New therapeutic agents such as bortezomib and lenolinomede have been introduced with impressive results, albeit a curative treatment constitutes an unmet need. This article explores at the preclinical level the use of immunostimulatory monoclonal antibodies for myeloma treatment. These agents act augmenting the antitumor immune response acting on molecules of the immune system. Such monoclonal antibodies either release the brakes of inhibitory activities or agonistically enhance tumor-rejecting functions. This article focuses on agonist anti-CD137 monoclonal antibodies that are known to enhance immunity against several tumor types in mice and are currently undergoing phase I and II clinical trials in patients with other malignancies. The in vivo effects of these antibodies in various myeloma models indicate the suitability of these agents for clinical trials in multiple myeloma.
REFERENCES
- 1.Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7:585–98. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- 2.Barlogie B, Shaughnessy J, Tricot G, Jacobson J, Zangari M, Anaissie E, et al. Treatment of multiple myeloma. Blood. 2004;103:20–32. doi: 10.1182/blood-2003-04-1045. Epub 2003 Sep 11. [DOI] [PubMed] [Google Scholar]
- 3.Armand JP, Burnett AK, Drach J, Harousseau JL, Lowenberg B, San Miguel J. The emerging role of targeted therapy for hematologic malignancies: update on bortezomib and tipifarnib. Oncologist. 2007;12:281–90. doi: 10.1634/theoncologist.12-3-281. [DOI] [PubMed] [Google Scholar]
- 4.Richardson PG, Mitsiades C, Hideshima T, Anderson KC. Lenalidomide in multiple myeloma. Expert Rev Anticancer Ther. 2006;6:1165–73. doi: 10.1586/14737140.6.8.1165. [DOI] [PubMed] [Google Scholar]
- 5.Mihelic R, Kaufman JL, Lonial S. Maintenance therapy in multiple myeloma. Leukemia. 2007;21:1150–7. doi: 10.1038/sj.leu.2404633. [DOI] [PubMed] [Google Scholar]
- 6.Chiriva-Internati M, Cobos E, Kast WM. Advances in immunotherapy of multiple myeloma: from the discovery of tumor-associated antigens to clinical trials. Int Rev Immunol. 2007;26:197–222. doi: 10.1080/08830180701365966. [DOI] [PubMed] [Google Scholar]
- 7.Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer. 2007;7:95–106. doi: 10.1038/nrc2051. [DOI] [PubMed] [Google Scholar]
- 8.Peggs KS, Segal NH, Allison JP. Targeting immunosupportive cancer therapies: accentuate the positive, eliminate the negative. Cancer Cell. 2007;12:192–9. doi: 10.1016/j.ccr.2007.08.023. [DOI] [PubMed] [Google Scholar]
- 9.Ribas A, Hanson DC, Noe DA, Millham R, Guyot DJ, Bernstein SH, et al. Tremelimumab (CP-675,206), a cytotoxic T lymphocyte associated antigen 4 blocking monoclonal antibody in clinical development for patients with cancer. Oncologist. 2007;12:873–83. doi: 10.1634/theoncologist.12-7-873. [DOI] [PubMed] [Google Scholar]
- 10.Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellstrom KE, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. 1997;3:682–5. doi: 10.1038/nm0697-682. [DOI] [PubMed] [Google Scholar]
- 11.Nam KO, Kang WJ, Kwon BS, Kim SJ, Lee HW. The therapeutic potential of 4-1BB (CD137) in cancer. Curr Cancer Drug Targets. 2005;5:357–63. doi: 10.2174/1568009054629681. [DOI] [PubMed] [Google Scholar]
- 12.Mittler RS, Foell J, McCausland M, Strahotin S, Niu L, Bapat A, et al. Anti-CD137 antibodies in the treatment of autoimmune disease and cancer. Immunol Res. 2004;29:197–208. doi: 10.1385/IR:29:1-3:197. [DOI] [PubMed] [Google Scholar]
- 13.Seo SK, Choi JH, Kim YH, Kang WJ, Park HY, Suh JH, et al. 4-1BB-mediated immunotherapy of rheumatoid arthritis. Nat Med. 2004;10:1088–94. doi: 10.1038/nm1107. [DOI] [PubMed] [Google Scholar]
- 14.Sun Y, Chen HM, Subudhi SK, Chen J, Koka R, Chen L, et al. Costimulatory molecule-targeted antibody therapy of a spontaneous autoimmune disease. Nat Med. 2002;8:1405–13. doi: 10.1038/nm1202-796. [DOI] [PubMed] [Google Scholar]
- 15.Sun Y, Chen JH, Fu Y. Immunotherapy with agonistic anti-CD137: two sides of a coin. Cell Mol Immunol. 2004;1:31–6. [PubMed] [Google Scholar]
- 16.Myers LM, Vella AT. Interfacing T-cell effector and regulatory function through CD137 (4-1BB) co-stimulation. Trends Immunol. 2005;26:440–6. doi: 10.1016/j.it.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 18.Wilcox RA, Tamada K, Strome SE, Chen L. Signaling through NK cell-associated CD137 promotes both helper function for CD8+ cytolytic T cells and responsiveness to IL-2 but not cytolytic activity. J Immunol. 2002;169:4230–6. doi: 10.4049/jimmunol.169.8.4230. [DOI] [PubMed] [Google Scholar]
- 19.Miller RE, Jones J, Le T, Whitmore J, Boiani N, Gliniak B, et al. 4-1BB-specific monoclonal antibody promotes the generation of tumor-specific immune responses by direct activation of CD8 T cells in a CD40-dependent manner. J Immunol. 2002;169:1792–800. doi: 10.4049/jimmunol.169.4.1792. [DOI] [PubMed] [Google Scholar]
- 20.Melero I, Johnston JV, Shufford WW, Mittler RS, Chen L. NK1.1 cells express 4-1BB (CDw137) costimulatory molecule and are required for tumor immunity elicited by anti-4-1BB monoclonal antibodies. Cell Immunol. 1998;190:167–72. doi: 10.1006/cimm.1998.1396. [DOI] [PubMed] [Google Scholar]
- 21.Ye Z, Hellstrom I, Hayden-Ledbetter M, Dahlin A, Ledbetter JA, Hellstrom KE. Gene therapy for cancer using single-chain Fv fragments specific for 4-1BB. Nat Med. 2002;8:343–8. doi: 10.1038/nm0402-343. [DOI] [PubMed] [Google Scholar]
- 22.Oyajobi BO, Munoz S, Kakonen R, Williams PJ, Gupta A, Wideman CL, et al. Detection of myeloma in skeleton of mice by whole-body optical fluorescence imaging. Mol Cancer Ther. 2007;6:1701–8. doi: 10.1158/1535-7163.MCT-07-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dallas SL, Garrett IR, Oyajobi BO, Dallas MR, Boyce BF, Bauss F, et al. Ibandronate reduces osteolytic lesions but not tumor burden in a murine model of myeloma bone disease. Blood. 1999;93:1697–706. [PubMed] [Google Scholar]
- 24.Radl J, Croese JW, Zurcher C, Van den Enden-Vieveen MH, de Leeuw AM. Animal model of human disease. Multiple myeloma. Am J Pathol. 1988;132:593–7. [PMC free article] [PubMed] [Google Scholar]
- 25.Wilcox RA, Flies DB, Zhu G, Johnson AJ, Tamada K, Chapoval AI, et al. Provision of antigen and CD137 signaling breaks immunological ignorance, promoting regression of poorly immunogenic tumors. J Clin Invest. 2002;109:651–9. doi: 10.1172/JCI14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–6. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 27.Melero I, Gabari I, Corbi AL, Relloso M, Mazzolini G, Schmitz V, et al. An anti-ICAM-2 (CD102) monoclonal antibody induces immune-mediated regressions of transplanted ICAM-2-negative colon carcinomas. Cancer Res. 2002;62:3167–74. [PubMed] [Google Scholar]
- 28.Melero I, Gabari I, Tirapu I, Arina A, Mazzolini G, Baixeras E, et al. Anti-ICAM-2 monoclonal antibody synergizes with intratumor gene transfer of interleukin-12 inhibiting activation-induced T-cell death. Clin Cancer Res. 2003;9:3546–54. [PubMed] [Google Scholar]
- 29.Arina A, Murillo O, Hervas-Stubbs S, Azpilikueta A, Dubrot J, Tirapu I, et al. The combined actions of NK and T lymphocytes are necessary to reject an EGFP+ mesenchymal tumor through mechanisms dependent on NKG2D and IFN gamma. Int J Cancer. 2007;121:1282–95. doi: 10.1002/ijc.22795. [DOI] [PubMed] [Google Scholar]
- 30.Mokyr MB, Kalinichenko T, Gorelik L, Bluestone JA. Realization of the therapeutic potential of CTLA-4 blockade in low-dose chemotherapy-treated tumor-bearing mice. Cancer Res. 1998;58:5301–4. [PubMed] [Google Scholar]
- 31.Niu L, Strahotin S, Hewes B, Zhang B, Zhang Y, Archer D, et al. Cytokine-mediated disruption of lymphocyte trafficking, hemopoiesis, and induction of lymphopenia, anemia, and thrombocytopenia in anti-CD137-treated mice. J Immunol. 2007;178:4194–213. doi: 10.4049/jimmunol.178.7.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kocak E, Lute K, Chang X, May KF, Jr., Exten KR, Zhang H, et al. Combination therapy with anti-CTL antigen-4 and anti-4-1BB antibodies enhances cancer immunity and reduces autoimmunity. Cancer Res. 2006;66:7276–84. doi: 10.1158/0008-5472.CAN-05-2128. [DOI] [PubMed] [Google Scholar]
- 33.Wilcox RA, Flies DB, Wang H, Tamada K, Johnson AJ, Pease LR, et al. Impaired infiltration of tumor-specific cytolytic T cells in the absence of interferon-gamma despite their normal maturation in lymphoid organs during CD137 monoclonal antibody therapy. Cancer Res. 2002;62:4413–8. [PubMed] [Google Scholar]
- 34.Lee HW, Nam KO, Park SJ, Kwon BS. 4-1BB enhances CD8+ T cell expansion by regulating cell cycle progression through changes in expression of cyclins D and E and cyclin-dependent kinase inhibitor p27kip1. Eur J Immunol. 2003;33:2133–41. doi: 10.1002/eji.200323996. [DOI] [PubMed] [Google Scholar]
- 35.Lee HW, Park SJ, Choi BK, Kim HH, Nam KO, Kwon BS. 4-1BB promotes the survival of CD8+ T lymphocytes by increasing expression of Bcl-xL and Bfl-1. J Immunol. 2002;169:4882–8. doi: 10.4049/jimmunol.169.9.4882. [DOI] [PubMed] [Google Scholar]
- 36.Shuford WW, Klussman K, Tritchler DD, Loo DT, Chalupny J, Siadak AW, et al. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med. 1997;186:47–55. doi: 10.1084/jem.186.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Myers L, Lee SW, Rossi RJ, Lefrancois L, Kwon BS, Mittler RS, et al. Combined CD137 (4-1BB) and adjuvant therapy generates a developing pool of peptide-specific CD8 memory T cells. Int Immunol. 2006;18:325–33. doi: 10.1093/intimm/dxh371. [DOI] [PubMed] [Google Scholar]
- 38.Arina A, Murillo O, Dubrot J, Azpilikueta A, Alfaro C, Perez-Gracia JL, et al. Cellular liaisons of natural killer lymphocytes in immunology and immunotherapy of cancer. Expert Opin Biol Ther. 2007;7:599–615. doi: 10.1517/14712598.7.5.599. [DOI] [PubMed] [Google Scholar]
- 39.Mazzolini G, Narvaiza I, Martinez-Cruz LA, Arina A, Barajas M, Galofre JC, et al. Pancreatic cancer escape variants that evade immunogene therapy through loss of sensitivity to IFNgamma-induced apoptosis. Gene Ther. 2003;10:1067–78. doi: 10.1038/sj.gt.3301957. [DOI] [PubMed] [Google Scholar]
- 40.Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–95. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 41.Choi BK, Kim YH, Kang WJ, Lee SK, Kim KH, Shin SM, et al. Mechanisms involved in synergistic anticancer immunity of anti-4-1BB and anti-CD4 therapy. Cancer Res. 2007;67:8891–9. doi: 10.1158/0008-5472.CAN-07-1056. [DOI] [PubMed] [Google Scholar]
- 42.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. 2006;90:297–339. doi: 10.1016/S0065-2776(06)90008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tirapu I, Huarte E, Guiducci C, Arina A, Zaratiegui M, Murillo O, et al. Low surface expression of B7-1 (CD80) is an immunoescape mechanism of colon carcinoma. Cancer Res. 2006;66:2442–50. doi: 10.1158/0008-5472.CAN-05-1681. [DOI] [PubMed] [Google Scholar]
- 44.DeBenedette MA, Shahinian A, Mak TW, Watts TH. Costimulation of CD28- T lymphocytes by 4-1BB ligand. J Immunol. 1997;158:551–9. [PubMed] [Google Scholar]
- 45.Saoulli K, Lee SY, Cannons JL, Yeh WC, Santana A, Goldstein MD, et al. CD28-independent, TRAF2-dependent costimulation of resting T cells by 4-1BB ligand. J Exp Med. 1998;187:1849–62. doi: 10.1084/jem.187.11.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guinn BA, Bertram EM, DeBenedette MA, Berinstein NL, Watts TH. 4-1BBL enhances anti-tumor responses in the presence or absence of CD28 but CD28 is required for protective immunity against parental tumors. Cell Immunol. 2001;210:56–65. doi: 10.1006/cimm.2001.1804. [DOI] [PubMed] [Google Scholar]
- 47.Diehl L, van Mierlo GJ, den Boer AT, van der Voort E, Fransen M, van Bostelen L, et al. In vivo triggering through 4-1BB enables Th-independent priming of CTL in the presence of an intact CD28 costimulatory pathway. J Immunol. 2002;168:3755–62. doi: 10.4049/jimmunol.168.8.3755. [DOI] [PubMed] [Google Scholar]
- 48.Melero I, Bach N, Hellstrom KE, Aruffo A, Mittler RS, Chen L. Amplification of tumor immunity by gene transfer of the co-stimulatory 4-1BB ligand: synergy with the CD28 co-stimulatory pathway. Eur J Immunol. 1998;28:1116–21. doi: 10.1002/(SICI)1521-4141(199803)28:03<1116::AID-IMMU1116>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 49.Murillo O, Arina A, Tirapu I, Alfaro C, Mazzolini G, Palencia B, et al. Potentiation of therapeutic immune responses against malignancies with monoclonal antibodies. Clin Cancer Res. 2003;9:5454–64. [PubMed] [Google Scholar]
- 50.Uno T, Takeda K, Kojima Y, Yoshizawa H, Akiba H, Mittler RS, et al. Eradication of established tumors in mice by a combination antibody-based therapy. Nat Med. 2006;12:693–8. doi: 10.1038/nm1405. [DOI] [PubMed] [Google Scholar]