

Abstract
The precise genetic and molecular defects underlying epithelial ovarian cancer (EOC) remain largely unknown, and treatment options for patients with advanced disease are limited. Cyclooxygenases (COX-1 and COX-2) catalyze the conversion of arachidonic acid to prostaglandins. Whereas overwhelming evidence suggests a role for COX-2 in a variety of cancers, the contribution of COX-1 remains much less explored. The expression status of COX isoforms in ovarian cancers also remains confusing. We have previously shown that human epithelial ovarian tumors have increased levels of COX-1 but not COX-2. To more carefully examine the role of COXs in ovarian cancer, we used a mouse model of EOC in which genetic and oncogenic modifications were experimentally engineered into ovarian surface epithelial cells (OSE) thought to be the cells of origin for human EOC. These OSE cells produce tumors when allografted into host mice. Using multiple approaches, we observed that OSE cells and the tumors comprised of these cells express high levels of COX-1 but not COX-2. Prostacyclin (PGI2) is the major prostaglandin generated downstream of COX-1 in these cells, and SC-560, a COX-1-selective inhibitor, dramatically inhibits PGI2 production. More importantly, SC-560 reduced the growth of tumors when OSE cells were allografted in nude female mice. In contrast, the COX-2-selective inhibitor celecoxib had little effect on tumor growth. The growth inhibitory effects of SC-560 result from reduced cell proliferation and/or accelerated apoptosis. Our results imply COX-1 as a target for the prevention and/or treatment of EOC.
Introduction
The cyclooxygenases, COX-1 and COX-2, catalyze arachidonic acid to prostaglandin H2 (PGH2), which is then converted to various prostaglandins by specific synthases (1). Whereas COX-1 and COX-2 isoforms are encoded by separate genes, they have very similar structural and kinetic properties and show distinct cell-specific expression and regulation (1). COX-1 is considered a constitutive enzyme, whereas COX-2 is highly inducible by diverse stimuli including cytokines, growth factors, mitogens, and tumor promoters and also regulates inflammation, differentiation, mitogenesis, and angiogenesis (1). Nonsteroidal anti-inflammatory steroids (NSAID) primarily interfere with prostaglandin biosynthesis by inhibiting COX enzymes. Research primarily focusing on colorectal cancer has provided mounting evidence that NSAIDs are effective in cancer prevention and possibly for adjuvant therapy in the treatment of established tumors (2). These drugs impede colorectal cancer growth primarily by inhibiting COX-2, although other non-COX-2 targets cannot be ruled out. Furthermore, COX-2 is up-regulated in a range of extracolonic cancers, and selective COX-2 inhibitors have potent antineoplastic effects in vivo in preclinical models of various solid malignancies (2). These findings led to several clinical trials examining the efficacy of COX-2-selective inhibitors in primary and/or secondary prevention of cancer or as part of a combination therapy regimen for established tumors (2). Unfortunately, recent clinical results indicate that prolonged use of highly selective COX-2 inhibitors leads to increased myocardial infractions and strokes.
Epidemiologic studies to examine whether NSAIDs prevent or delay the development of ovarian cancers remain inconclusive. Although several population- and hospital-based case-control studies present evidence for time- and dose-dependent decreases in the risk of developing ovarian cancers with the consumption of several NSAIDs (3-5), other studies failed to find any significant correlation or found that the reduction in risk was associated only with the use of particular NSAIDs (6, 7). Whether treatment with COX-2-selective inhibitors is beneficial for the prevention and/or treatment of ovarian cancer remains unknown.
The expression profiles of COX isotypes in ovarian cancer remain puzzling, as most of the published reports mainly focused on COX-2 expression rather than COX-1 using the rationale that COX-2 is overexpressed in most tumors, whereas COX-1 is expressed primarily in normal tissues (Table 1). Furthermore, these studies mainly employed a single approach of immunologic detection of COX expression. It is possible that some of these studies used COX-2 antibodies that are nonspecific. Using multiple approaches, we recently showed that human epithelial ovarian cancer (EOC) manifests heightened expression of COX-1 but not COX-2 (8).
Table 1.
Published reports of COX-1 and COX-2 expressions in human ovarian tumors or tumor cell lines
| COX-1 | COX-2 | Ref. | ||
|---|---|---|---|---|
| 1 | NE | +(RT) | Cell line | (38) |
| 2 | +(RT, WB) | +(NB, WB) | Cell line | (39) |
| NE | +/-(IHC) | Tumor | ||
| 3 | NE | +(WB) | Cell line | (40) |
| NE | +/-(IHC) | Tumor | ||
| 4 | NE | +/-(IHC) | Tumor | (41) |
| 5 | NE | +/-(IHC) | Tumor | (42) |
| 6 | +(WB) | -(WB) | Cell line | (43) |
| +/-(IHC) | +/-(IHC) | Tumor | ||
| 7 | NE | +(IHC) | Tumor | (44) |
| 8 | NE | +(IHC) | Tumor | (45) |
| 9 | +/-(WB) | +/-(WB) | Cell line | (46) |
| NE | +/-(IHC) | Tumor | ||
| 10 | +(WB) | +(WB induced by ILβ, RT) | Cell line | (26) |
| 11 | NE | +(WB induced by ILβ) | Cell line | (47) |
| 12 | NE | +(IHC) | Tumor | (48) |
| 13 | NE | +(IHC) | Cell line | (49) |
| 14 | NE | +(IHC) | Tumor | (50) |
| 15 | +/-(WB) | -(WB) | Cell line | (8) |
| +(WB, IHC, NB, ISH) | -(WB, IHC, NB, ISH) | Tumor | ||
| 16 | NE | +(WB) | Tumor | (51) |
| 17 | NE | +/-(IHC) | Tumor | (52) |
| 18 | NE | +(RT) | Cell line | (53) |
| 19 | NE | +(RT) | Tumor | (54) |
| 20 | NE | +(WB), (IHC) | Tumor | (55) |
| 21 | +(RT, WB) | +/-(RT, WB) | Cell line | (56) |
| +(RT), -(IHC) | +(RT, IHC) | Tumor | ||
| 22 | +(WB, IHC) | +(IHC, WB) | Cell line | (57) |
| 23 | NE | +(IHC) | Tumor | (58) |
| 24 | NE | +/-(IHC) | Tumor | (59) |
| 25 | NE | +(IHC) | Tumor | (60) |
| 26 | NE | +(IHC) | Tumor | (61) |
| 27 | +(IHC, WB) | NE | Tumor | (62) |
| 28 | NE? | -(NB) | Tumor | (63) |
Abbreviations: NE, not examined; +, positive; RT, RT-PCR; -, negative; WB, Western blotting; +/-, mixed; IHC, immunohistochemistry; NB, Northern blotting; ISH, in situ hybridization.
More than 85% of human ovarian cancers are EOCs, which originates from ovarian surface epithelial (OSE) cells (9). EOC is lethal, and effective treatment options for patients who present with the advanced disease are very limited. The high mortality rate is due to the lack of effective screening strategies identifying patients at high risk or those with early neoplastic lesions still amenable to treatment (10-12). The hallmark of EOC is the rapid growth and spread of solid i.p. tumors, often leading to ascitic fluid formation. It is the major cause of death among patients with gynecologic malignancies and is the fifth most common cause of death from cancer in the United States (13). EOCs are morphologically and biologically heterogeneous, making it difficult to define the molecular events underlying the disease development and progression. Morphologic criteria classify EOCs into four major types: serous, mucinous, endometriod, and clear cell (14). Although the causes of EOCs remain elusive, reproductive, endocrine, and hereditary factors play contributing roles in their genesis (9). Evaluation of human ovarian cancer specimens reveals several genetic alterations, including mutations in tumor suppressor genes, including p53, BRCA1, or BRCA2, and/or activating mutations or amplification of proto-oncogenes such as c-myc, K-ras, and Akt. Thus, mutation or loss of tumor suppressor genes, amplification of growth stimulatory and/or suppression of death signaling pathways make certain individuals more susceptible to various cancers, including breast and ovarian cancers in women (12). However, the molecular mechanism by which these genes contribute to the initiation and/or progression of EOC remains poorly defined. Thus, the underlying causes and early events required for the progression of EOCs are among the least understood of all major human cancers.
Understanding the initiation and progression of EOC has been slow primarily because of the lack of suitable experimental models. Recently, an experimental model in which defined multiple genetic alterations can be introduced into mouse OSE cells in culture and in mouse ovaries has been developed (15). This system is based on avian RCAS virus delivery to the cells that are programmed to express the avian TVA receptor (16). Thus, genetically defined aberrations that are known to be involved in the genesis and growth of epithelial ovarian carcinomas can be introduced into the mouse ovarian epithelial cells ex vivo. For example, the minimal genetic requirements for induction of a tumorigenic state in primary mouse ovarian epithelial cells were determined by introducing combinations of c-myc, K-ras, and Akt into ovarian cells from p53 null mice. It was shown that a loss of the p53 gene, and the addition of any two of the c-myc, K-ras, and Akt oncogenes are sufficient to induce transformation of mouse primary OSE cells. Orthotopic implantation of infected OSE cells results in metastatic ovarian tumors that resemble human ovarian serous papillary carcinomas. Mouse OSE cell lines with defined genetic alterations have been generated from the ex vivo-infected cells and the i.p. tumors. More recently, three other groups have generated genetically engineered mouse models of ovarian epithelial cancers (17-19). One study revealed that female transgenic mice expressing the transforming region of SV40 under control of the Mullerian inhibitory substance type II receptor gene promoter develop bilateral ovarian tumors (17), whereas the other two studies, using Cre-lox-mediated gene alterations in the ovarian surface epithelium, show that concurrent inactivation of p53 and Rb1 (18) or simultaneous activation of K-ras and inactivation of Pten (19) leads to the development of ovarian epithelial cancers in mice.
In this study, we used the mouse OSE cell lines and tumors described by Orsulic et al. (15) to examine the expression of COX isoforms and the effects of selective COX inhibitors on OSE tumor growth. We found that mouse OSE tumors express mainly COX-1 but not COX-2. More importantly, a COX-1-selective inhibitor SC-560 dramatically reduces tumor growth in vivo. The most abundant prostaglandin produced by OSE cells is prostacyclin (PGI2), the production of which is dose-dependently inhibited by SC-560. A COX-2-selective inhibitor celecoxib was ineffective in either reducing prostaglandin biosynthesis or tumor growth. These results suggest that COX-1 is an important player in this model of ovarian cancer and that selective COX-1 inhibitors may have potential for use in the treatment and prevention of EOC in humans.
Materials and Methods
Cell lines and culture
To generate C1 (genotype: p53-/-, myc, K-ras) and C2 (genotype: p53-/-, myc, Akt) cell lines, ovarian explants from K5-TVA/p53-/- mice were infected with different combinations of RCAS viruses carrying human c-myc, mouse K-rasG12D, and mouse myr-Akt1 oncogenes. The cell cultures were passaged in the presence of viruses for 3 weeks, after which they consisted of a pure population of transformed OSE cells. T1 and T2 cell lines were derived by isolating cells from tumors that were generated by orthotopic placement of C1 and C2 cells into nude mice, respectively. The C1, C2, T1, and T2 cell lines readily developed tumors when transplanted into immunocompromised mice. These OSE cells were propagated in DMEM containing 10% fetal bovine serum (FBS), 100 units/mL of penicillin, and 100 Ag/mL streptomycin.
Allografting of ovarian surface epithelial cells and tumor growth
T2 OSE cells were used for tumor growth studies in vivo. A suspension of 2.5 × 106 T2 cells was placed under the dorsal skin of female athymic nude mice (nu/nu, 5-6 weeks old). Tumor growth was measured every 2 to 4 days by direct measurement of tumor dimensions. COX-1-selective inhibitor (SC-560) or COX-2-selective inhibitor (celecoxib) was suspended in 0.5% (w/v) methylcellulose and 0.1% (v/v) polysorbate 80 suspended in water by constant stirring. Drugs were given by oral gavage twice a day. Tumor volume was determined by external measurement and calculated according to the equation (V = 0.5 × [LW2]), where V = volume, L = length, and W = width (20).
Preparation of lysates and Western blot analysis
Tissue samples and cultured cells were homogenized in lysis buffer [150 mmol/L NaCl, 1% NP40, 0.5% deoxycholate, 0.1% SDS, and 50 mmol/L Tris (pH 8)] containing proteinase and phosphatase inhibitors. The lysates were centrifuged at 9,880 × g for 10 minutes at 4°C. After measuring protein concentration, the supernatants (25 μg protein) were boiled for 5 minutes in SDS sample buffer. The samples were run on 10% SDS-PAGE gels under reducing conditions and transferred onto nitrocellulose membranes. The membranes were blocked with 10% milk in TBST and probed with goat polyclonal antibodies against COX-1, COX-2, or PGIS (Cayman, Ann Arbor, MI) overnight at 4°C. After thorough washing, the blots were incubated in peroxidase-conjugated donkey/anti-goat IgG or donkey/anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), followed by washing in TBST. Protein signals were detected using chemiluminescent reagents (Amersham, Piscataway, NJ).
RNA isolation and Northern blot analysis
Total RNA was extracted from cultured cells or tissue specimens using Trizol reagent (Invitrogen, Carlsbad, CA). Total RNA (6 μg) was denatured and separated by formaldehyde/agarose gel electrophoresis, transferred to nylon membranes, and UV cross-linked. Blots were prehybridized, hybridized, and washed as previously described by us (21).
In situ hybridization
In situ hybridization was done as previously described (21). In brief, frozen sections (10 μm) were mounted onto poly-l-lysine-coated slides and fixed in cold 4% paraformaldehyde in PBS. The sections were prehybridized and hybridized at 45°C for 4 hours in 50% formamide hybridization buffer containing the 35S-labeled antisense or sense cRNA probes. RNase A-resistant hybrids were detected by autoradiography. Sections were post-stained with H&E. Sections hybridized with the sense probes did not exhibit any positive signals and served as negative controls.
Hybridization probes
The cDNA clones for Cox-1 and Cox-2 have been previously described (22). A mouse specific cDNA for Pgis was kindly provided by Mark Geraci (University of Colorado Health Sciences, Denver, CO). For in situ hybridization, sense and antisense 35S-labeled cRNA probes were generated using Sp6 and T7 polymerases, respectively. For Northern hybridization, antisense 32P-labeled cRNA probes for Cox-1, Cox-2, Pgis, and rPL7 (a housekeeping gene) were generated. Probes had specific activities of ×2 × 109 dpm/μg.
Prostaglandin measurements
OSE cells were inoculated in 24-well plates at a density of 105 cells per well and incubated in 10% FBS/DMEM overnight. The medium was changed to serum-free medium and cells were incubated for another 12 hours. After changing to medium containing arachidonic acid and/or COX-1 inhibitor (SC-560) or COX-2 inhibitor (celecoxib), the cells were incubated for 4 hours. To determine the effects of COX inhibitors on the basal levels of prostaglandins, cells were cultured for 4 and 16 hours without arachidonic acid. The medium was analyzed for prostaglandin levels by gas chromatography/negative ion chemical ionization mass spectrometric assay (21). PGI2 was measured as its stable metabolite 6-keto-PGF1α. Arachidonic acid was dissolved in ethanol, whereas the COX inhibitors were dissolved in DMSO. Cells cultured in vehicle contained 0.1% ethanol and 0.1% DMSO.
Cell growth assay
Cell viability was measured by trypan blue exclusion assay. Briefly, T2 cells (0.5 × 105 per well) were plated in 6-well plates. After culture overnight, the cells were washed twice with PBS, and treatment was initiated with serum-free medium containing either 10% FBS, 0.1% DMSO, SC-560, or celecoxib for the indicated times. Viable cells were counted by the trypan blue exclusion assay.
Apoptosis assays
The protocol used to assess apoptosis has previously been published by us (23). T2 cells (1.5 × 105 per well) were plated in 6-well plates. After culture overnight, the cells were washed twice with PBS and incubated in serum-free medium containing either 10% FBS, 0.1% DMSO, SC-560, or celecoxib for 3 days. The assay for apoptotic cells was done using a TACS Annexin V-FITC Apoptosis Detection Kit according to the manufacturer’s instructions (R&D Systems, Inc., Minneapolis, MN). For this assay, the cells were harvested, washed, and incubated with Annexin V-FITC and propidium iodide. After washing, the cells were analyzed by flow cytometry. The combination of Annexin V-FITC and propidium iodide allowed us to differentiate between early apoptotic cells (Annexin V-FITC positive), late apoptotic cells (Annexin V-FITC and propidium iodide positive), necrotic cells (propidium iodide positive), and viable cells (both negative).
Immunostaining of proliferating cell nuclear antigen and Ki67
Formalin-fixed paraffin-embedded tumor sections (10 μm) were subjected to immunostaining using a proliferating cell nuclear antigen (PCNA) or Ki67 antigen staining kit according to the manufacturer’s instructions (Zymed Laboratories, Inc., San Francisco, CA and Novocastra Laboratories, Ltd., Newcastle, United Kingdom). Brown color indicates the sites of positive staining for both PCNA and Ki67 nuclei.
Terminal deoxynucleotidyl transferase-mediated nick-end labeling staining
Formalin-fixed paraffin-embedded tumor sections (10 μm) were subjected to terminal deoxynucleotidyl transferase-mediated nick-end labeling (TUNEL) assay using the DeadEnd Colorimetric TUNEL system (Promega, Madison, WI) according to the manufacturer’s instructions. This system end labels the fragmented DNA of apoptotic cells, and the apoptotic nuclei are visualized as dark brown.
Results
COX-1 levels are increased in oncogenic mouse ovarian surface epithelial cells and tumors
Our first objective was to determine whether mouse OSE cells lacking functional p53 with activation of oncogenic pathways express COX-1 and/or COX-2. Several mouse ovarian cancer cell lines that contain combinations of defined genetic alterations were generated. C1 (genotype: p53-/-, myc, K-ras) and C2 (genotype: p53-/-, myc, Akt) mouse ovarian cancer cell lines were generated by infecting ovarian explants from K5-TVA/p53-/- mice with combinations of RCAS viruses carrying coding sequences for human c-myc, mouse K-ras with the G12D activating mutation, and HA-tagged and myristoylated mouse Akt1. T1 (genotype: p53-/-, myc, K-ras) and T2 (genotype: p53-/-, myc, Akt) cell lines were derived from i.p. tumors that developed after orthotopic implantation of C1 and C2 cell lines into nude mice.
All four cell lines are capable of producing tumors when allografted into immunocompromised mice. We examined levels of COX-1 and COX-2 expression in these cell lines. The results of Western blotting showed that COX-1 protein levels are higher in C2 and T2 cells compared with those in C1 and T1 cells. In contrast, the levels of COX-2 protein were low to undetectable (Fig. 1A). Analysis of Northern hybridization are comparable with those of Western blotting (Fig. 1B), except that the levels of Cox-1 mRNA were low to undetectable in T1 cells. Interestingly, Cox-2 mRNA was undetectable in all four cell lines. These results provide evidence that COX-1 but not COX-2 is the predominant pathway for generating prostaglandins in OSE cell lines. These results are in agreement with our previous report showing elevation of COX-1 levels in human EOCs (8).
Figure 1.
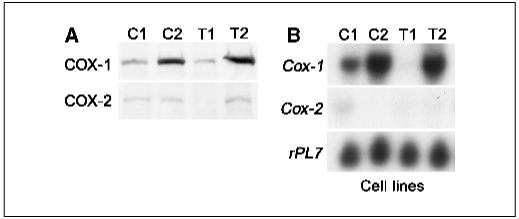
COX-1 is the predominant isoform expressed in OSE cancer cell lines. A, Western blot analysis of COX-1 and COX-2 expression in C1, C2, T1, and T2 OSE cell lines. B, Northern hybridization of Cox-1 and Cox-2 mRNA in C1, C2, T1, and T2 cancer cell line samples. rPL7 is a housekeeping gene.
We next asked whether tumors generated in vivo from these OSE cell lines show similar expression patterns of COX-1 and COX-2 as cells grown in vitro. C1, C2, T1, or T2 OSE cells were allografted under the dorsal skin of nude mice and allowed to grow for 8 weeks. These tumors were analyzed for expression of both COX isoforms (Fig. 2). The results of Western blot analysis show that the levels of COX-1 protein are substantially higher than the levels of COX-2 in these tumor samples (Fig. 2A). Northern hybridization results were even more dramatic. Whereas the presence of Cox-1 mRNA was distinctly evident in all tumor samples, Cox-2 mRNA was undetectable in the same samples (Fig. 2B). Because tumors grown in vivo are comprised of host stroma and of originally allografted carcinoma cells, we examined cell-specific expression of COX-1 and COX-2 in tumor sections by in situ hybridization. Again, the expression of COX-1 was dramatically elevated in all tumor samples but was restricted to the epithelial components of the solid tumors; the expression was undetectable in the stroma (Fig. 2C). In contrast, COX-2 expression was undetectable in tumor samples, except for some focal expression in tumor tissues comprised of allografted C1 cells. It is surprising to note that even OSE cells or OSE tumors overexpressing K-ras have low to undetectable levels of COX-2 (Fig. 2C); K-ras amplification/mutation induces COX-2 in other epithelial cells (24). Whereas COX-1 is overexpressed in OSE tumors in mice, we found that in normal mouse ovaries both COX-1 and COX-2 are expressed primarily in granulosa cells of the developing follicles before ovulation in a temporal and genetic background-dependent manner (22); no evidence of COX expression was noted in the ovarian surface epithelium (Fig. 2D). This suggests that COX-1 expression in ovarian epithelial cells is associated with their oncogenic transformation. Collectively, our results provide evidence that COX-1 is the predominant pathway for generating prostaglandins in EOCs in mice, and these results support our previous observations in human EOCs (8).
Figure 2.
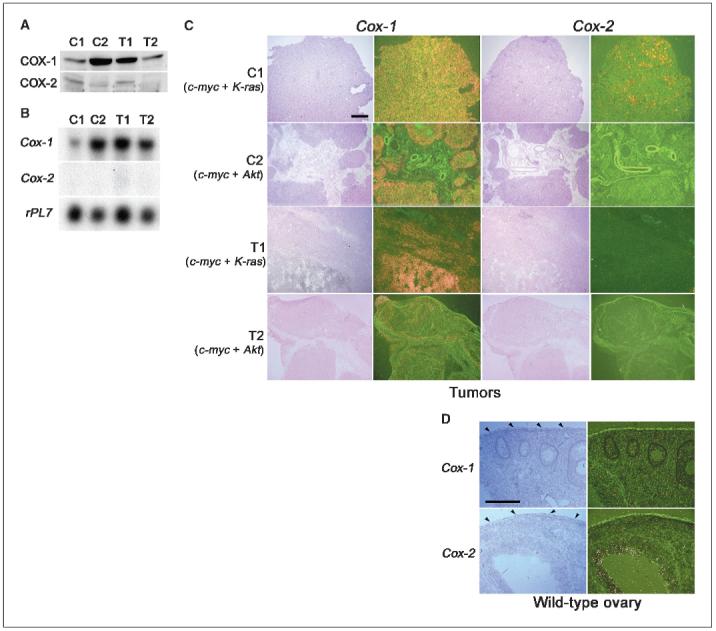
Tumors generated from OSE cancer cell lines show similar expression of COX-1 and COX-2 as observed for cell lines. A, Western blot analysis of COX-1 and COX-2 expression in tumors. B, Northern hybridization of Cox-1 and Cox-2 in tumor samples. rPL7 is a housekeeping gene. C, in situ hybridization of Cox-1 and Cox-2 in tumor sections. Bar, 500 μm. D, in situ hybridization of Cox-1 and Cox-2 in normal ovaries. Wild-type mice were treated with 5 IU of pregnant mare serum gonadotropin for 48 hours followed by 5 IU of human chorionic gonadotropin to stimulate follicular growth for preparation of ovulation at a defined time point. Ovarian sections from representative mice at 8 hours after hCG treatment. Arrowheads, surface epithelium. Bar, 200 μm.
Prostacyclin is the major prostaglandin produced in ovarian surface epithelial cells and its level is attenuated by inhibition of COX-1 activity
Prostaglandin E2 (PGE2) is the major prostaglandin produced by OVCAR-3 cells, a human EOC cell line, although PGI2 production was also substantial (8). Thus, we sought to characterize the prostaglandin profile in mouse OSE cells expressing COX-1. The levels of prostaglandins were measured by gas chromatography/mass spectrometry in serum-free medium in which cells were cultured in the presence or absence of arachidonic acid. PGI2 is the major prostaglandin produced and its levels in T2 cells increase in an arachidonic acid concentration-dependent manner (Fig. 3A and B). PGE2 is also detected in the medium but at levels much lower than PGI2. Levels of PGF2α and TBX2 were very low or below detection levels. A COX-1-selective inhibitor SC-560 dramatically reduced the PGI2 levels in a dose-dependent manner, whereas celecoxib, a COX-2-selective inhibitor, had little effect (Fig. 3C); levels of PGE2 were not altered by either SC-560 or celecoxib. We observed similar prostaglandin profiles in C1 and C2 OSE cell lines (data not shown). T1 cells have low levels of PGI2 (data not shown), which is not surprising because these cells express low levels of COX-1, and an undetectable level of COX-2 (Fig. 1A). To determine whether the basal levels of PGI2 without arachidonic acid are altered by the COX-1 inhibitor with time, T2 cells were cultured for 4 or 16 hours in the presence or absence of SC-560. We observed that the basal levels at 4 hours were higher than those at 16 hours (3,900 ± 200 versus 1,200 ± 200 pg/mL, n = 3). However, SC-560 at 2, 5, or 10 μmol/L was equipotent at 4 hours in further lowering the levels of PGI2 (3,900 ± 200 versus ∼200 ± 20 pg/mL, n = 3 for each concentration), whereas at 16 hours, SC-560 at 2 μmol/L was less potent than 5 or 10 μmol/L in reducing such levels (400 ± 70 versus ∼200 ± 20 pg/mL, n = 3 for each concentration). These results suggest that a relatively higher concentration of SC-560 is necessary to sustain a reduced basal level of PGI2 for an extended period. We next examined the expression of prostacyclin synthase (PGIS), the enzyme that catalyzes conversion of PGH2 to prostacyclin, in OSE cell lines and allografted tumors. The results of Western blot analysis showed that these cell lines express PGIS protein (Fig. 4A). The expression of PGIS was also evident in tumor tissues as determined by Western blotting and in situ hybridization. The presence of PGIS protein was clearly evident in tumor samples, but the levels were lower in tumors derived from C2 and T1 cell lines (Fig. 4B). The results of in situ hybridization provide evidence that PGIS is expressed in these tumors. The expression was primarily restricted to the growing epithelial cells but not in the host stroma (Fig. 4C). Collectively, these results provide evidence that PGI2 is the major prostaglandin produced by OSE cells and tumors, and this production is sensitive to inhibition by SC-560, but not celecoxib.
Figure 3.
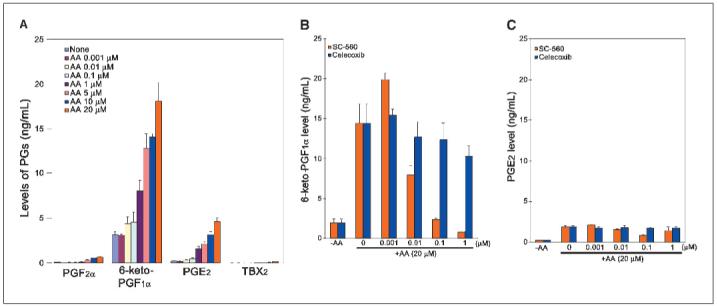
PGI2 is the predominant prostaglandin produced in T2 OSE cell lines and is inhibited by a COX-1 selective inhibitor SC-560 in a dose-dependent manner. T2 cells were cultured in 24-well plates and treated with arachidonic acid (AA) with or without COX inhibitors. Prostaglandin (PG) levels were detected by GS/MS. A, prostaglandin synthesis in mouse OSE cancer cells is a function of arachidonic acid concentration. B, effects of COX inhibitors on 6-keto-PGF1α formation as an index of PGI2 levels. C, effects of COX inhibitors on PGE2 levels.
Figure 4.
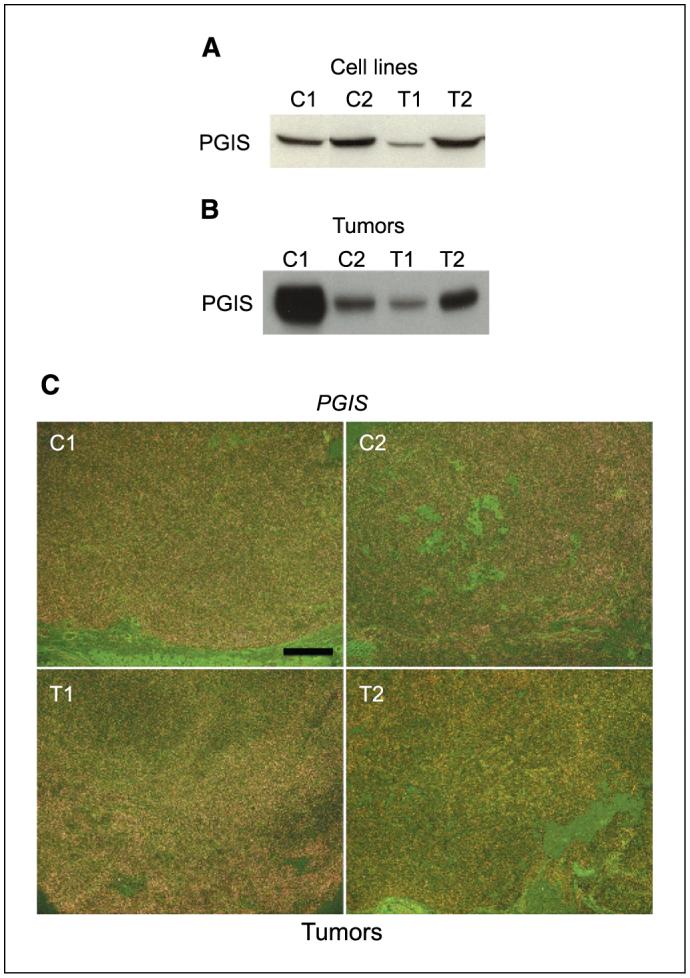
PGIS is expressed in OSE cells and tumors. A, Western blotting of PGIS in C1, C2, T1, and T2 OSE cell line samples. B, Western blotting of PGIS in tumor samples. C, in situ hybridization of Pgis in tumor sections. Representative darkfield photomicrographs of tumor sections show hybridization signals. Bar, 500 μm.
SC-560, a COX-1-selective inhibitor, suppresses ovarian surface epithelial tumor growth in vivo
Our observation of predominant expression of COX-1 in oncogenic OSE cell lines and tumors derived from these cells suggested that COX-1 inhibition would inhibit or retard tumor growth of allografted OSE cells in vivo. To address this issue, we initiated further studies with T2 cells, which primarily express COX-1. These cells were allografted under the dorsal skin of female nude mice. After tumor engraftment (7 days), these mice were gavaged with vehicle, SC-560 (3 mg/kg body weight) or celecoxib (50 mg/kg body weight) twice a day for 18 days or 50 days. These doses were chosen for their specificity in inhibiting COX isotypes (25). Tumor volume was measured every 2 to 4 days for 59 and 64 days for the short-term and long-term treatment groups, respectively. As shown in Fig. 5, whereas the tumor growth increased throughout the period examined in the vehicle-treated or celecoxib-treated group, the growth was substantially suppressed in the SC-560-treated group even when the drug treatment was withdrawn after 18 days. Tumor growth was also suppressed in allografted mice treated with SC-560 for a longer period, but the reduction in tumor growth was less dramatic than the short-term treated group. Nonetheless, these results suggest that COX-1 is a potential target for the prevention and treatment of ovarian cancer.
Figure 5.
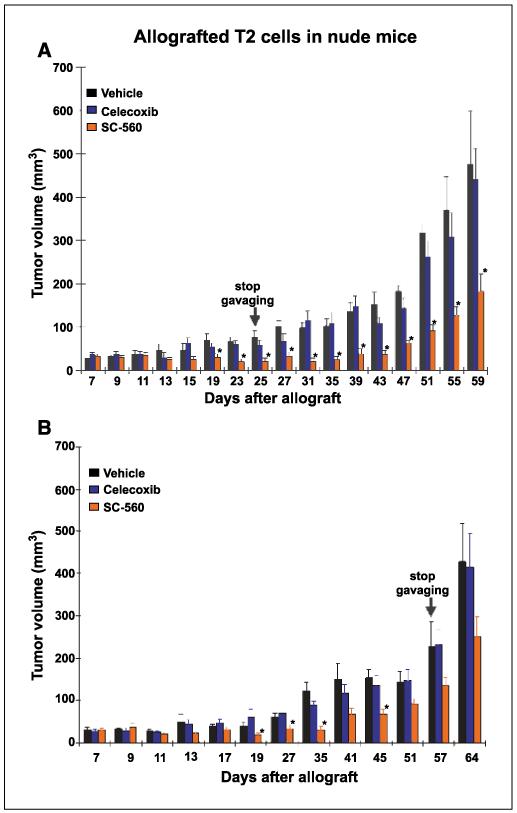
COX-1 selective inhibitor SC-560 attenuates OSE tumor growth. Nude mice were allografted with T2 OSE cells and after establishment of the tumors 7 days later they were gavaged orally with the vehicle (0.1 mL), SC-560 (3 mg/kg bd) or celecoxib (50 mg/kg bd) for (A) 18 days (nine mice each for vehicle or SC-560 treated group and six mice for celecoxib treated group; these experiments were repeated at two different times) and (B) for 50 days (5-6 mice per group). Tumor growth was recorded every 2 to 4 days for a period of 59 and 64 days, respectively. Columns, mean; bars, ±SE. *, P < 0.05, significantly different from the vehicle-treated group (unpaired t test).
COX-1-selective inhibitor SC-560 attenuates cell proliferation and promotes apoptosis
Our next objective was to better understand the mechanism of inhibition of tumor growth by SC-560. We employed cell growth and apoptosis assays by culturing T2 OSE cells in serum-free medium in either the presence or absence of SC-560 or celecoxib. As shown in Fig. 6A, the COX-1-selective inhibitor SC-560 at 5 or 10 μmol/L substantially decreased the number of cells, whereas celecoxib at 10 μmol/L was not effective in this response. It is to be recalled that these concentrations of SC-560 are required to sustain the reduced basal levels of PGI2 for an extended time period. Furthermore, apoptosis assays of T2 OSE cells, cultured in serumfree medium in the presence and absence of specific COX inhibitors, revealed that SC-560 increased the rate of apoptosis, whereas celecoxib failed to show such an effect (Fig. 6B and C). These results suggest that inhibition of COX-1 not only inhibits cell proliferation but also accelerates apoptosis, resulting in attenuated tumor growth. This was also reflected in allografted tumors in nude mice treated with vehicle, SC-560 or celecoxib, as assessed by PCNA, proliferation-associated nuclear antigen (Ki67), and TUNEL staining. The assessment of cell proliferation by PCNA and Ki67 staining is an indicator of neoplastic growth, whereas TUNEL assay provides information on the status of apoptotic cells in tissue. We observed that the population of PCNA- and Ki67-positive cells in tumor sections was substantially lower when the mice were exposed to SC-560 than in those receiving the vehicle or celecoxib (Fig. 6D). In contrast, the number of TUNEL-positive (apoptotic) cells was more frequent in tumor sections of SC-560-treated mice than those from the vehicle- or celecoxib-treated mice (Fig. 6D). However, the possibility that some of the effects of SC-560 are mediated independent of its effects on COX-1 activity cannot be ruled out, because there is evidence that certain NSAIDs can influence cell growth independent of their effects on COX activity (26). However, the failure of celecoxib at equimolar concentrations with similar chemical structure as SC-560 to influence prostaglandin synthesis or cell growth suggests that the effects of SC-560 are mostly due to inhibition of COX-1 activity.
Figure 6.

COX-1 selective inhibitor SC-560 attenuates OSE tumor cell proliferation and accelerates apoptosis. A, cell growth assay. T2 Cells (0.5 × 105) were seeded in 12-well plates and incubated with DMSO (vehicle, 0.1%), SC-560 or celecoxib at different concentrations for indicated days. Cell numbers were counted from triplicate wells. Points, mean of three independent experiments; bars, ±SE. B and C, apoptosis assay. Cells were treated with DMSO, SC-560, or celecoxib at indicated concentrations for 3 days after removal of the serum from the culture media. Number of apoptotic cells was determined by flow cytometry using an Annexin V-FITC kit. Columns, mean of percent apoptotic cells in three separate experiments; bars, ±SE. D, immunostaining of PCNA, Ki67, and TUNEL in tumors. Tumor samples used in this study were derived from nude mice in Fig. 5 legend. Bar, 50 μm.
Discussion
This investigation reveals that COX-1 is the dominant pathway responsible for generating prostaglandins in EOCs in mice. These results support our recent findings of COX-1 overexpression in human EOCs (8). Because oncogenes, cytokines, and growth factors are known to up-regulate COX-2, it is often assumed that COX-2 derived prostanoids function as downstream signaling molecules in tumorigenesis. Whereas this is true for colorectal cancer and many other extracolonic cancers including breast, prostrate, uterus, lung, head, and neck cancers (2, 27-29), the story of COX expression in ovarian cancer has become highly controversial and requires careful reevaluation. To our knowledge, there are 28 published reports (Table 1) that investigated COX expression in ovarian cancers. Interestingly, 20 of 28 reports did not examine COX-1 levels; attempts were made to examine only COX-2 expression and among these studies, not all of the samples examined were positive for COX-2 expression. In addition, 9 of 28 reported studies only used human ovarian cancer cell lines but not tumor samples. More importantly, except our study in which multiple approaches were used to confirm COX-1 expression (8), all other studies on human tumor samples used a singular approach of immunodetection of COX expression (Table 1). The results of immunodetection of COX-1 and COX-2 may lack specificity due to known cross-reactivity of many available antibodies and may lead to erroneous interpretation. Furthermore, it is not clearly stated in many of the reports whether samples were excluded from patients that had undergone cytoreductive therapy, a treatment commonly employed for advanced ovarian malignancy. It is well established that drugs used in cytoreductive therapy would likely induce COX-2 (30), and it is unclear whether elevated COX-2 levels observed in these studies is primarily due to the oncogenic transformation of ovarian epithelial cells or secondary to the use of cytotoxic agents for the treatment of the primary disease. Our previous report on human EOCs (8), and our present finding of COX-1 expression in mouse OSE tumors lead us to seriously consider that COX-1 is the dominant pathway for generating prostaglandins in ovarian cancers.
It is interesting to note that COX-1 is expressed at much higher levels than COX-2 in OSE cells and tumors overexpressing c-myc and K-ras (C1 and T1) or c-myc and Akt (C2 and T2). This is surprising because K-ras is known to regulate COX-2 expression in several other systems (31, 32). This raises a question as to the underlying mechanism for regulation of COX-1 in OSE tumors. As shown previously (22) and reported here (Fig. 2D), COX-1 and COX-2 are expressed in granulosa cells of mouse ovarian follicles before ovulation in a temporal manner, but neither COX isoform is detectable in the ovarian surface epithelium. This suggests that oncogenic transformation leads to the expression of COX-1 in the ovarian surface epithelium. One might also argue that mRNA stability accounts for increased COX-1 and depressed COX-2 expression in ovarian tumors. Our preliminary experiments of Northern hybridization with OSE cells in the presence of Actinomycin D argue against this possibility (data not shown). Thus, the mechanism by which COX-1 expression is regulated in both OSE cells and tumors is under active investigation in the laboratory.
A lack of suitable animal models for ovarian cancer has been a major drawback in obtaining useful mechanistic information on ovarian tumorigenesis. Although there are several human ovarian carcinoma cell lines that generate tumors upon xenografting into immunocompromised mice, the ovarian origin of these cell lines is not well characterized. Most human ovarian tumors are believed to arise from the single layer of epithelial cells that covers the ovarian surface (9). However, a recent report describes a genetically defined model of human EOC and should be useful for studying this disease (33). In this respect, mouse OSE cell lines with defined genetic alterations provide alternative approaches for studying EOCs that could be relevant to humans. It would be interesting to see whether COX-1 expression is common to other models of EOCs in mice produced by alteration of different genes (17-19).
Unrestricted cell proliferation and reduced apoptosis are hallmarks of transformed cells. A plethora of signaling pathways and molecules influences these processes. Our results of reduced tumor growth with decreased cell proliferation and accelerated apoptosis following SC-560 treatment, not with celecoxib, suggest that COX-1 derived prostaglandins promote OSE tumor growth at least by influencing cell proliferation and apoptosis. Because PGI2 is the major prostaglandin produced by mouse OSE cells and tumors, we suspect that this prostaglandin is involved in these processes. The next question concerns the downstream signaling pathways affected by PGI2. PGI2 normally binds to and activates its cognate G-protein-coupled cell surface receptor IP. The activation of IP results in increased levels of intracellular cyclic AMP, the second messenger system linked to PKA signaling (34). There is also evidence that PGI2 functions as an endogenous ligand for the nuclear receptor PPARδ (21). We have previously provided evidence for the participation of both PGI2 and PGE2 in the activation of PPARδ involved in colorectal and intestinal cancers (23, 35). Although both IP and PPARδ are expressed in mouse OSE cells and tumors (data not shown), whether each of these pathways singularly or cooperatively participates in tumor growth is the subject for future research. Recently, a COX-1 splice variant coined as COX-3 has been identified and acetaminophen, an analgesic/antipyretic agent, inhibits its activity (36). Whether this isoform is involved in ovarian cancer is not known.
The effects of SC-560 in attenuating tumor growth in vivo are remarkable even after withdrawal of the drug treatment. Our preliminary results also show that C2 OSE cells allografted under the dorsal skin of immunocompromised γc/Rag2 double mutant mice, lacking natural killer cells and T and B lymphocytes, also produce tumors, and the tumor growth is attenuated after short-term (14 days) treatment with SC-560 but not celecoxib (data not shown). We speculated that long-term treatment with SC-560 would be even more dramatic but were surprised to see that continuous treatment was less effective than short-term therapy. This may reflect a tumor’s ability to develop drug resistance, a well-known trait of ovarian cancers (37). Because long-term use of certain COX-2 inhibitors are linked to hypertension, peripheral edema, and an increased risk of serious heart problems in some patients, aspirin as a preferred COX-1 inhibitor may have more meaningful application in combating ovarian tumors. In conclusion, our previous study on human EOCs and the present investigation on mouse EOCs using multiple approaches provide convincing evidence that COX-1 not COX-2 is the major prostanoid generating pathway operative in ovarian cancers of epithelial origin. These results present COX-1 as a potential target for the prevention and treatment of human EOCs.
Acknowledgments
Grant support: USPHS grants, P01-CA-77839, R37 HD12304, R37 DK47297, and GM42056; Mary Kay Ash Charitable Foundation; American Cancer Society Institutional (T. Daikoku); and National Cancer Institute grant CA103924-01 (S. Orsulic).
We thank Mark Geraci for providing us with the PGIS cDNA and Jennifer Jones for her help in PGIS Western blotting.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem. 1996;271:33157–60. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 2.Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer. 2001;1:11–21. doi: 10.1038/35094017. [DOI] [PubMed] [Google Scholar]
- 3.Cramer DW, Harlow BL, Titus-Ernstoff L, Bohlke K, Welch WR, Greenberg ER. Over-the-counter analgesics and risk of ovarian cancer. Lancet. 1998;351:104–7. doi: 10.1016/S0140-6736(97)08064-1. [DOI] [PubMed] [Google Scholar]
- 4.Moysich KB, Mettlin C, Piver MS, Natarajan N, Menezes RJ, Swede H. Regular use of analgesic drugs and ovarian cancer risk. Cancer Epidemiol Biomarkers Prev. 2001;10:903–6. [PubMed] [Google Scholar]
- 5.Akhmedkhanov A, Toniolo P, Zeleniuch-Jacquotte A, Kato I, Koenig KL, Shore RE. Aspirin and epithelial ovarian cancer. Prev Med. 2001;33:682–7. doi: 10.1006/pmed.2001.0945. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg L, Palmer JR, Rao RS, et al. A case-control study of analgesic use and ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2000;9:933–7. [PubMed] [Google Scholar]
- 7.Tavani A, Gallus S, La Vecchia C, Conti E, Montella M, Franceschi S. Aspirin and ovarian cancer: an Italian case-control study. Ann Oncol. 2000;11:1171–3. doi: 10.1023/a:1008373616424. [DOI] [PubMed] [Google Scholar]
- 8.Gupta RA, Tejada LV, Tong BJ, et al. Cyclooxygenase-1 is overexpressed and promotes angiogenic growth factor production in ovarian cancer. Cancer Res. 2003;63:906–11. [PubMed] [Google Scholar]
- 9.Auersperg N, Wong AS, Choi KC, Kang SK, Leung PC. Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr Rev. 2001;22:255–88. doi: 10.1210/edrv.22.2.0422. [DOI] [PubMed] [Google Scholar]
- 10.Nasi ML, Castiglione M. Cyclooxygenase-2 (COX-2) a new prognostic and predictive factor for ovarian cancer? Are all the criteria fulfilled? Ann Oncol. 2002;13:1169–71. doi: 10.1093/annonc/mdf312. [DOI] [PubMed] [Google Scholar]
- 11.Ozols RF. Recurrent ovarian cancer: evidence-based treatment. J Clin Oncol. 2002;20:1161–3. doi: 10.1200/JCO.2002.20.5.1161. [DOI] [PubMed] [Google Scholar]
- 12.Wenham RM, Lancaster JM, Berchuck A. Molecular aspects of ovarian cancer. Best Pract Res Clin Obstet Gynaecol. 2002;16:483–97. doi: 10.1053/beog.2002.0298. [DOI] [PubMed] [Google Scholar]
- 13.Jemal A, Tiwari RC, Murray T, et al. Cancer statistics. CA Cancer J Clin. 2004;54:8–29. doi: 10.3322/canjclin.54.1.8. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz PE. Current diagnosis and treatment modalities for ovarian cancer. Cancer Treat Res. 2002;107:99–118. doi: 10.1007/978-1-4757-3587-1_4. [DOI] [PubMed] [Google Scholar]
- 15.Orsulic S, Li Y, Soslow RA, Vitale-Cross LA, Gutkind JS, Varmus HE. Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell. 2002;1:53–62. doi: 10.1016/s1535-6108(01)00002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orsulic S. An RCAS-TVA-based approach to designer mouse models. Mamm Genome. 2002;13:543–7. doi: 10.1007/s00335-002-4003-4. [DOI] [PubMed] [Google Scholar]
- 17.Connolly DC, Bao R, Nikitin AY, et al. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res. 2003;63:1389–97. [PubMed] [Google Scholar]
- 18.Flesken-Nikitin A, Choi KC, Eng JP, Shmidt EN, Nikitin AY. Induction of carcinogenesis by concurrent inactivation of p53 and Rb1 in the mouse ovarian surface epithelium. Cancer Res. 2003;63:3459–63. [PubMed] [Google Scholar]
- 19.Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med. 2005;11:63–70. doi: 10.1038/nm1173. [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Sun L, Myeroff L, et al. Demonstration that mutation of the type II transforming growth factor β receptor inactivates its tumor suppressor activity in replication error-positive colon carcinoma cells. J Biol Chem. 1995;270:22044–9. doi: 10.1074/jbc.270.37.22044. [DOI] [PubMed] [Google Scholar]
- 21.Lim H, Gupta RA, Ma WG, et al. Cyclo-oxygenase-2-derived prostacyclin mediates embryo implantation in the mouse via PPARδ. Genes Dev. 1999;13:1561–74. doi: 10.1101/gad.13.12.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Ma WG, Tejada L, et al. Rescue of female infertility from the loss of cyclooxygenase-2 by compensatory up-regulation of cyclooxygenase-1 is a function of genetic makeup. J Biol Chem. 2004;279:10649–58. doi: 10.1074/jbc.M312203200. [DOI] [PubMed] [Google Scholar]
- 23.Wang D, Wang H, Shi Q, et al. Prostaglandin E(2) promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor δ. Cancer Cell. 2004;6:285–95. doi: 10.1016/j.ccr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 24.Maciag A, Sithanandam G, Anderson LM. Mutant K-rasV12 increases COX-2, peroxides, and DNA damage in lung cells. Carcinogenesis. 2004;25:2231–7. doi: 10.1093/carcin/bgh245. [DOI] [PubMed] [Google Scholar]
- 25.Reese J, Zhao X, Ma WG, Brown N, Maziasz TJ, Dey SK. Comparative analysis of pharmacologic and/or genetic disruption of cyclooxygenase-1 and cyclooxygenase-2 function in female reproduction in mice. Endocrinology. 2001;142:3198–206. doi: 10.1210/endo.142.7.8307. [DOI] [PubMed] [Google Scholar]
- 26.Denkert C, Furstenberg A, Daniel PT, et al. Induction of G0/G1 cell cycle arrest in ovarian carcinoma cells by the anti-inflammatory drug NS-398, but not by COX-2-specific RNA interference. Oncogene. 2003;22:8653–61. doi: 10.1038/sj.onc.1206920. [DOI] [PubMed] [Google Scholar]
- 27.Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94:252–66. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- 28.Altorki NK, Subbaramaiah K, Dannenberg AJ. COX-2 inhibition in upper aerodigestive tract tumors. Semin Oncol. 2004;31:30–6. doi: 10.1053/j.seminoncol.2004.03.043. [DOI] [PubMed] [Google Scholar]
- 29.Chang SH, Liu CH, Conway R, et al. Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc Natl Acad Sci U S A. 2004;101:591–6. doi: 10.1073/pnas.2535911100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subbaramaiah K, Hart JC, Norton L, Dannenberg AJ. Microtubule-interfering agents stimulate the transcription of cyclooxygenase-2. Evidence for involvement of ERK1/2 AND p38 mitogen-activated protein kinase pathways. J Biol Chem. 2000;275:14838–45. doi: 10.1074/jbc.275.20.14838. [DOI] [PubMed] [Google Scholar]
- 31.Araki Y, Okamura S, Hussain SP, et al. Regulation of cyclooxygenase-2 expression by the Wnt and ras pathways. Cancer Res. 2003;63:728–34. [PubMed] [Google Scholar]
- 32.Sheng H, Shao J, Dubois RN. K-Ras-mediated increase in cyclooxygenase 2 mRNA stability involves activation of the protein kinase B1. Cancer Res. 2001;61:2670–5. [PubMed] [Google Scholar]
- 33.Liu J, Yang G, Thompson-Lanza JA, et al. A genetically defined model for human ovarian cancer. Cancer Res. 2004;64:1655–63. doi: 10.1158/0008-5472.can-03-3380. [DOI] [PubMed] [Google Scholar]
- 34.Wise H. Multiple signalling options for prostacyclin. Acta Pharmacol Sin. 2003;24:625–30. [PubMed] [Google Scholar]
- 35.Gupta RA, Tan J, Krause WF, et al. Prostacyclin-mediated activation of peroxisome proliferator-activated receptor y in colorectal cancer. Proc Natl Acad Sci U S A. 2000;97:13275–80. doi: 10.1073/pnas.97.24.13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chandrasekharan NV, Dai H, Roos KL, et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci U S A. 2002;99:13926–31. doi: 10.1073/pnas.162468699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer. 2003;3:502–16. doi: 10.1038/nrc1123. [DOI] [PubMed] [Google Scholar]
- 38.Zhu G, Saed GM, Deppe G, Diamond MP, Munkarah AR. Hypoxia up-regulates the effects of prostaglandin E2 on tumor angiogenesis in ovarian cancer cells. Gynecol Oncol. 2004;94:422–6. doi: 10.1016/j.ygyno.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 39.Spinella F, Rosano L, Di Castro V, Nicotra MR, Natali PG, Bagnato A. Inhibition of cyclooxygenase-1 and -2 expression by targeting the endothelin a receptor in human ovarian carcinoma cells. Clin Cancer Res. 2004;10:4670–9. doi: 10.1158/1078-0432.CCR-04-0315. [DOI] [PubMed] [Google Scholar]
- 40.Sakamoto A, Yokoyama Y, Umemoto M, et al. Clinical implication of expression of cyclooxygenase-2 and peroxisome proliferator activated-receptor γ in epithelial ovarian tumours. Br J Cancer. 2004;91:633–8. doi: 10.1038/sj.bjc.6602009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seo SS, Song YS, Kang DH, et al. Expression of cyclooxygenase-2 in association with clinicopathological prognostic factors and molecular markers in epithelial ovarian cancer. Gynecol Oncol. 2004;92:927–35. doi: 10.1016/j.ygyno.2003.11.055. [DOI] [PubMed] [Google Scholar]
- 42.Raspollini MR, Amunni G, Villanucci A, et al. Expression of inducible nitric oxide synthase and cyclooxygenase-2 in ovarian cancer: correlation with clinical outcome. Gynecol Oncol. 2004;92:806–12. doi: 10.1016/j.ygyno.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 43.Li S, Miner K, Fannin R, Barrett CJ, Davis BJ. Cyclooxygenase-1 and 2 in normal and malignant human ovarian epithelium. Gynecol Oncol. 2004;92:622–7. doi: 10.1016/j.ygyno.2003.10.053. [DOI] [PubMed] [Google Scholar]
- 44.Denkert C, Weichert W, Pest S, et al. Overexpression of the embryonic-lethal abnormal vision-like protein HuR in ovarian carcinoma is a prognostic factor and is associated with increased cyclooxygenase 2 expression. Cancer Res. 2004;64:189–95. doi: 10.1158/0008-5472.can-03-1987. [DOI] [PubMed] [Google Scholar]
- 45.Raspollini MR, Amunni G, Villanucci A, et al. COX-2 status in relation to tumor microvessel density and VEGF expression: analysis in ovarian carcinoma patients with low versus high survival rates. Oncol Rep. 2004;11:309–13. [PubMed] [Google Scholar]
- 46.Roland IH, Yang WL, Yang DH, et al. Loss of surface and cyst epithelial basement membranes and preneo-plastic morphologic changes in prophylactic oophorectomies. Cancer. 2003;98:2607–23. doi: 10.1002/cncr.11847. [DOI] [PubMed] [Google Scholar]
- 47.Erkinheimo TL, Lassus H, Sivula A, et al. Cytoplasmic HuR expression correlates with poor outcome and with cyclooxygenase 2 expression in serous ovarian carcinoma. Cancer Res. 2003;63:7591–4. [PubMed] [Google Scholar]
- 48.Ali-Fehmi R, Che M, Khalifeh I, et al. The effect of cyclooxygenase-2 expression on tumor vascularity in advanced stage ovarian serous carcinoma. Cancer. 2003;98:1423–9. doi: 10.1002/cncr.11650. [DOI] [PubMed] [Google Scholar]
- 49.Wang CY, Wang M, Wang XY, Zhang SL, Wang YX, Li LK. Inhibitory effect of NS398 on the proliferation of human ovarian cancer cell lines CAOV3 and OVCAR3 in vitro. Zhonghua Fu Chan Ke Za Zhi. 2003;38:415–8. [PubMed] [Google Scholar]
- 50.Landen CN, Jr, Mathur SP, Richardson MS, Creasman WT. Expression of cyclooxygenase-2 in cervical, endometrial, and ovarian malignancies. Am J Obstet Gynecol. 2003;188:1174–6. doi: 10.1067/mob.2003.284. [DOI] [PubMed] [Google Scholar]
- 51.Tang L, Wang M, Ma J. Relationship between cyclooxygenase-2 protein expression, prostaglandins levels and biologic behavior in ovarian carcinoma tissue. Zhonghua Fu Chan Ke Za Zhi. 2002;37:687–90. [PubMed] [Google Scholar]
- 52.Ferrandina G, Lauriola L, Distefano MG, et al. Increased cyclooxygenase-2 expression is associated with chemotherapy resistance and poor survival in cervical cancer patients. J Clin Oncol. 2002;20:973–81. doi: 10.1200/JCO.2002.20.4.973. [DOI] [PubMed] [Google Scholar]
- 53.Munkarah AR, Morris R, Baumann P, et al. Effects of prostaglandin E(2) on proliferation and apoptosis of epithelial ovarian cancer cells. J Soc Gynecol Investig. 2002;9:168–73. [PubMed] [Google Scholar]
- 54.Chu S, Rushdi S, Zumpe ET, et al. FSH-regulated gene expression profiles in ovarian tumours and normal ovaries. Mol Hum Reprod. 2002;8:426–33. doi: 10.1093/molehr/8.5.426. [DOI] [PubMed] [Google Scholar]
- 55.Ferrandina G, Ranelletti FO, Lauriola L, et al. Cyclooxygenase-2 (COX-2), epidermal growth factor receptor (EGFR), and Her-2/neu expression in ovarian cancer. Gynecol Oncol. 2002;85:305–10. doi: 10.1006/gyno.2002.6620. [DOI] [PubMed] [Google Scholar]
- 56.Denkert C, Kobel M, Pes S, et al. Expression of cyclooxygenase 2 is an independent prognostic factor in human ovarian carcinoma. Am J Pathol. 2002;160:893–903. doi: 10.1016/S0002-9440(10)64912-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rodriguez-Burford C, Barnes MN, Oelschlager DK, et al. Effects of nonsteroidal anti-inflammatory agents (NSAIDs) on ovarian carcinoma cell lines: preclinical evaluation of NSAIDs as chemopreventive agents. Clin Cancer Res. 2002;8:202–9. [PubMed] [Google Scholar]
- 58.Deguchi M, Ishiko O, Sumi T, Yoshida H, Yamamoto K, Ogita S. Expression of angiogenic factors in extrapelvic endometriosis. Oncol Rep. 2001;8:1317–9. doi: 10.3892/or.8.6.1317. [DOI] [PubMed] [Google Scholar]
- 59.Sumi T, Ishiko O, Yoshida H, et al. Expression of cyclooxygenase-2 in ovarian mature cystic teratomas with malignant transformation. Int J Mol Med. 2001;8:495–8. doi: 10.3892/ijmm.8.5.495. [DOI] [PubMed] [Google Scholar]
- 60.Klimp AH, Hollema H, Kempinga C, van der Zee AG, de Vries EG, Daemen T. Expression of cyclooxygenase-2 and inducible nitric oxide synthase in human ovarian tumors and tumor-associated macrophages. Cancer Res. 2001;61:7305–9. [PubMed] [Google Scholar]
- 61.Dore M, Cote LC, Mitchell A, Sirois J. Expression of prostaglandin G/H synthase type 1, but not type 2, in human ovarian adenocarcinomas. J Histochem Cytochem. 1998;46:77–84. doi: 10.1177/002215549804600110. [DOI] [PubMed] [Google Scholar]
- 62.Matsumoto Y, Ishiko O, Deguchi M, Nakagawa E, Ogita S. Cyclooxygenase-2 expression in normal ovaries and epithelial ovarian neoplasms. Int J Mol Med. 2001;8:31–6. doi: 10.3892/ijmm.8.1.31. [DOI] [PubMed] [Google Scholar]
- 63.Ristimaki A, Honkanen N, Jankala H, Sipponen P, Harkonen M. Expression of cyclooxygenase-2 in human gastric carcinoma. Cancer Res. 1997;57:1276–80. [PubMed] [Google Scholar]