

Abstract
Retinal dystrophy in Bardet–Biedl Syndrome (BBS) is caused by defective genes that are expressed within ciliated cells such as photoreceptors. The purpose of this study was to characterize and compare the retinal structure and lamination of two groups of patients, carrying mutations in BBS1 or BBS10. Eight patients with BBS (ages 11.9–28.5 years) and mutations in BBS1 (4/8) or BBS10 (4/8) were tested. A high-resolution hand-held probe Fourier-domain optical coherence tomography system (Fd-OCT) was used for retinal image acquisition. Macular scans were evaluated with respect to structure, retinal layering and photoreceptor integrity. Micro-structural in-vivo analysis showed abnormalities within retinal layers but preserved retinal lamination. Photoreceptor integrity was disrupted in all patients. Macular scans from patients with BBS10 mutations most often showed ‘deposits’ adjacent and anterior to Bruch's membrane. Age, genotype and presence of macular changes did not correlate with the structural changes observed. Retinal dystrophy in BBS is reflected by major changes in the outer retinal layers. This is the first report of in-vivo micro-structural analysis of retinal layers in patients with BBS. Mutations in different BBS genes seem to be associated with similar micro-structural changes in retinal layers.
Keywords: Bardet–Biedl Syndrome, BBS1, BBS10, Fourier-domain OCT, Retinal lamination
1. Introduction
Bardet–Biedl Syndrome (BBS)1, first described by Bardet (1920) and Biedl (1922), is defined as an autosomal recessive multi-systemic disorder. Cardinal features include digit anomalies, obesity, cognitive impairment, hypogonadism, kidney malformation or dysfunction and retinal dystrophy (Churchill, McManamon, & Hurley, 1981; Schachat & Maumenee, 1982). BBS is now thought of as a ‘ciliopathy’ with the underlying defects affecting the basal body of ciliated cells, such as the connecting cilium in the outer retina (Ansley et al., 2003; Ross et al., 2005). Twelve different genes have been associated with the disease, with BBS1 and BBS10 being the most frequently involved (Beales et al., 2003; Stoetzel et al., 2006).
The retinal dystrophy in BBS is progressive and shows variable severity. Retinal layer analysis using time-domain optical coherence tomography (OCT) in patients with homozygous or compound heterozygous mutations in BBS1 was performed by Azari et al. (2006). Definable lamination was observed with no extreme retinal thickening and a normal nerve fiber layer around the optic nerve for most of the 10 patients tested. No genotype–phenotype correlation or age-matched correlation was identified.
Fourier-domain OCT (Fd-OCT) permits faster retinal image acquisition with significantly higher axial resolution than commercial time-domain OCT systems (Wojtkowski, Leitgeb, Kowalczyk, Bajraszewski, & Fercher, 2002; Nassif et al., 2004). Application of Fd-OCT in patients with retinal diseases has provided an opportunity to delineate details of retinal structure and lamination (Gerth et al., 2007; Sun et al., 2007).
Here, we describe retinal structure characteristics using a custom high-resolution imaging instrument in younger patients with BBS1 or BBS10 mutations. We found preserved inner retinal layers but disrupted outer retinal structure without definable connecting cilium (inner/outer segment junction) in most of the patients studied. Retinal structure abnormalities were not correlated with either genotype, age or disease severity.
2. Materials and methods
2.1. Subjects
Patients diagnosed with Bardet–Biedl Syndrome carrying either a homozygous or compound heterozygous mutation in the BBS1 or BBS10 genes were recruited through the Ocular Genetics Clinic at The Hospital for Sick Children (Sick Kids) in Toronto, Canada. A comprehensive ocular assessment as well as a detailed phenotype review of systems; body mass index, kidney structure and function, liver function and digit anomalies were completed. Presence of cognitive impairment was assessed through parental interviews. Written informed consent and/or assent were obtained from all participants and/or their guardians. The project was approved by the Research Ethics Board at Sick Kids and conducted in accordance with the Tenets of Helsinki.
2.2. Ocular function and morphology assessment
The comprehensive eye evaluation included dilated fundus examination, visual acuity (VA), kinetic perimetry and full-field electroretinography (ERG). Best-corrected monocular distant visual acuity (VA) was measured on the backlit Early Treatment Diabetic Retinopathy Study Charts using a logMAR scale (Ferris, Kassoff, Bresnick, & Bailey, 1982). A VA of ‘counting fingers’ was converted into 1.85 logMAR (Schulze-Bonsel, Feltgen, Burau, Hansen, & Bach, 2006). Monocular kinetic visual fields (VF) were performed on the Goldman perimeter using test target III-4e. VF area was measured as a solid angle using the VisFields 1.3.2. (Weleber & Tobler, 1986). Full-field electroretinograms (ERG) were recorded according to the ISCEV standard (Marmor, Holder, Seeliger, & Yamamoto, 2004) and compared with age-matched control data.
2.3. Fourier-domain optical coherence tomography
In-vivo high-resolution retinal image acquisition was performed using a Fourier-domain high-speed, high-resolution optical coherence tomography (Fd-OCT) system (axial resolution: 4.5 μm; acquisition speed: 9 frames/second, 1000 A – scans/frame) constructed at the UC Davis Medical Center (Alam et al., 2006; Zawadzki, Bower et al., 2005) with a sample arm replaced by a hand-held scanner (Bioptigen Inc. Durham, NC). Horizontal scans of 5 mm were obtained through the macular area. Images were post-processed as described in detail elsewhere (Zawadzki, Bower et al., 2005; Zawadzki et al., 2007).
Retinal layers were identified based on previously published data (Zawadzki, Jones et al., 2005) and compared with control data as shown in Fig. 1. We (Zawadzki, Jones et al., 2005) and others (Wojtkowski et al., 2005; Wojtkowski et al., 2004) have identified three distinct bands in the outer retina using high-resolution Fd-OCT, which are not visible with lower-resolution systems. Direct overlay of OCT and histological sections from the monkey retina (Macaca fascicularis), following correction for shrinkage, led to positive identification of the inner/outer segment junction corresponding to the connecting cilia (Anger et al., 2004). Similar results were found in the porcine retina (Gloesmann et al., 2003).
Fig. 1.
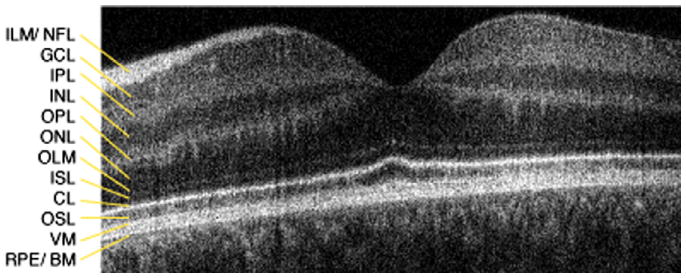
Macular OCT imaging of a 16 year old control subject. 5 mm horizontal scan through the macula: CL, connecting cilia; GCL, ganglion cell layer; ILM/NFL, internal limiting membrane/nerve fiber layer; INL, inner nuclear layer; IPL, inner plexiform layer; ISL, inner segment layer; OLM, outer limiting membrane; ONL, outer nuclear layer; OPL, outer plexiform layer; OSL, outer segment layer; RPE/BM, retinal pigment epithelium/Bruch's membrane; VM, Verhoeff's membrane.
2.4. Mutational analysis
Mutation screening was performed for all coding exons and splice junctions of BBS1 (NM_024649) and BBS10 (NM_024685). Mutational analysis was performed by direct sequencing of purified polymerase chain reaction (PCR) products amplified from genomic DNA, using standard protocols. A complete list of primers used for PCR and sequencing is available upon request. Sequence changes were verified with family segregation, when the family was available. A minimum of 150 healthy control individuals were screened to assess the allele frequency of novel sequence changes.
3. Results
Eight patients (5 females, 3 males) between ages 11.9 and 28.5 years with mutations in the BBS1 (n = 4) or in the BBS10 gene (n = 4) were included in the study. Systemic features of patients studied are summarized in Table 1.
Table 1.
Genotype–phenotype Summary
| Case # | Gender | Gene | Mutation | Age (years) | Kidney anomaly | Liver anomaly | Digit anomaly | Limb # | Weight | Cognitive impairment |
|---|---|---|---|---|---|---|---|---|---|---|
| 2597 | F | BBS1 | M390R/M390R | 20.6 | No | No | Polydactyly | 1 | Overweight | Learning disability |
| 2512 | F | BBS1 | M1V†/M1V† | 28.5 | No | No | Brachydactyly | NA | Obese | No |
| 2301 | F | BBS1 | D8D/R483X | 15.8 | Yes | Yes | Polydactyly | 4 | Obese | Dev delayed |
| 2293 | M | BBS1 | M390R/N524del† | 20.8 | No | No | NA | NA | Obese | Dev delayed |
| 2296 | M | BBS10 | C91W/A474fs483X | 11.9 | Yes | No | Polydactyly | 4 | Overweight | Dev delayed/Autistic |
| 2621 | M | BBS10 | C91W/A474fs483X | 13.6 | Yes | No | Clinodactyly | 2 | Obese | NA |
| 2213 | F | BBS10 | C91W/V707fs708X | 13.5 | Yes | No | Polydactyly | 1 | Overweight | Dev delayed |
| 2294 | F | BBS10 | C91fs95X/R103fs110X† | 15.5 | Yes | No | Polydactyly | 4 | Overweight | Dev delayed |
novel mutation; Age, age at time of kidney and liver blood work assessment; Kidney anomaly, defined as either abnormal function as per electrolyte and elevated creatinine and urea levels and/or abnormal kidney ultrasound; Liver anomaly, defined as either elevated liver enzymes and/or abnormal liver ultrasound; Weight, based on percentile for body mass index-for-age; Obese: >95th percentile and Overweight: 85th–95th percentile; NA, not available; Limb#, refers to the numbers of limbs affected with a digit anomaly.
Results of ocular assessments are summarized in Table 2. Visual acuity ranged from 0.2 to 1.85 logMAR. Despite their younger age, most of the patients carrying mutations in BBS10 had more severely reduced VA and smaller visual field areas than patients with mutation in BBS1.
Table 2.
Ocular phenotype and OCT findings
| Case # | Gender | Age | VA | GVF | ERG (age at test) | Macula | Fd-OCT | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ILM wrinkling | Inner retina layer abnormal | Enlarged space within ONL in fovea | ISL/OSL present | RPE thinned | Deposits above Bruch's membrane | |||||||
| 2597 | F | 20.6 | 0.2 | 1.2 | RCD (20.5) | Abnormal | + | − | + | + | + | − |
| 2512 | F | 28.5 | 1.6 | 0 | NR (5.7) | Abnormal | − | + | − | (+) | + | + |
| 2301 | F | 15.8 | 0.54 | 0.23 | NR (12.3) | Abnormal | − | − | − | − | + | − |
| 2293 | M | 20.8 | 0.6 | 1.7 | RCD (20.5) | Abnormal | + | − | + | + | + | − |
| 2296* | M | 11.9 | 1.85† | NT | NT | Abnormal | − | − | + | + | + | + |
| 2621* | M | 13.6 | 1.6† | 0.13 | NR (8.1) | Abnormal | + | − | + | (+) | + | + |
| 2213 | F | 13.5 | 1.85† | 0.01 | RCD (11.4), NR (13.5) | Normal | − | − | − | − | + | + |
| 2294 | F | 16.6 | 0.6 | 0.08 | RCD (4.4) | Normal | − | − | − | (+) | + | + |
siblings; VA, visual acuity (log MAR);
nystagmus; GVF, Goldman visual field (III4e, solid angle); RCD, rod-cone dysfunction; NR, non-recordable; NT, not tested; ILM, inner limiting membrane; ONL, outer nuclear layer; ISL/OSL, inner/outer segment layer; RPE, retinal pigment epithelium; +, yes; −, no.
3.1. Retinal morphology
All patients with mutations in BBS1 showed some degree of maculopathy, whereas this was present in only 2 of 4 patients in the BBS10 group. Fd-OCT image acquisition was possible in all patients including those with nystagmus and fixation instability associated with low VA. Microstructural findings of retinal layers imaged are shown in Fig. 2 (BBS1) and Fig. 3 (BBS10) and summarized in Table 2.
Fig. 2.
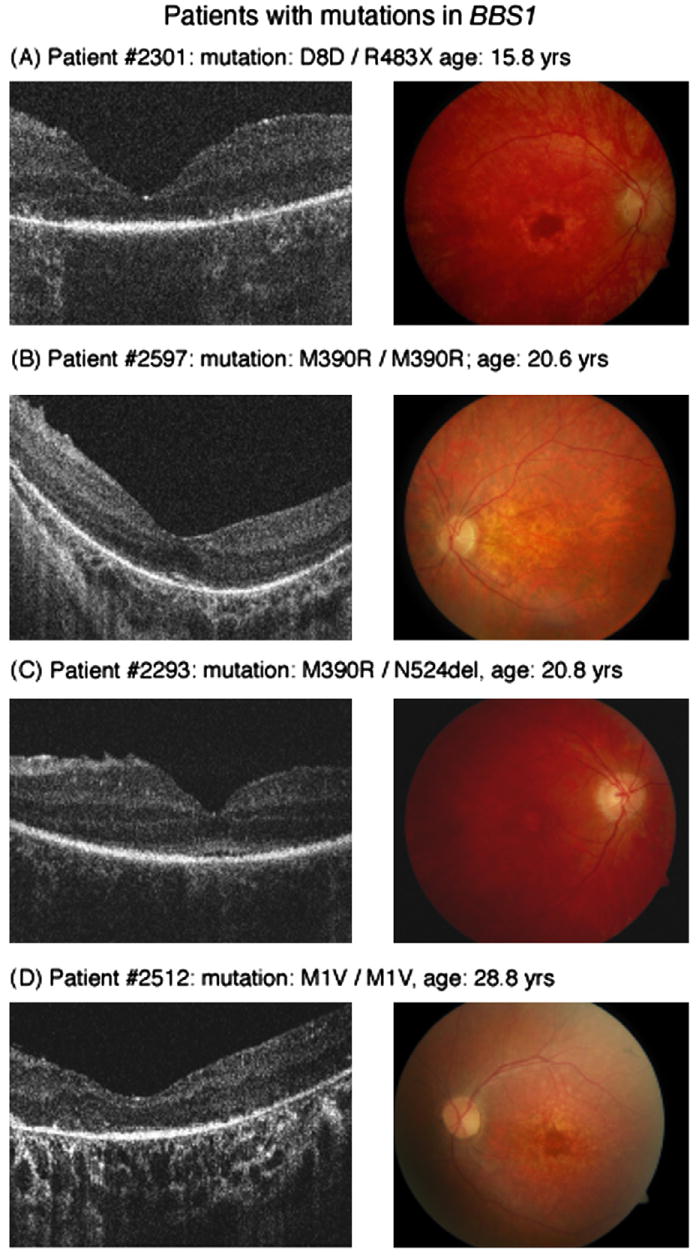
Horizontal Fd-OCT scans (5 mm) (left panel) and corresponding fundus photograph (right panel) of patients with mutations in BBS1. Not identifiable ISL or OSL in the Fd-OCT scan were associated with atrophic macular changes in patient #2301 (A). Present ISL/OSL layer, ILM wrinkling and enlarged space within the ONL were visible in patient #2597 (B) and # 2293 (C) with clinically early maculopathy. Deposits above Bruch's membrane are visible in macular scan of patient #2512 (D).
Fig. 3.
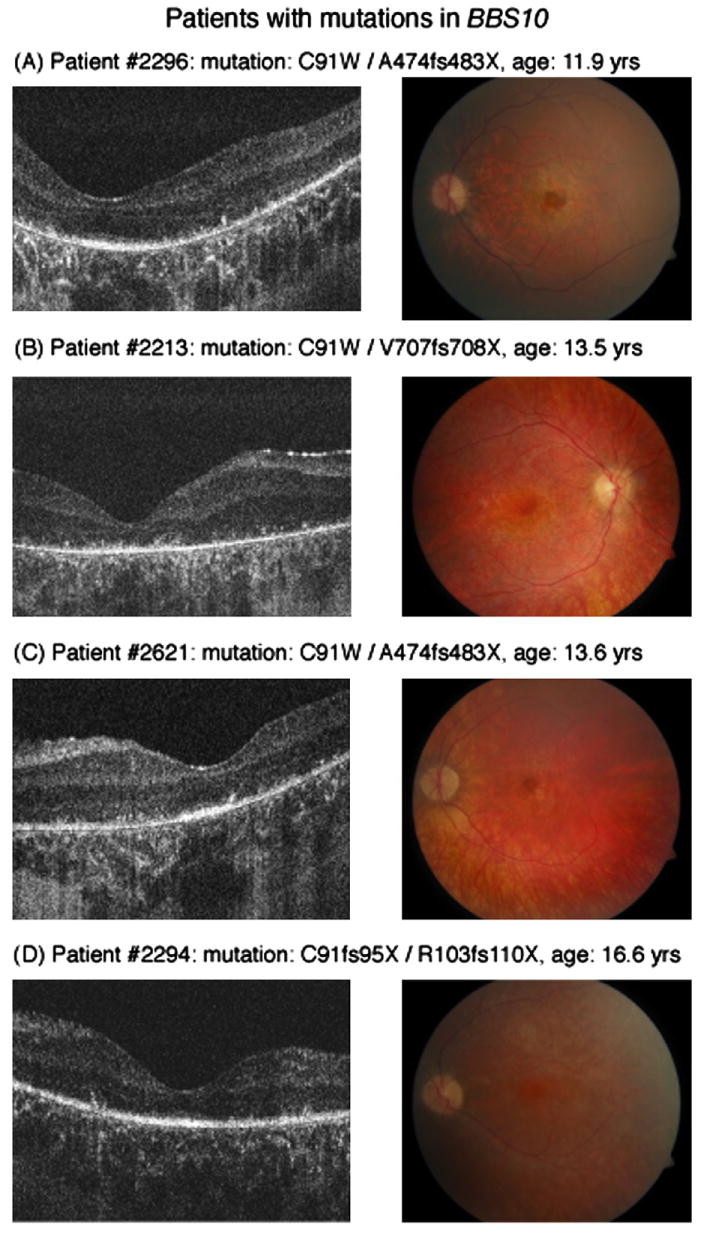
Horizontal Fd-OCT scans (5 mm) (left panel) and corresponding fundus photograph (right panel) of patients with mutations in BBS10. Maculopathy is early to moderate in patient #2296 (A) and #2621 (C) but absent in the other 2 patients (B) (D). ISL/OSL are identifiable in all but patient #2213 (B). Deposits above Bruch's membrane are visible in all patients (A–D, left panel).
Four cases were selected based on the genetic heterogeneity
BBS1: Case 2597, homozygous mutation in M390R (Fig. 2B)
This patient with polydactyly, obesity and retinopathy was diagnosed with BBS at age 17.7 years and was re-evaluated 3 years later. Examination first showed a VA of 0.1 logMAR, slightly constricted visual field with paracentral scotoma and an early maculopathy in both eyes. Repeated ERG assessment demonstrated a progressive rod-cone dysfunction. Ophthalmic examination revealed stable VA and retinopathy at follow up visits (Table 2, Fig. 2). Fd-OCT macular scan demonstrates intact inner, but disrupted outer retinal layers. The space within the ONL or the inner and outer layers in the foveal area appears to be enlarged when compared with the control scan. Analysis of outer retinal layers was made difficult because of the disruption and possibly layer re-organization. It could not be determined whether the layer posterior to the OLM was consistent with the inner or outer segment layer because of non-visible connecting cilia. The photoreceptor inner or outer segment layer was only seen in the foveal area with gradual thinning centrifugal of the fovea. The RPE layer showed drastic thinning with a resulting higher reflectivity from the choroid.
BBS1: Case 2512, homozygous mutation in M1V (Fig. 2D)
This patient with obesity, polydactyly, retinopathy and learning disability was diagnosed with BBS at age 5. Her ERG was non-recordable above noise at that time. Visual function progressed from 0.3 logMAR at age 8 to 1.6 logMAR at age 28. VF constriction was progressive from 20 degrees at age 13 to non-detectable with the III4e test target by age 28. Her most recent fundus examination showed an atrophic maculopathy, constricted vessels and general retinopathy with some minor spiculae pigmentation. Unlike the other patients studied, analysis of inner retinal layers demonstrated thinning. The outer retinal layers, including inner and outer segment layer, were not definable. The subfoveal increase in reflectivity above the RPE layer may have indicated a remaining inner or outer segment layer. The RPE was thinner than in Case 2597. Areas of increased reflectivity, almost like deposits, were seen above Bruch's membrane.
BBS10: Case 2213, heterozygous mutation in C91W and V707fs708X (Fig. 3B)
This patient demonstrated polydactyly, overweight, polycystic kidneys, and learning disabilities and a progressive retinal dysfunction. VA dropped from 0.3 logMAR at age 9 to 1.85 logMAR at age 13.5, when she developed nystagmus. Retinal function deteriorated over those 4 years from a recordable rod-cone dysfunction with an electronegative waveform at age 9 to being non-recordable at age 13.5 years. VFs were constricted to less than 5 degrees at that time. Fundus examination did not show a maculopathy at her last visit. Fd-OCT revealed defined inner retinal layers, but thinned outer retinal layers and RPE layer. Inner and outer segment layers were not distinguishable. Multiple small round ‘deposits’ were localized adjacent and anterior to Bruch's membrane.
BBS10: case 2621, heterozygous mutation in C91W and A474fs483X (Fig. 3C)
This patient had clinodactyly, obesity, polycystic kidneys, learning disability and retinopathy and was diagnosed at age 8. VA dropped from 0.5 log-MAR at the first visit to 1.6 logMAR at age 13.6. Nystagmus occurred from age 10 onwards. Fundus examination demonstrated a moderate maculopathy, optic atrophy and thinned retinal vessels. Retinal layer analysis demonstrated internal limiting membrane wrinkling, intact but thinned inner retinal layers and an enlarged space within the outer nuclear layer. Inner and/or outer segment layers appeared to be present within the fovea. ‘Deposits’ above Bruch's membrane were similar to the patient described above.
4. Discussion
High-speed and high-resolution Fd-OCT allowed a detailed analysis of retinal layers in all patients with BBS tested. Unstable fixation or nystagmus did not interfere with image quality. The common characteristics of the macular scans were preserved inner retinal layers and outer nuclear layer, disrupted inner and outer segment layer and thinned RPE. Reduced, but present photoreceptor inner or outer segment layer within the foveal area was identifiable in most of the patients. A differentiation between inner and outer segment layers was not always possible due to reduced thickness and non-identifiable connecting cilium. Data from younger patients with less advanced retinopathy might yield insight into inner segment/outer segment photoreceptor layer changes including the connecting cilia.
No significant genotype–phenotype correlation was observed. To our knowledge, this is the first in vivo high-resolution retinal layer analysis in patients with mutations in BBS1 or BBS10. Preservation of inner retinal layers agrees with the recent in-vivo study by Azari et al. (2006) and with histopathological studies of a patient with BBS (Lahav et al., 1977; Runge, Calver, Marshall, & Taylor, 1986). Our observation supports a retinal electron microscopic study from an 18-year old patient demonstrating nuclei and inner segments but no outer segments within the macular area (Lahav et al., 1977). Animal models of Bbs4 mice showed a general retinopathy, which was detected earlier with histopathology than with ophthalmoscopy. ERGs in these animals revealed a cone-rod dysfunction by week 4 (Eichers et al., 2006). Histopathology demonstrated thinned photoreceptors with loss of nuclei and shortening of inner and outer segment layer.
Deposits adjacent and anterior to Bruch's membrane, which were more often evident in patients with mutations in BBS10 than with mutations in BBS1, might be similar to the abnormal material described in patients with RP (Brosnahan, Kennedy, Converse, Lee, & Hammer, 1994; Duvall, McKechnie, Lee, Rothery, & Marshall, 1986). Those data from patients with autosomal dominant RP depict deposits of abnormal material between the RPE and inner collagenous layer of Bruch's membrane containing PAS positive material, lipids, calcium and iron. Whether the deposits are signs of more advanced retinopathy or a faster rate of retinal layer degeneration is not known. Clinical assessment of a larger patient cohort with mutations in BBS1 and BBS10 indicate a parallel rate of progression in the two groups, but a more advanced deterioration at an earlier age in the BBS10 group (Héon, Gerth, Elia, & Munier, 2007).
Internal limiting membrane wrinkling, which is not an uncommon sign in early stages of retinal dystrophies, was identified in 3 patients. The macula in those patients showed an enlarged space within the outer nuclear layer or the inner and outer layers in the foveal area. This effect might be caused by retinal thinning and ‘collapsing’ of retinal layers.
In summary, Fd-OCT allowed excellent imaging of retinal changes seen in BBS and did not reveal any significant difference between patients carrying mutation in BBS1 or BBS10. Intraretinal ‘deposits’ seen mostly in BBS10 are possibly related to retinal re-organization. More advanced imaging techniques such as adaptive optics OCT (Zawadzki, Jones et al., 2005) could help further characterize the nature of retinal changes in advanced retinal dystrophy associated with BBS mutations. The in-vivo imaging of degenerative changes related to BBS will contribute to a better understanding of the processes, which leads to photoreceptor loss. Characterization of those changes is important to best design novel therapeutic approaches.
Acknowledgments
The study was supported by FFB Canada (E.H.), the Sick Kids Research Institute's Restracomp Fund (C.G.), NIH (NEI 014743) (J.S.W.), the Albrecht Fund (J.S.W.) in collaboration with Bioptigen, Inc. We thank Yesmino Elia for study coordination, Richard Weleber for providing the Visual Field analysis program, the Héon lab for genotyping, Tom Wright and Fil Cortese for help with data analysis.
Footnotes
Abbreviations: BBS, Bardet–Biedl Syndrome; ERG, electroretinogram; Fd-OCT, Fourier-domain optical coherence tomography; RPE, retinal pigment epithelium; VA, visual acuity.
References
- Alam S, Zawadzki R, Choi S, Gerth C, Park S, Morse L, et al. Clinical application of rapid serial fourier-domain optical coherence tomography for macular imaging. Ophthalmology. 2006;113(8):1425–1431. doi: 10.1016/j.ophtha.2006.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anger EM, Unterhuber A, Hermann B, Sattmann H, Schubert C, Morgan JE, et al. Ultrahigh resolution optical coherence tomography of the monkey fovea. Identification of retinal sublayers by correlation with semithin histology sections. Experimental Eye Research. 2004;78:1117–1125. doi: 10.1016/j.exer.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet–Biedl syndrome. Nature. 2003;425(6958):628–633. doi: 10.1038/nature02030. [DOI] [PubMed] [Google Scholar]
- Azari AA, Aleman TS, Cideciyan AV, Schwartz SB, Windsor EA, Sumaroka A, et al. Retinal disease expression in Bardet–Biedl syndrome-1 (BBS1) is a spectrum from maculopathy to retina-wide degeneration. Investigative Ophthalmology & Visual Science. 2006;47(11):5004–5010. doi: 10.1167/iovs.06-0517. [DOI] [PubMed] [Google Scholar]
- Bardet G. Sur un syndrome d'obésité congénitale avec polydactylie et réunite pigmentaire (contribution á l'etude des formes cliniques de l'obésite hypophysaire) Thése de Paris. 1920;170:107. [Google Scholar]
- Beales PL, Badano JL, Ross AJ, Ansley SJ, Hoskins BE, Kirsten B, et al. Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet–Biedl syndrome. American Journal of Human Genetics. 2003;72(5):1187–1199. doi: 10.1086/375178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biedl A. Ein Geschwisterpaar mit adiposo-genitaler dystrophie. Dtsch Med Wschr. 1922;48:1630. [Google Scholar]
- Brosnahan DM, Kennedy SM, Converse CA, Lee WR, Hammer HM. Pathology of hereditary retinal degeneration associated with hypobetalipoproteinemia. Ophthalmology. 1994;101(1):38–45. doi: 10.1016/s0161-6420(94)31358-3. [DOI] [PubMed] [Google Scholar]
- Churchill DN, McManamon P, Hurley RM. Renal disease-a sixth cardinal feature of the Laurence–Moon–Biedl syndrome. Clinical Nephrology. 1981;16(3):151–154. [PubMed] [Google Scholar]
- Duvall J, McKechnie NM, Lee WR, Rothery S, Marshall J. Extensive subretinal pigment epithelial deposit in two brothers suffering from dominant retinitis pigmentosa. A histopathological study. Graefes Archive for Clinical and Experimental Ophthalmology. 1986;224(3):299–309. doi: 10.1007/BF02143075. [DOI] [PubMed] [Google Scholar]
- Eichers ER, Abd-El-Barr MM, Paylor R, Lewis RA, Bi W, Lin X, et al. Phenotypic characterization of Bbs4 null mice reveals age-dependent penetrance and variable expressivity. Human Genetics. 2006;120(2):211–226. doi: 10.1007/s00439-006-0197-y. [DOI] [PubMed] [Google Scholar]
- Ferris FL, 3rd, Kassoff A, Bresnick GH, Bailey I. New visual acuity charts for clinical research. American Journal of Ophthalmology. 1982;94(1):91–96. [PubMed] [Google Scholar]
- Gerth C, Zawadzki RJ, Choi SS, Keltner JL, Park SS, Werner JS. Visualization of lipofuscin accumulation in Stargardt macular dystrophy by high-resolution Fourier-domain optical coherence tomography. Archives of Ophthalmology. 2007;125(4):575. doi: 10.1001/archopht.125.4.575. [DOI] [PubMed] [Google Scholar]
- Gloesmann M, Hermann B, Schubert C, Sattmann H, Ahnelt PK, Drexler W. Histologic correlation of pig retina radial stratification with ultrahigh-resolution optical coherence tomography. Investigative Ophthalmology & Visual Science. 2003;44(4):1696–1703. doi: 10.1167/iovs.02-0654. [DOI] [PubMed] [Google Scholar]
- Héon E, Gerth C, Elia Y, Munier F. Ocular phenotype comparison between patients with Bardet–Biedl Syndrome with identified BBS1 and BBS10 mutations. Investigative Ophthalmology & Visual Science. 2007:3698. ARVO E-Abstract. [Google Scholar]
- Lahav M, Albert DM, Buyukmihci N, Jampol L, McLean EB, Howard R, et al. Ocular changes in Lawrence Moon Bardet Biedl Syndrome: a clinical and histopathologic study of a case. Advances in Experimental Medicine and Biology. 1977;77:51–84. doi: 10.1007/978-1-4899-5010-9_7. [DOI] [PubMed] [Google Scholar]
- Marmor MF, Holder GE, Seeliger MW, Yamamoto S. Standard for clinical electroretinography (2004 update) Documenta Ophthalmologica. 2004;108(2):107–114. doi: 10.1023/b:doop.0000036793.44912.45. [DOI] [PubMed] [Google Scholar]
- Nassif N, Cense B, Park B, Pierce M, Yun S, Bouma B, et al. In vivo high-resolution video-rate spectral-domain optical coherence tomography of the human retina and optic nerve. Optics Express. 2004;12:367–376. doi: 10.1364/opex.12.000367. [DOI] [PubMed] [Google Scholar]
- Ross AJ, May-Simera H, Eichers ER, Kai M, Hill J, Jagger DJ, et al. Disruption of Bardet–Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nature Genetics. 2005;37(10):1135–1140. doi: 10.1038/ng1644. [DOI] [PubMed] [Google Scholar]
- Runge P, Calver D, Marshall J, Taylor D. Histopathology of mitochondrial cytopathy and the Laurence–Moon–Biedl syndrome. British Journal of Ophthalmology. 1986;70(10):782–796. doi: 10.1136/bjo.70.10.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachat AP, Maumenee IH. Bardet–Biedl syndrome and related disorders. Archives of Ophthalmology. 1982;100(2):285–288. doi: 10.1001/archopht.1982.01030030287011. [DOI] [PubMed] [Google Scholar]
- Schulze-Bonsel K, Feltgen N, Burau H, Hansen L, Bach M. Visual acuities “hand motion” and “counting fingers can be quantified with the freiburg visual acuity test. Investigative Ophthalmology & Visual Science. 2006;47(3):1236–1240. doi: 10.1167/iovs.05-0981. [DOI] [PubMed] [Google Scholar]
- Stoetzel C, Laurier V, Davis EE, Muller J, Rix S, Badano JL, et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nature Genetics. 2006;38(5):521–524. doi: 10.1038/ng1771. [DOI] [PubMed] [Google Scholar]
- Sun W, Gerth C, Maeda A, Lodowski DT, Van Der Kraak L, Saperstein DA, et al. Novel RDH12 mutations associated with Leber congenital amaurosis and cone-rod dystrophy: Biochemical and clinical evaluations. Vision Research. 2007;47:2055–2066. doi: 10.1016/j.visres.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weleber R, Tobler W. Computerized quantitative analysis of kinetic visual fields. American Journal of Ophthalmology. 1986;101(4):461–468. doi: 10.1016/0002-9394(86)90648-3. [DOI] [PubMed] [Google Scholar]
- Wojtkowski M, Leitgeb R, Kowalczyk A, Bajraszewski T, Fercher AF. In vivo human retinal imaging by Fourier domain optical coherence tomography. Journal of Biomedical Optics. 2002;7(3):457–463. doi: 10.1117/1.1482379. [DOI] [PubMed] [Google Scholar]
- Wojtkowski M, Srinivasan V, Fujimoto JG, Ko T, Schuman JS, Kowalczyk A, et al. Three-dimensional retinal imaging with high-speed ultrahigh-resolution optical coherence tomography. Ophthalmology. 2005;112(10):1734–1746. doi: 10.1016/j.ophtha.2005.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtkowski M, Srinivasan V, Ko T, Fujimoto JG, Kowalczyk A, Duker J. Ultrahigh-resolution, high-speed, Fourier domain optical coherence tomography and methods for dispersion compensation. Optics Express. 2004;12:2402–2422. doi: 10.1364/opex.12.002404. [DOI] [PubMed] [Google Scholar]
- Zawadzki RJ, Fuller AR, Wiley DF, Hamann B, Choi SS, Werner JS. Adaptation of a support vector machine algorithm for segmentation and visualization of retinal structures in volumetric optical coherence tomography data sets. Journal of Biomedical Optics. 2007;12(4):041206-1–041206-7. doi: 10.1117/1.2772658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawadzki RJ, Bower B, Zhao M, Sarunic MV, Laut S, Werner JS, et al. Exposure time dependence of image quality in high-speed retinal in vivo Fourier domain OCT. Proceedings of SPIE. 2005;5688:45–52. [Google Scholar]
- Zawadzki RJ, Jones SM, Olivier SS, Zhao M, Bower BA, Izatt JA, et al. Adaptive-optics optical coherence tomography for high-resolution and high-speed 3D retinal in-vivo imaging. Optics Express. 2005;13:8532–8546. doi: 10.1364/opex.13.008532. [DOI] [PMC free article] [PubMed] [Google Scholar]