

Abstract
OBJECTIVE— To characterize normal and malformed embryos within the same litters from control and diabetic rats for expression of genes related to metabolism of reactive oxygen species (ROS) or glucose as well as developmental genes.
RESEARCH DESIGN AND METHODS— Embryos from nondiabetic and streptozotocin-induced diabetic rats were collected on gestational day 11 and evaluated for gene expression (PCR) and distribution of activated caspase-3 and glutathione peroxidase (Gpx)-1 by immunohistochemistry.
RESULTS— Maternal diabetes (MD group) caused growth retardation and an increased malformation rate in the embryos of MD group rats compared with those of controls (N group). We found decreased gene expression of Gpx-1 and increased expression of vascular endothelial growth factor-A (Vegf-A) in malformed embryos of diabetic rats (MDm group) compared with nonmalformed littermates (MDn group). Alterations of messenger RNA levels of other genes were similar in MDm and MDn embryos. Thus, expression of copper zinc superoxide dismutase (CuZnSOD), manganese superoxide dismutase (MnSOD), and sonic hedgehog homolog (Shh) were decreased, and bone morphogenetic protein-4 (Bmp-4) was increased, in the MD embryos compared with the N embryos. In MDm embryos, we detected increased activated caspase-3 immunostaining in the first visceral arch and cardiac area and decreased Gpx-1 immunostaining in the cardiac tissue; both findings differed from the caspase/Gpx-1 immunostaining of the MDn and N embryos.
CONCLUSIONS— Maternal diabetes causes growth retardation, congenital malformations, and decreased general antioxidative gene expression in the embryo. In particular, enhanced apoptosis of the first visceral arch and heart, together with decreased cardiac Gpx-1 levels, may compromise the mandible and heart and thus cause an increased risk of developing congenital malformation.
The cellular and molecular mechanisms of diabetic embryopathy are not completely clear. Previous experimental studies have suggested that the teratological impact of a diabetic environment partly depends on excess of reactive oxygen species (ROS) in the embryo (1) as a consequence of either increased free oxygen radical formation (2–4), decreased capacity of ROS-scavenging enzymes (5–9), or both. Furthermore, previous work has demonstrated that supplementation of antioxidative agents such as copper zinc superoxide dismutase (CuZnSOD) (1,2), N-acetylcysteine (10), vitamins E and C (8), and folic acid (11) in vitro, as well as butylated hydroxytoluene (12), vitamin E (13–19), vitamin C (18,20), N-acetylcysteine (21), and folic acid (11) in vivo, attenuate malformation rate and diminish markers of oxidative stress (e.g., by normalizing tissue levels of thiobarbituric acid reactive substances [15], isoprostane 8-iso-PGF2α [22,23], and carbonylated proteins [24]).
The driving cellular force behind diabetes-induced oxidative stress is likely associated with enhanced glucose metabolism (25–27) in the embryonic/fetal cells exposed to increased ambient levels of glucose. One putative primary source of reactive radical compounds is mitochondria receiving a high influx of pyruvate and oxygen and, subsequently, producing a large amount of ROS (mainly superoxide) (3) in the oxidative processes of the electron transport chain. The ensuing leakage of superoxide into other compartments of the mitochondria and the cytosol, and the further formation of hydrogen peroxide and hydroxyl radicals, should yield mitochondrial alterations (28) as well as lipid peroxidation (22) and DNA damage (29) in the embryo. There are several observations in support of this notion. The structural alterations, mainly high-amplitude swelling of the embryonic mitochondria (28), are diminished by maternal antioxidative treatment (16), thereby supporting the notion of a ROS-related etiology of the structural changes. Enhanced lipid peroxidation, as evidenced by increased levels of the isoprostane 8-iso-PGF2α (22,23,30), may induce several teratogenic pathways in addition to the developmental disturbance caused by peroxidation of structural lipids in mitochondrial and cellular membranes. For instance, it has been demonstrated that 8-iso-PGF2α, which is produced nonenzymatically by ROS-mediated oxidation of arachidonic acid in the offspring (30), has its own teratogenic potency (23). In addition, an excessive peroxidation of arachidonic acid may hamper prostaglandin biosynthesis by depleting precursor pools and, in particular, yield decreased concentration of prostaglandin E2 (31), which could obstruct neural tube closure (22).
ROS-induced DNA damage (29) may directly disrupt development via altered expression of key genes. In addition, cellular DNA repair processes may activate poly(ADP-ribose) polymerase (PARP), which may cause glyceraldehyde-3-phosphate dehydrogenase (GAPDH) inhibition by poly(ADP-ribosylation) (32). The net result would be diminished embryonic GAPDH activity, which has been demonstrated in rat embryos subjected to diabetes in vivo and high glucose in vitro (33). Furthermore, decreased glycolytic flux proximal to GAPDH (32) and the presence of increased ambient glucose levels will yield enhanced flux in the polyol (34,35) and hexosamine (36) pathways. An increased availability of proximal glycolytic intermediaries would increase diacylglycerol production and cause activation of several protein kinase C isoforms (37,38), as well as enhance the flux in the advanced glycosylation end product pathway (39). All of the consequences of inhibited GAPDH activity may thus contribute to the teratogenic outcome of diabetic pregnancy. Evidently, there are multiple ways for a diabetes-induced state of oxidative stress in the embryo to disturb embryonic/fetal development, several of which enjoy considerable experimental support.
Another consequence of a state of oxidative stress would be enhanced apoptosis in embryonic/fetal tissue (40), which has been described (26,41–43) and has been suggested to be mediated by enhanced Jun-amino-terminal kinase-1 and -2 activity (19,44). It has also been suggested that maternal diabetes induces an inflammatory state in the embryo, where proinflammatory cytokines (i.e., tumor necrosis factor-α [TNF-α] [45,46]) act to downregulate the principal ROS-scavenging enzymes via increased activity of mitogen-activated protein kinases (47). The exact relation between enhanced apoptosis and induction of malformations is still unclear, mainly since we do not fully comprehend the specific transmission of a general increase in programmed cell death into precisely restricted developmental damage to embryonic organs or organ systems (42).
Based on earlier studies (7,48,49) and on a question that has been raised several times—which genes are involved in diabetes-induced embryonic dysmorphogenesis? (5,8,26,42,50)—we wanted to add to the teratological knowledge by identifying differences in gene expression between the malformed and nonmalformed offspring within the same litters of diabetic animals.
Our working hypothesis was that genes of the malformed offspring with an expression pattern different from that of genes in the nonmalformed offspring within the same litter may be associated, either directly or indirectly, with the teratogenic process. We thus decided to compare gene expression of embryos that were morphologically normal and those that were malformed in litters of normal and diabetic rats. By comparing gene expression and protein distribution in embryos of the same age and with exposure to an identical intrauterine milieu, we aimed to control for possible confounding factors of the teratogenic process. From earlier work, we knew that the two tissues of the rat fetuses that are particularly vulnerable to the diabetic state are the mandible and the heart (17) (Fig. 1). We also knew that a susceptible period in diabetic rat pregnancy occurs during gestational days 6–10 (51), which corresponds to weeks 2–4 in human pregnancy. Bearing in mind that the size and relative immaturity of day-10 embryos makes them difficult to evaluate for developmental defects, we chose to interrupt the rat pregnancies on gestational day 11 in order to be able to clearly distinguish between malformed and nonmalformed embryos. We decided to relate embryonic (mal)development to expression of the major oxidative defense genes (SODs, glutathione peroxidase [Gpx]-1, Gpx-2, and catalase), to key genes of glucose metabolism (aldose reductase [AR] and GAPDH), to developmental/teratological genes [poly(ADP-ribose) polymerase (PARP)], to tumor protein 53 (p53), to bone morphogenetic protein-4 (Bmp-4), to Ret proto-oncogene (Ret), to sonic hedgehog homolog (Shh), to vascular endothelial growth factor-A (VEGF-A), to TNF-α, to interleukin-6 (IL-6), and to the tissue distribution of activated caspase-3 (denoting apoptotic rate) and Gpx-1.
FIG. 1.

A: Fetuses displaying micrognathia (left fetus) and normal morphology (right fetus). B: Outcome of pregnancy in the control (N) and manifestly diabetic (MD) groups, distributed as normal (□), malformed (▪), and resorbed (▒) embryos on gestational day 11. *P < 0.05 vs. N (χ2 statistics). (Please see http://dx.doi.org/10.2337/db08-0830 for a high-quality digital representation of this figure.)
RESEARCH DESIGN AND METHODS
Nondiabetic and diabetic rats from a local Sprague-Dawley strain (bred in the animal facility of the Biomedical Center of Uppsala University) with an increased incidence of congenital malformations in diabetic pregnancy were used as embryo donors (12). The rats had free access to food (AB Analycen, Lidköping, Sweden) and tap water and were maintained at an ambient temperature of 22°C with a 12-h light/dark cycle.
Diabetes was induced with a single injection of 40 mg/kg streptozotocin (STZ) (Sigma-Aldrich Chemie, Stenheim, Germany) into a tail vein (12). Within 1 week after STZ injection, blood glucose concentration was measured with a Free Style Mini Glucose Meter (Thera Sense, Alameda, CA). Rats with a glucose concentration >20 mmol/l were considered to be manifestly diabetic (MD group), and female rats without STZ-induced diabetes were denoted as nondiabetic controls (N group). After establishing diabetes status, the MD and N rats were mated overnight with nondiabetic male rats. A positive vaginal smear (containing sperm) the following morning designated gestational day 0. On gestational day 11, the MD and N rats were killed by cervical dislocation after light ether anesthesia. This gestational day was chosen because it corresponds with an early stage of embryogenesis during which malformations of the heart, neural tube, and general rotation pattern can be identified and, consequently, their severity can be assessed. The embryos were dissected and evaluated with regard to presence of malformations (cardiac hypertrophy, neural tube defects, and somatic malrotation), crown rump length, and somite number. Viability of the embryos was confirmed by the presence of a heartbeat. Embryos were prepared for immunostaining (four N and four MD litters) and gene expression determination (four N and five MD litters).
Preparation of total RNA.
Total RNA from each embryo was isolated with an RNeasy mini kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions. Briefly, each embryo was lysed in 350 μl buffer. The samples were DNase treated (RNase-free DNase; Qiagen) and incubated at room temperature for 25 min. The isolated total RNA was washed twice with 50 μl RNase-free water, and the accumulated flow-through was collected, yielding the total RNA sample. Lastly, 1 μl of an RNase inhibitor (RNA Guard; Amersham Pharmacia, Uppsala, Sweden) was added to each sample.
Preparation of complementary DNA.
One microgram of total RNA was used for complementary DNA (cDNA) preparation. For cDNA synthesis, we used first-strand beads (Ready To Go; Amersham Pharmacia Biotech), according to the manufacturer's instructions. The resulting cDNA was diluted threefold with RNase-free water.
Analysis of messenger RNA levels.
Four litters with 32 embryos yielded the messenger RNA (mRNA) samples of the N group, whereas five litters with 35 embryos produced the mRNA samples of the MD group. One microliter of cDNA purified from each embryo with 10 ng of converted total RNA was amplified and measured in duplicate with real-time PCR using the MiniOpticon system (Bio-Rad Laboratories, Hercules, CA), with SYBR green used to detect the PCR product. Specific primers for CuZnSOD, MnSOD, Gpx-1, Gpx-2, catalase, AR, GAPDH, p53, PARP, Shh, Ret, Bmp-4, VEGF-A, TNF-α, and IL-6 were constructed with the aid of the Primer3 free software (available at http://primer3.sourceforge.net/) and subsequently purchased from TIB Molbiol (Berlin, Germany) (Table 1). We have previously assessed the stability of expression of various housekeeping genes and found the glucose-6-phosphate dehydrogenase (G6PDH) gene to be constant in day-10 and day-11 embryos exposed to high glucose in vitro or diabetes in vivo (data not shown); therefore, we chose the G6PDH gene as a reference in the real-time PCR protocol.
TABLE 1.
Primers used in the study
| Gene | Forward primer | Reverse primer |
|---|---|---|
| G6PDH | gTCATgCAgAACCACCTCCT | ACATACTggCCAAggACCAC |
| CuZnSOD | AAgCggTgAACCAgTTgTg | CCAggTCTCCAACATgCC |
| MnSOD | ggTggAgAACCCAAAggAgA | AgCAgTggAATAAggCCTgT |
| Gpx-1 | TgAgAAgTgCgAggTgAATg | AACACCgTCTggACCTACCA |
| Gpx-2 | TgCCCTACCCTTATgACgAC | ggAgATTCCTAggCTgAgCA |
| Catalase | TTATgTTACCTCACAgCCTggT | gTgTTgTgTgTTCTgTgTgTgTAg |
| Aldose reductase | CgCCAggATCTCTTCATTgT | CTCCATAgCCgTCCAAgTgT |
| GAPDH | ggCATTgCTCTCAATgACAA | TgTgAgggAgATgCTCAgTg |
| PARP | CTggACAACCTCCTggACAT | CgCgTgAgTgTTCTTCACAT |
| p53 | CCCTgAAgACTggATAACTgTCAT | CTCAgTTCCAggTTCCTgTg |
| Shh | TTAAATgCCTTggCCATCTC | TTTCACAgAgCAgTggATgC |
| Ret | CTggAgCCAACAAggAgAAg | CCACATCTgCATCAAACACC |
| Bmp-4 | CgTTACCTCAAgggAgTggA | AgTCCACgTAgAgCgAATgg |
| VEGF-A | gCCCTgAgTCAAgAggACAg | CAggCTCCTgATTCTTCCAg |
| TNF-α | ggCATggATCTCAAAgACAACC | gAggCTgACTTTCTCCTggTAT |
| IL-6 | TgATggATgCTTCCAAACTg | gAgCATTggAAgTTggggTA |
Controls were included in each run of the real-time PCR assay; for each primer pair, one sample with no cDNA (with only RNase-free water) was included. To exclude the possibility of remaining DNA fragments being present in the samples, 10 ng of the total RNA of each sample was amplified in the LightCycler. We found no PCR product in the water or the total RNA samples. Furthermore, we excluded the avian myoblastosis virus-RT enzyme in the cDNA preparation and found no amplified PCR product.
Results were analyzed for each sample with relative quantification comparing the difference between sample and reference crossing point (Cp) values. The relative abundance of mRNA/G6PDH in each sample was determined using the following equation to yield the ratio sample to G6PDH:
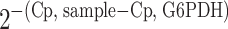 |
Immunohistochemistry of activated (cleaved) caspase-3 and Gpx-1.
Thirty-five embryos from four litters and 28 embryos from four litters were used for immunohistochemistry in the N and MD groups, respectively. Embryos from different rats were fixed in 4% paraformaldehyde for 24 h and stored in 70% ethanol before they were embedded in paraffin and sectioned in 5-μm-thick sections. The slides were deparaffinized and rehydrated in accordance with standard procedures. Primary antibody; cleaved caspase-3; asp 175, 1:50 (1.2 μg); or Gpx-1, 1:25 (2.5 μg) (Cell Signaling Technology, Beverly, MA) were applied. The slides were incubated with secondary antibody (Dako EnVision+Peroxidase, goat anti-rabbit; Dako, Glostrup, Denmark). Thereafter, the slides were incubated with chromogenic substrate solution Sigma Fast 3,3′-diaminobenzidine tablet sets (Sigma-Aldrich Chemie). Lastly, the slides were counterstained (Mayer hematoxylin; Histolab, Gothenburg, Sweden) and mounted with coverslips. The slides were viewed and photographed in a Leitz DMRB Leica fluorescence microscope (Leica, Stockholm, Sweden).
Statistical and ethical considerations.
The Uppsala Regional Ethical Committee on Animal Experiments approved the research protocol, including all experimental procedures involving animals. The comparisons between different experimental groups were based on individual embryos. Differences between means were evaluated by Student's two-tailed t test as well as by ANOVA with Fisher's protected least significant difference as a post hoc test. The rates of normal embryos and embryos with malformations were compared with χ2 statistics. A value of P < 0.05 was considered to denote a statistically significant difference between groups. The calculations were performed with the aid of the Macintosh version of the statistical program Statview.
RESULTS
Maternal diabetes increased malformation and resorption rates in embryos of diabetic pregnancy compared with embryos of normal pregnancy (Fig. 1). We also found decreased crown rump length as well as diminished somite number in embryos from diabetic rats compared with embryos from nondiabetic rats (Table 2).
TABLE 2.
Crown rump length and somite number in embryos of control (N group) and manifestly diabetic (MD group) rats
| n | Crown rump length (mm) | Somites | |
|---|---|---|---|
| N group (normal) | 66 | 3.85 ± 0.03 | 29.8 ± 0.2 |
| N group (malformed) | 1 | 1.50 | 17.0 |
| MD group (normal) | 36 | 2.95 ± 0.07* | 25.7 ± 0.3* |
| MD group (malformed) | 27 | 2.20 ± 0.11*† | 15.8 ± 1.2*† |
Data are means ± SE. The N and MD offspring are also subdivided into nonmalformed (N/MD normal) and malformed (N/MD malformed).
P < 0.05 vs. N and
P < 0.05 vs. N/MD normal (Student's t test).
The expression patterns of the antioxidative enzymes (AOEs) are shown in Fig. 2. We found decreased gene expression of CuZnSOD, MnSOD, and Gpx-1 in MD embryos (both nonmalformed embryos of STZ-induced diabetic rats [MDn group] and malformed littermates [MDm group]) compared with N embryos. In addition, Gpx-1 mRNA levels were more decreased in MDm than in MDn embryos, and Gpx-2 and catalase expression did not differ between the groups (Fig. 2). No significant differences were found with regard to gene expression of AR, GAPDH, p53, and PARP (Fig. 3).
FIG. 2.

Gene expression of CuZnSOD, MnSOD, Gpx-1, Gpx-2, and catalase on gestational day 11 in the control (N) and manifestly diabetic (MD) groups containing 29 and 32 embryos, respectively. Nonmalformed and malformed N and MD offspring are denoted Nn (28 embryos), MDn (20 embryos), Nm (1 embryo), and MDm (12 embryos), respectively. Data are means ± SE. *P < 0.05 vs. N and #P < 0.05 vs. MDn (ANOVA).
FIG. 3.

Gene expression of aldose reductase (AR), GAPDH, p53, and PARP on gestational day 11 in the control (N) and manifestly diabetic (MD) groups containing 29 and 32 embryos, respectively. Nonmalformed and malformed N and MD offspring are denoted Nn (28 embryos), MDn (20 embryos), Nm (1 embryo), and MDm (12 embryos), respectively. Data are means ± SE.
Shh was decreased, Ret was unaltered, and Bmp-4 mRNA levels were increased in MD embryos compared with N embryos (Fig. 4). VEGF-A mRNA levels were increased in both the MDn and MDm embryos compared with N embryos, and, in addition, the MDm embryos had higher mRNA levels than the MDn embryos (Fig. 4). The TNF-α mRNA levels were decreased in the MD embryos (Fig. 4), whereas we could not demonstrate IL-6 mRNA in the embryos (data not shown).
FIG. 4.

Gene expression of Shh, Ret, Bmp-4, VEGF-A, and TNF-α on gestational day 11 in the control (N) and manifestly diabetic (MD) groups containing 29 and 32 embryos, respectively. Nonmalformed and malformed N and MD offspring are denoted Nn (28 embryos), MDn (20 embryos), Nm (1 embryo), and MDm (12 embryos), respectively. Data are means ± SE. *P < 0.05 vs. N and #P < 0.05 vs. MDn (ANOVA).
We detected increased activated caspase-3 immunostaining in the first visceral arch and cardiac area in the MDm embryos compared with the MDn and N embryos (Fig. 5). The immunostaining of Gpx-1 displayed a general accumulation of positive cells in the cardiac tissue of all embryos; however, the MDn embryos had less staining than the N embryos, and the MDm embryos had less staining than the MDn embryos (Fig. 6).
FIG. 5.

Distribution of activated caspase-3 protein in nonmalformed N embryo (A), nonmalformed MD embryo (B), and malformed MD embryo (C). The first visceral arch (square) and cardiac area (circle) are marked. Note differences in caspase-3 staining between the embryos. Space bar = 400 μm. (Please see http://dx.doi.org/10.2337/db08-0830 for a high-quality digital representation of this figure.)
FIG. 6.

Distribution of Gpx-1 protein in whole embryo (A–C) and first visceral arch (D–F). A and D: Shows an N embryo. B and E: Shows a nonmalformed MD embryo. C and F: Shows a malformed MD embryo. Note differences in staining of cardiac area. (Please see http://dx.doi.org/10.2337/db08-0830 for a high-quality digital representation of this figure.)
DISCUSSION
One important finding in the present study was that maternal diabetes diminishes the antioxidative defense gene expression in the embryos, as evidenced by decreased expression of CuZnSOD, MnSOD, and Gpx-1 in MDn and MDm offspring, which may be responsible for a decrease in AOE activity (52). This observation is in concert with previous reports (7–9,48) of diminished AOE activity in embryonic tissues exposed to diabetes; however, it is not in concert with all previous reports (2,7,9), which may be related to differences in mode and time of exposure to the diabetes milieu. In addition, in the present study we detected decreased Gpx-1 mRNA levels in MDm embryos and a corresponding reduction of Gpx-1 protein in the cardiac tissues of the MDm embryos, both findings differing from MDn embryos of the same litters, as well as from Gpx-1 estimations in N embryos. In a similar rat model, Gpx activity was found decreased in embryos with neural tube defects compared with nonmalformed embryos from the same litters of diabetic rats (7).
The specific decrease in Gpx-1 expression/occurrence in MDm embryos may suggest an association between the abundance of this isoform of glutathione peroxidase and the teratogenicity of diabetes, tentatively, with the processes behind cardiac malformations. Such an association has not been suggested before, to our knowledge, and the localization of Gpx-1 protein in the cardiac wall on day 11 may suggest the occurrence of a protective antioxidative mechanism. The net effect of the Gpx-1 deficiency (further enhanced by the decreased levels of the other AOEs) would be enhanced cellular damage, as suggested by mice with a targeted Gpx-1 mutation; they display mitochondrial dysfunction (53) and apoptosis (54)—changes that are in line with a teratological disturbance of normal development. Evidently, however, the association between malformation and reduced Gpx-1 expression may also be the opposite; the embryonic maldevelopment may actually induce the alteration in Gpx-1 mRNA levels and, putatively, decreased activity of the enzyme (7).
In previous work, we have only been able to secure suggestive evidence of enhanced regional apoptotic activity in embryos subjected to a diabetic environment. We thus regard the results of the immunohistochemistry efforts of the present study with great interest. We thus found increased caspase-3 immunostaining, in the first visceral arch and in cardiac tissue of the MDm embryos compared with that of MDn and N embryos, indicative of enhanced apoptosis in the two regions corresponding to the two major types of diabetes-induced malformations in this rat model, micrognathia and cardiac outflow defects (Figure 1).
We found only one malformed N embryo, which displayed gene expression patterns similar to the MDm embryos, further supporting the view that generalized changes of the gene clusters pave the way for specific morphogenetic alterations. In particular, the SODs, Gpx-1, and Shh genes decreased and VEGF increased in the Nm embryo, thus indicating a coupling to oxidative stress and neural crest cell development. However, the small sample size (n = 1) precludes any far-reaching conclusions.
The effect on AOE gene expression was strikingly similar in CuZnSOD, MnSOD, and Gpx-1, which were all decreased. The finding of these diabetes-related genes suggests, in concert with previous studies (4,5,7,9), that maternal diabetes causes a general downregulation of the principal AOEs in rat embryos prone to develop congenital malformations and allows the speculation that this putative weakening of the antioxidative defense may be involved in the teratogenic process. The physiological reasons for diminished ROS-scavenging gene expression in the diabetes-exposed embryos are not clear and may, therefore, currently be subject to speculation only. It is noteworthy that in the present study, we found decreased TNF-α mRNA levels and could not detect IL-6 in the embryos; therefore, a “generalized inflammation” causing the diminished AOE gene expression does not seem to be likely, at least not at this embryonic stage (45,46).
The metabolic state of the severely STZ-induced diabetic non–insulin-treated rat has been characterized previously by us (10,25). In general, the diabetic state yields blood glucose levels in the range of 20–25 mmol/l, as well as increased concentrations of ketone bodies (10) and increased levels of branched chain amino acids (25). The particular characteristics of the diabetic state, which may be primarily affecting gene expression, are not clear, but increased glucose levels should be of importance because in vitro exposure of embryos to high glucose levels promotes gene expression changes analogous to those found in vivo. Furthermore, existence of a functioning glucose transporter gene has been shown to be of fundamental importance for diabetes-induced embryonic (mal)development (27), illustrating the importance of high intracellular levels of glucose in diabetic embryopathy. Regarding the genes controlling glucose metabolism, we and others have previously identified an association between increased sorbitol accumulation and dysmorphogenesis in the embryo (34,35). However, attempting to diminish such accumulation by using aldose reductase inhibitors did not prevent the maldevelopment in the offspring (34,35).
Another reason for the interest in the AR and GAPDH genes is a recent linkage study of skeletal malformation of diabetic rats, in which we found linkage to the chromosomal regions harboring these two genes (U.J.E., N. Nordquist, H. Luthman, U. Pettersson, unpublished data). In the present study, however, we found no significant changes of mRNA levels of AR and GAPDH in the MD embryos, as noted previously with regard to the GAPDH expression (33), a difference we are unable to explain. The previously demonstrated diabetes-induced decrease in GAPDH enzyme activity (33) may thus be a ROS-mediated alteration of the GAPDH enzyme itself (either directly or via DNA damage–induced PARP activity) (32), thereby causing a block in the glycolytic pathway with a number of metabolic effects with teratological importance, such as diacylglycerol-mediated activation of (several isoforms of) protein kinase C (37,38), increased hexosamine flux (36), and enhanced production of glycosylation precursors/products (39).
Maternal diabetes did not alter embryonic p53 (as noted previously [43]) and PARP expression in the present study, despite a putative PARP activity increase due to the presence of DNA damage in the MD embryos (29). The increased Bmp-4 levels of the embryos of STZ-induced diabetic rats suggested specific affection of the precursors of the skeletal system, which backs earlier findings of delayed and defective chondrogenesis and osteogenesis in the offspring (55).
With regard to the diabetes-induced malformations of this rat model (Fig. 1), suggested to have a neural crest cell-derived origin (17), the two neural crest-associated genes, Shh and Ret, were expected to be affected. Whereas the Ret gene was unaltered by maternal diabetes, we found a diabetes-induced decrease in the Shh mRNA levels in the MD embryos. Despite the absence of a significant difference in Shh mRNA levels between the MDn and MDm embryos, the decreased Shh expression in the MD offspring is of particular interest in relation to the findings of embryonic craniofacial (56) and aortic (57) anomalies resulting from direct inhibition of Shh, as well as the findings of an early role for Shh in securing the neural crest cell survival in the early development of the lower jaw (58). Taken together, these results support a diabetes-induced disturbance in the neural crest cell development (17,59) of the embryos of the present model and, putatively, in human diabetic pregnancy as well. However, since isolated neural stem cells exposed to glucose in vitro show increased gene expression of both Shh and Bmp-4 (50), the exact relationship(s) between gene expression changes in the surrounding tissues and the neural crest cells themselves caused by exposure to a diabetic environment will have be subject to further studies.
We found markedly increased VEGF-A expression in the MD embryos and further increased expression in the malformed embryos compared with the nonmalformed MD embryos. This is of particular interest with regard to the coupling between VEGF alterations and hampered embryonic development (60), although an increased VEGF expression has not been previously reported in association with embryonic dysmorphogenesis. We suggest that the enhanced embryonic VEGF-A levels in the present study may be interpreted as a compensatory change in response to a diabetes-induced general developmental delay. The demonstrated gene expression changes in malformed (and nonmalformed) embryos of diabetic rats will evidently demand future work efforts in order to clarify the association to embryonic maldevelopment (cause, effect, or no relation at all—efforts that, to some extent, are ongoing.
One major, and slightly surprising, observation was that there was no clear individual pattern of gene expression in the embryos for any of the genes studied (online appendix [available at http://dx.doi.org/10.2337/db08-0830]). Thus, when examining individual gene expression values, there was no evident relation to uterine position, growth, or morphological status (with the possible exceptions of Gpx-1 and Vegf-A). The Gpx-1 mRNA was markedly decreased and the VEGF-A mRNA was markedly increased in malformed compared with nonmalformed embryos from the same litter of diabetic rats. Further efforts in the search for the etiology of diabetes-induced congenital malformations may therefore benefit from considering a putative impact of a decrease in embryonic Gpx-1 expression and, possibly, function as well as considering the importance of enhanced VEGF-A expression for embryonic development.
We conclude that maternal diabetes causes growth retardation, congenital malformations, and decreased general antioxidative gene expression in the embryo. In particular, enhanced apoptosis of the first visceral arch and heart, together with decreased cardiac Gpx-1 levels, may cause the mandible and heart to have an increased risk of developing congenital malformation.
Supplementary Material
Acknowledgments
Financial support was obtained from the Ernfors Family Fund, the Swedish Diabetes Association, the Novo Nordisk Foundation, and the Swedish Research Council (grant no. 12X-7475).
The authors are indebted to Ing-Marie Mörsare for valuable technical support.
Published ahead of print at http://diabetes.diabetesjournals.org on 26 August 2008.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
See accompanying commentary, p. 3187.
REFERENCES
- 1.Eriksson UJ, Borg LAH: Protection by free oxygen radical scavenging enzymes against glucose-induced embryonic malformations in vitro. Diabetologia 34 :325 –331,1991 [DOI] [PubMed] [Google Scholar]
- 2.Eriksson UJ, Borg LAH: Diabetes and embryonic malformations: role of substrate-induced free-oxygen radical production for dysmorphogenesis in cultured rat embryos. Diabetes 42 :411 –419,1993 [DOI] [PubMed] [Google Scholar]
- 3.Yang X, Borg LAH, Eriksson UJ: Altered metabolism and superoxide generation in neural tissue of rat embryos exposed to high glucose. Am J Physiol 272 :E173 –E180,1997 [DOI] [PubMed] [Google Scholar]
- 4.Sakamaki H, Akazawa S, Ishibashi M, Izumino K, Takino H, Yamasaki H, Yamaguchi Y, Goto S, Urata Y, Kondo T, Nagataki S: Significance of glutathione-dependent antioxidant system in diabetes-induced embryonic malformations. Diabetes 48 :1138 –1144,1999 [DOI] [PubMed] [Google Scholar]
- 5.Trocino RA, Akazawa S, Ishibashi M, Matsumoto K, Matsuo H, Yamamoto H, Goto S, Urata Y, Kondo T, Nagataki S: Significance of glutathione depletion and oxidative stress in early embryogenesis in glucose-induced rat embryo culture. Diabetes 44 :992 –998,1995 [DOI] [PubMed] [Google Scholar]
- 6.Hagay ZJ, Weiss Y, Zusman I, Peled-Kamar M, Reece EA, Eriksson UJ, Groner Y: Prevention of diabetes-associated embryopathy by overexpression of the free radical scavenger copper zinc superoxide dismutase in transgenic mouse embryos. Am J Obstet Gynecol 173 :1036 –1041,1995 [DOI] [PubMed] [Google Scholar]
- 7.Sivan E, Lee Y, Wu Y, Reece E: Free radical scavenging enzymes in fetal dysmorphogenesis among offspring of diabetic rats. Teratology 56 :343 –349,1997 [DOI] [PubMed] [Google Scholar]
- 8.Zaken V, Kohen R, Ornoy A: Vitamins C and E improve rat embryonic antioxidant defense mechanism in diabetic culture medium. Teratology 64 :33 –44,2001 [DOI] [PubMed] [Google Scholar]
- 9.Weksler-Zangen S, Yaffe P, Ornoy A: Reduced SOD activity and increased neural tube defects in embryos of the sensitive but not of the resistant Cohen diabetic rats cultured under diabetic conditions. Birth Defects Res A Clin Mol Teratol 67 :429 –437,2003 [DOI] [PubMed] [Google Scholar]
- 10.Wentzel P, Thunberg L, Eriksson UJ: Teratogenic effect of diabetic serum is prevented by supplementation of superoxide dismutase and N-acetylcysteine in rat embryo culture. Diabetologia 40 :7 –14,1997 [DOI] [PubMed] [Google Scholar]
- 11.Wentzel P, Gareskog M, Eriksson UJ: Folic acid supplementation diminishes diabetes- and glucose-induced dysmorphogenesis in rat embryos in vivo and in vitro. Diabetes 54 :546 –553,2005 [DOI] [PubMed] [Google Scholar]
- 12.Eriksson UJ, Simán CM: Pregnant diabetic rats fed the antioxidant butylated hydroxytoluene show decreased occurrence of malformations in the offspring. Diabetes 45 :1497 –1502,1996 [DOI] [PubMed] [Google Scholar]
- 13.Viana M, Herrera E, Bonet B: Teratogenic effects of diabetes mellitus in the rat. Prevention with vitamin E. Diabetologia 39 :1041 –1046,1996 [DOI] [PubMed] [Google Scholar]
- 14.Sivan E, Reece EA, Wu YK, Homko CJ, Polansky M, Borenstein M: Dietary vitamin E prophylaxis and diabetic embryopathy: morphologic and biochemical analysis. Am J Obstet Gynecol 175 :793 –799,1996 [DOI] [PubMed] [Google Scholar]
- 15.Simán CM, Eriksson UJ: Vitamin E decreases the occurrence of malformations in the offspring of diabetic rats. Diabetes 46 :1054 –1061,1997 [DOI] [PubMed] [Google Scholar]
- 16.Yang X, Borg LAH, Simán CM, Eriksson UJ: Maternal antioxidant treatments prevent diabetes-induced alterations of mitochondrial morphology in rat embryos. Anat Rec 251 :303 –315,1998 [DOI] [PubMed] [Google Scholar]
- 17.Simán CM, Gittenberger-De Groot AC, Wisse B, Eriksson UJ: Malformations in offspring of diabetic rats: morphometric analysis of neural crest-derived organs and effects of maternal vitamin E treatment. Teratology 61 :355 –367,2000 [DOI] [PubMed] [Google Scholar]
- 18.Cederberg J, Eriksson UJ: Antioxidative treatment of pregnant diabetic rats diminishes embryonic dysmorphogenesis. Birth Defects Res A Clin Mol Teratol 73 :498 –505,2005 [DOI] [PubMed] [Google Scholar]
- 19.Reece EA, Wu YK, Zhao Z, Dhanasekaran D: Dietary vitamin and lipid therapy rescues aberrant signaling and apoptosis and prevents hyperglycemia-induced diabetic embryopathy in rats. Am J Obstet Gynecol 194 :580 –585,2006 [DOI] [PubMed] [Google Scholar]
- 20.Simán CM, Eriksson UJ: Vitamin C supplementation of the maternal diet reduces the rate of malformation in the offspring of diabetic rats. Diabetologia 40 :1416 –1424,1997 [DOI] [PubMed] [Google Scholar]
- 21.Roest PA, van Iperen L, Vis S, Wisse LJ, Poelmann RE, Steegers-Theunissen RP, Molin DG, Eriksson UJ, Gittenberger-De Groot AC: Exposure of neural crest cells to elevated glucose leads to congenital heart defects, an effect that can be prevented by N-acetylcysteine. Birth Defects Res A Clin Mol Teratol 79 :231 –235,2007 [DOI] [PubMed] [Google Scholar]
- 22.Wentzel P, Welsh N, Eriksson UJ: Developmental damage, increased lipid peroxidation, diminished cyclooxygenase-2 gene expression, and lowered PGE2 levels in rat embryos exposed to a diabetic environment. Diabetes 48 :813 –820,1999 [DOI] [PubMed] [Google Scholar]
- 23.Wentzel P, Eriksson UJ: 8-Iso-PGF(2alpha) administration generates dysmorphogenesis and increased lipid peroxidation in rat embryos in vitro. Teratology 66 :164 –168,2002 [DOI] [PubMed] [Google Scholar]
- 24.Cederberg J, Basu S, Eriksson UJ: Increased rate of lipid peroxidation and protein carbonylation in experimental diabetic pregnancy. Diabetologia 44 :766 –774,2001 [DOI] [PubMed] [Google Scholar]
- 25.Styrud J, Thunberg L, Nybacka O, Eriksson UJ: Correlations between maternal metabolism and deranged development in the offspring of normal and diabetic rats. Pediatr Res 37 :343 –353,1995 [DOI] [PubMed] [Google Scholar]
- 26.Fine EL, Horal M, Chang TI, Fortin G, Loeken MR: Evidence that elevated glucose causes altered gene expression, apoptosis, and neural tube defects in a mouse model of diabetic pregnancy. Diabetes 48 :2454 –2462,1999 [DOI] [PubMed] [Google Scholar]
- 27.Li R, Thorens B, Loeken MR: Expression of the gene encoding the high-Km glucose transporter 2 by the early postimplantation mouse embryo is essential for neural tube defects associated with diabetic embryopathy. Diabetologia 50 :682 –689,2007 [DOI] [PubMed] [Google Scholar]
- 28.Yang X, Borg LAH, Eriksson UJ: Altered mitochondrial morphology of rat embryos in diabetic pregnancy. Anat Rec 241 :255 –267,1995 [DOI] [PubMed] [Google Scholar]
- 29.Lee AT, Reis D, Eriksson UJ: Hyperglycemia induced embryonic dysmorphogenesis correlates with genomic DNA mutation frequency in vitro and in vivo. Diabetes 48 :371 –376,1999 [DOI] [PubMed] [Google Scholar]
- 30.Gerber RT, Holemans K, O'Brien-Coker I, Mallet AI, van Bree R, Van Assche FA, Poston L: Increase of the isoprostane 8-isoprostaglandin f2alpha in maternal and fetal blood of rats with streptozotocin-induced diabetes: evidence of lipid peroxidation. Am J Obstet Gynecol 183 :1035 –1040,2000 [DOI] [PubMed] [Google Scholar]
- 31.Piddington R, Joyce J, Dhanasekaran P, Baker L: Diabetes mellitus affects prostaglandin E2 levels in mouse embryos during neurulation. Diabetologia 39 :915 –920,1996 [DOI] [PubMed] [Google Scholar]
- 32.Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, Brownlee M: Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest 112 :1049 –1057,2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wentzel P, Ejdesjo A, Eriksson UJ: Maternal diabetes in vivo and high glucose in vitro diminish GAPDH activity in rat embryos. Diabetes 52 :1222 –1228,2003 [DOI] [PubMed] [Google Scholar]
- 34.Hod M, Star S, Passonneau JV, Unterman TG, Freinkel N: Effect of hyperglycemia on sorbitol and myo-inositol content of cultured rat conceptus: failure of aldose reductase inhibitors to modify myo-inositol depletion and dysmorphogenesis. Biochem Biophys Res Commun 140 :974 –980,1986 [DOI] [PubMed] [Google Scholar]
- 35.Eriksson UJ, Naeser P, Brolin SE: Increased accumulation of sorbitol in offspring of manifest diabetic rats. Diabetes 35 :1356 –1363,1986 [DOI] [PubMed] [Google Scholar]
- 36.Horal M, Zhang Z, Stanton R, Virkamaki A, Loeken MR: Activation of the hexosamine pathway causes oxidative stress and abnormal embryo gene expression: involvement in diabetic teratogenesis. Birth Defects Res A Clin Mol Teratol 70 :519 –527,2004 [DOI] [PubMed] [Google Scholar]
- 37.Hiramatsu Y, Sekiguchi N, Hayashi M, Isshiki K, Yokota T, King GL, Loeken MR: Diacylglycerol production and protein kinase C activity are increased in a mouse model of diabetic embryopathy. Diabetes 51 :2804 –2810,2002 [DOI] [PubMed] [Google Scholar]
- 38.Gareskog M, Wentzel P: Altered protein kinase C activation associated with rat embryonic dysmorphogenesis. Pediatr Res 56 :849 –857,2004 [DOI] [PubMed] [Google Scholar]
- 39.Eriksson UJ, Wentzel P, Minhas HS, Thornalley PJ: Teratogenicity of 3-deoxyglucosone and diabetic embryopathy. Diabetes 47 :1960 –1966,1998 [DOI] [PubMed] [Google Scholar]
- 40.Salas-Vidal E, Lomeli H, Castro-Obregon S, Cuervo R, Escalante-Alcalde D, Covarrubias L: Reactive oxygen species participate in the control of mouse embryonic cell death. Exp Cell Res 238 :136 –147,1998 [DOI] [PubMed] [Google Scholar]
- 41.Pampfer S, Vanderheyden I, McCracken JE, Vesela J, De Hertogh R: Increased cell death in rat blastocysts exposed to maternal diabetes in utero and to high glucose or tumor necrosis factor-alpha in vitro. Development 124 :4827 –4836,1997 [DOI] [PubMed] [Google Scholar]
- 42.Sun F, Kawasaki E, Akazawa S, Hishikawa Y, Sugahara K, Kamihira S, Koji T, Eguchi K: Apoptosis and its pathway in early post-implantation embryos of diabetic rats. Diabetes Res Clin Pract 67 :110 –118,2005 [DOI] [PubMed] [Google Scholar]
- 43.Gareskog M, Cederberg J, Eriksson UJ, Wentzel P: Maternal diabetes in vivo and high glucose concentration in vitro increases apoptosis in rat embryos. Reproductive Toxicology 23 :63 –74,2007 [DOI] [PubMed] [Google Scholar]
- 44.Yang P, Zhao Z, Reece EA: Involvement of c-Jun N-terminal kinases activation in diabetic embryopathy. Biochem Biophys Res Commun 357 :749 –754,2007 [DOI] [PubMed] [Google Scholar]
- 45.Pampfer S, Wuu YD, Vanderheyden I, De Hertogh R: Expression of tumor necrosis factor-alpha (TNF alpha) receptors and selective effect of TNF alpha on the inner cell mass in mouse blastocysts. Endocrinology 134 :206 –212,1994 [DOI] [PubMed] [Google Scholar]
- 46.Torchinsky A, Brokhman I, Shepshelovich J, Orenstein H, Savion S, Zaslavsky Z, Koifman M, Dierenfeld H, Fein A, Toder V: Increased TNF-alpha expression in cultured mouse embryos exposed to teratogenic concentrations of glucose. Reproduction 125 :527 –534,2003 [DOI] [PubMed] [Google Scholar]
- 47.Afonso V, Santos G, Collin P, Khatib AM, Mitrovic DR, Lomri N, Leitman DC, Lomri A: Tumor necrosis factor-alpha down-regulates human Cu/Zn superoxide dismutase 1 promoter via JNK/AP-1 signaling pathway. Free Radic Biol Med 41 :709 –721,2006 [DOI] [PubMed] [Google Scholar]
- 48.Cederberg J, Eriksson UJ: Decreased catalase activity in malformation-prone embryos of diabetic rats. Teratology 56 :350 –357,1997 [DOI] [PubMed] [Google Scholar]
- 49.Pani L, Horal M, Loeken MR: Polymorphic susceptibility to the molecular causes of neural tube defects during diabetic embryopathy. Diabetes 51 :2871 –2874,2002 [DOI] [PubMed] [Google Scholar]
- 50.Fu J, Tay SS, Ling EA, Dheen ST: High glucose alters the expression of genes involved in proliferation and cell-fate specification of embryonic neural stem cells. Diabetologia 49 :1027 –1038,2006 [DOI] [PubMed] [Google Scholar]
- 51.Eriksson RS, Thunberg L, Eriksson UJ: Effects of interrupted insulin treatment on fetal outcome of pregnant diabetic rats. Diabetes 38 :764 –772,1989 [DOI] [PubMed] [Google Scholar]
- 52.Tiedge M, Lortz S, Drinkgern J, Lenzen S: Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 46 :1733 –1742,1997 [DOI] [PubMed] [Google Scholar]
- 53.Esposito LA, Kokoszka JE, Waymire KG, Cottrell B, MacGregor GR, Wallace DC: Mitochondrial oxidative stress in mice lacking the glutathione peroxidase-1 gene. Free Rad Biol Med 28 :754 –766,2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Flentjar NJ, Crack PJ, Boyd R, Malin M, de Haan JB, Hertzog P, Kola I, Iannello R: Mice lacking glutathione peroxidase-1 activity show increased TUNEL staining and an accelerated inflammatory response in brain following a cold-induced injury. Exp Neurol 177 :9 –20,2002 [DOI] [PubMed] [Google Scholar]
- 55.Styrud J, Eriksson UJ: Effects of D-glucose and beta-hydroxybutyric acid on the in vitro development of (pre)chondrocytes from embryos of normal and diabetic rats. Acta Endocrinologica (Kbh) 122 :487 –498,1990 [DOI] [PubMed] [Google Scholar]
- 56.Nagase T, Nagase M, Osumi N, Fukuda S, Nakamura S, Ohsaki K, Harii K, Asato H, Yoshimura K: Craniofacial anomalies of the cultured mouse embryo induced by inhibition of sonic hedgehog signaling: an animal model of holoprosencephaly. J Craniofac Surg 16 :80 –88,2005 [DOI] [PubMed] [Google Scholar]
- 57.Nagase T, Nagase M, Yoshimura K, Machida M, Yamagishi M: Defects in aortic fusion and craniofacial vasculature in the holoprosencephalic mouse embryo under inhibition of sonic hedgehog signaling. J Craniofac Surg 17 :736 –744,2006 [DOI] [PubMed] [Google Scholar]
- 58.Brito JM, Teillet MA, Le Douarin NM: An early role for sonic hedgehog from foregut endoderm in jaw development: ensuring neural crest cell survival. Proc Natl Acad Sci U S A 103 :11607 –11612,2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suzuki N, Svensson K, Eriksson UJ: High glucose concentration inhibits migration of rat cranial neural crest cells in vitro. Diabetologia 39 :401 –411,1996 [DOI] [PubMed] [Google Scholar]
- 60.Pinter E, Haigh J, Nagy A, Madri JA: Hyperglycemia-induced vasculopathy in the murine conceptus is mediated via reductions of VEGF-A expression and VEGF receptor activation. Am J Pathol 158 :1199 –1206,2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.