

Abstract
Populations of the teleost fish Fundulus heteroclitus inhabit and have adapted to highly polluted Superfund sites that are contaminated with persistent toxic chemicals. Populations inhabiting different Superfund sites provide independent contrasts for studying mechanisms of toxicity and resistance due to exposure to environmental pollutants. To identify both shared and unique responses to chronic pollutant exposure, liver, metabolic gene expression in F. heteroclitus populations from each of three Superfund sites (New Bedford Harbor, MA, Newark Bay, NJ, and Elizabeth River, VA) were compared to two flanking reference site populations (9 populations in total). In comparisons to their two clean reference sites, the three Superfund sites had 8 to 32% of genes with altered expression patterns. Between any two Superfund populations, up to 9 genes (4%) show a conserved response, yet among all three populations, there was no gene which had a conserved, altered pattern of expression. Across all three Superfund sites in comparison to all six reference populations, the most significant gene was fatty acid synthase. Fatty acid synthase is involved in the storage of excess energy as fat, and its lesser expression in the polluted populations suggests that the polluted populations may have limited energy stores. In contrast to previous studies of metabolic gene expression in F. heteroclitus, body weight was a significant covariate for many of the genes which could reflect accumulation and different body burdens of pollutants. Overall, the altered gene expression in these populations likely represents both induced and adaptive changes in gene expression.
Keywords: Fundulus heteroclitus, pollution, natural populations, Superfund site, gene expression, microarray
Introduction
F. heteroclitus are widely distributed estuarine fish found in polluted waters. Liver gene expression was measured in three independent, polluted populations to determine similarities and differences in gene expression. These populations inhabit three different Superfund sites (sites identified by the U. S. Environmental Protection Agency (EPA) that contain high levels of a variety of lipophilic, persistent and toxic contaminants and are worthy of remediation using Federal funds) and are exposed to some of the highest concentrations of aromatic hydrocarbon pollutants of any vertebrate species (Wirgin and Waldman 2004). The overall goal is to elucidate the molecular mechanisms underlying biological effects of environmental chemical exposure. Such a mechanistic understanding is important both to understand the responses of animals to chronic chemical exposure and to identify molecular markers of susceptibility associated with increased risk in populations of animals, including humans.
The most northern F. heteroclitus population examined inhabits New Bedford Harbor, MA, a federal Superfund site that is heavily contaminated with polychlorinated biphenyls (PCBs) and other halogenated aromatic hydrocarbons (HAHs) (Pruell et al. 1990, Lake et al. 1995). F. heteroclitus from this site have accumulated extraordinarily high concentrations of PCBs (272 μg/g dry weight) and have reduced sensitivity to aryl hydrocarbon receptor agonists compared with fish collected from a reference site (Bello et al. 2001). The reduced sensitivity is systemic and exhibits compound-specific differences in magnitude. These fish also exhibit heritable resistance to toxic effects of planar halogenated aromatic hydrocarbons (PHAHs), including 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and PCBs (Bello et al. 2001).
A F. heteroclitus population from Newark Bay, New Jersey also was examined. Newark Bay is part of a large, highly urbanized estuarine system that is chemically impacted from wastewater treatment plant discharge, combined sewer overflow, urban runoff, petrochemical factories, illegal dumping and accidental spills. Contaminants include polycyclic aromatic hydrocarbons (PAHs), PCBs, pesticides, and metals (Long et al. 1995, Wolfe et al. 1996). Both adult and larval fish from Newark Bay have altered response to CYP1A inducers that is persistent and possibly heritable (Elskus et al. 1999). In addition, a subpopulation of F. heteroclitus from Newark Bay was found to be resistant to TCDD: they did not exhibit TCDD-dependent lesions or death (Prince and Cooper 1995).
The third polluted F. heteroclitus population examined was from the Southern Branch of the Elizabeth River in Virginia. This Superfund site is highly contaminated with PAHs. PAHs occur in extremely high concentrations in the sediments (2200 mg/kg dry weight) in the vicinity of a site where creosote historically was used to treat wood for use in the marine environment (Greaves 1990, Alden and Winfield 1995). Grossly visible liver lesions were present in 93% of the F. heteroclitus collected at this site and 33% of the fish had liver cancers (Vogelbein et al. 1990). F. heteroclitus from sites with low levels of PAHs (730 and 35 times less) in the sediments showed no indication of disease (Vogelbein et al. 1990). In the majority of hepatic neoplasms, over-expression and altered patterns of the xenobiotic transporter P-glycoprotein were observed (Cooper et al. 1999). Like New Bedford Harbor and Newark Bay fish, Elizabeth River F. heteroclitus from highly polluted sites are resistant to CYP1A inducers (Vogelbein et al. 1996), and they appear to have heritable tolerance to contaminated sediments (Ownby et al. 2002).
To begin to understand what changes allow these three F. heteroclitus populations to inhabit and even thrive in these highly polluted environments, metabolic gene expression patterns were measured. Gene expression often is altered as a result of toxicant exposure (Thomas et al. 2001, Hamadeh et al. 2002) and thus is a sensitive, measurable endpoint for toxicity that can serve as an early warning of compromised health. Natural populations exposed to complex suites of pollutants likely will show a polygenic response involving changes in gene expression, and microarrays can be used to discover which genes respond. Fundulus cDNA microarrays were used to compare metabolic gene expression patterns in the livers of individuals from nine populations: three independent, polluted Superfund populations and two genetically similar, reference populations flanking each Superfund population (9 populations in total, Table 1).
Table 1. Sample locations.
Site descriptions (Reference or Superfund) and geographical locations for F. heteroclitus populations.
| Reference/Superfund | Location | Latitude (N) | Longitude (W) |
|---|---|---|---|
| Reference | Sandwich, MA | 41°44.0′ | 70°23.0′ |
| Superfund | New Bedford, MA | 41°34.0′ | 70°54.9′ |
| Reference | Point Judith, RI | 41°21.7′ | 71°28.9′ |
| Reference | Clinton, CT | 41°15.3′ | 72°32.8′ |
| Superfund | Newark, NJ | 40°41.2′ | 74°06.7′ |
| Reference | Tuckerton, NJ | 39°32.2′ | 74°19.4′ |
| Reference | Magotha, VA | 37°10.6′ | 75°56.5′ |
| Superfund | Elizabeth River, VA | 36°48.5′ | 76°17.7′ |
| Reference | Manteo, NC | 35°53.8′ | 75°36.9′ |
Materials and Methods
Field fish collection, care and sampling
F. heteroclitus were collected using minnow traps in the spring of 2003. Collection sites included three Superfund Sites (New Bedford Harbor, MA; Newark Bay, NJ; Elizabeth River, VA, [EPA ID MAD980731335, NJD980528996, and VAD990710410, respectively]), and six reference sites. Pairs of reference sites were located north and south of each polluted site (Sandwich, MA and Pt. Judith, RI for New Bedford Harbor, MA; Tuckerton NJ and Clinton, CT for Newark, NJ: and Magnatha, VA and Manteo, NC for Elizabeth River, VA; Table 1). Fish were kept in a common re-circulating aquarium system with controlled temperature and salinity of 20°C and 15 ppt salinity for four months before experiments in order to minimize physiologically induced differences, particularly differences due to reproductive status. Thus, six weeks after field collection, fish were subjected to pseudo-winter (6:18, light: dark cycle) for four weeks, then maintained for 6 weeks with a light cycle of 16:8, light:dark. After the pseudo-winter, Fundulus came into reproductive condition and spawned, and reproductive tissues regressed. The reproductive tissues were in regression when fish were assayed. Effluent from tanks was passed through an activated charcoal filter and 20% water changes were performed weekly. Tanks were cleaned and fish checked for health status on a daily basis. Fish were fed once daily a 33% mixture of brine shrimp flake, blood meal flake and Spirulina flake (FOD, Aquatic Ecosystems).
Populations and individuals within a population were chosen randomly for sacrifice and sampled in the morning and early afternoon in order to minimize physiological changes due to diurnal cycles. Five fish from each of 9 populations were sacrificed by cervical dislocation. Fish were mixed sex and ranged in weight, with an average weight of 5.6 ± 0.41g. Sex was not a significant variable in the mixed models used to discern treatment (location) effects discussed below, and analysis of covariance (ANCOVA) with log10 body weight as the covariate was used to control for effects of weight in the analyses. Livers were dissected, immediately frozen in a dry ice/ethanol bath and stored at −80° C for subsequent RNA extraction. This experiment was performed according to an approved Institutional Animal Care and Use Committee at North Carolina State University.
RNA extraction, amplification, hybridization, scanning
Total RNA was isolated from each liver using a guanidinium thiocyanate buffer (Chomczynski and Sacchi 1987) followed by purification using the Qiagen RNeasy Mini kit (Qiagen Inc., Valencia, CA, USA) according to the manufacturer's protocols. Purified RNA was quantified with a spectrophotometer, and RNA quality was assessed by gel electrophoresis. RNA for hybridization was prepared by one round of amplification (aRNA) using Ambion's Amino Allyl MessageAmp aRNA Kit to form copy template RNA by T7 amplification. Amino-allyl UTP was incorporated into targets during T7 transcription, and resulting amino-allyl aRNA was coupled to Cy3 and Cy5 dyes (GE Healthcare, Piscataway, NJ, USA).
Labeled aRNA samples (5 pmol/ul) were hybridized to slides in 12 ul of hybridization buffer (50% formamide buffer, 5× SSPE, 1% sodium dodecyl sulfate, 0.2 mg/ml bovine serum albumin, 1 mg/ml denatured salmon sperm DNA (Sigma), and 1 mg/ml RNAse free poly(A) RNA (Sigma) for 42.5 hours at 42° C. Slides were prepared by blocking according to the manufacturer's recommendations with an additional treatment of 66 mM sodium borohydride to minimize background autofluorescence (Raghavachari et al. 2003). After hybridization, non-specifically bound probe was washed off with SSC and the slides were spun dry and scanned using a ScanArray Express 4000 (Perkin Elmer). Resulting 16 bit Tiff Images were quantified using IconoClust® (CLONDIAG, Jena, Germany) spotfinding software.
Metabolic arrays
Amplified cDNA sequences for 384 metabolic genes from F. heteroclitus heart and liver libraries were spotted onto CodeLink activated slides (GE Healthcare, Piscataway, NJ) at the University of Miami core microarray facility (slides were the kind gift of D. Crawford). Each slide contained 4 spatially separated arrays, and each array contained 4 replicates of 384 spots (genes) including controls. Printed cDNAs encode essential proteins for cellular metabolism based on KEGG (Kyoto Encyclopedia of Genes and Genomes; http://www.genome.ad.jp/kegg). Sequence information, annotation and gene ontology are available for Fundulus on the FunnyBase website http://genomics.rsmas.miami.edu/sandbox22/sandbox22.html.
Not all 384 gene-spots were analyzed. Unanalyzed spots included negative controls (random genomic amplification or Ctenophore specific cDNAs) and genes that either saturated the photomultiplier tube or had signals less than the negative controls. The number of genes examined were 296, 260, and 239 for the New Bedford Harbor, Newark Bay, and Elizabeth River comparisons, respectively, and the analysis combining all locations (9 populations) used 216 common genes.
Experimental design
A loop design was used for the microarray hybridizations where each sample is hybridized to 2 arrays using both Cy3 and Cy5 labeled fluorophores (Kerr and Churchill 2001). In this experiment, each loop consisted of Cy3 and Cy5 labeled liver aRNA from 5 individuals from a polluted site (P) hybridized to Cy3 and Cy5 labeled liver aRNA from 5 individuals from each of 2 adjacent reference sites (R1 and R2), for a total among the 3 loops of 45 individuals hybridized to 45 microarrays. Each array had different combinations of individuals, and each loop formed was P1→ R1.1→ R2.1→ P2 → R1.2→ R2.2→ P3→ R1.3→ R2.3→ P4→ R1.4→ R2.4→ P5 → R1.5→ R2.5→ P1 where each arrow represents a separate hybridization (array) with the individual at the base of the arrow labeled with Cy3 and the individual at the head of the arrow labeled with Cy5.
Statistical analysis
Log2 measures of gene expression were normalized using a linear mixed model in SAS (JMP v6.0.2 with a microarray platform) to remove the effects of dye (fixed effect) and array (random effect) following a joint regional and spatial Lowess transformation in MAANOVA Version 0.98.8 for R to account for both intensity and spatial bias (Wu et al. 2003). The model was of the form yij = μ + Ai + Dj + (AxD)ij + eij, where, yij is the signal from the ith array with dye j, μ is the sample mean, Ai and Dj are the overall variation in arrays (1-15) and dyes (Cy3 and Cy5), (AxD)ij is the array x dye interaction and eij is the stochastic error (Wolfinger et al. 2001). When comparing all nine sites (3 polluted and 6 reference populations hybridized across 3 separate loops), a loop term (Ll) was added in the mixed model normalization according to the model yijl = μ + Ai + Dj + (AxD)ij + Ll + eijl. Residuals from these models were used for gene-by-gene analyses (below).
Normalized data were modeled by analysis of covariance (ANCOVA) with log10 body weight as the covariate on a gene-by-gene basis using a linear mixed model in SAS using PROC MIXED. To test for a treatment effect (effect of chronic exposure to pollution), normalized data (residuals from the mixed model normalization) were modeled using treatment and dye as fixed effects and array and spot nested in array as random effects according to the model rijkm = μ + Ai + Dj + Tk + S(A)mi + eijkm, where Tk is the kth treatment (polluted or reference) and S(A)mi is the mth spot nested in array. To compare all nine populations, population was used as a fixed effect in the gene-by-gene model according to the model rijgm = μ + Ai + Dj + Pg+ S(A)mi + eijgm. To examine individual variation among locations, the mixed model gene-by-gene analysis was analyzed by location and used individual and dye as fixed effects according to the model rijhm = μ + Ai + Dj + Ih + S(A)mi + eijhm.
Mixed model analyses were performed for each loop or combined loop analysis, and a nominal p-value cut-off for significant genes of p < 0.01 was used. Using this p-value reveals more genes that may be differentially expressed at the risk of identifying genes that may be false positives. Genes identified as also being significant after false positives are reduced by a more stringent multiple comparison correction were analyzed using a Bonferroni correction (p = 0.05).
Results
Differences within and among populations
To establish the variation within a population, a mixed model was used to test for variation in gene expression among individuals within each of the nine populations. This analysis showed that gene expression varied significantly among individuals within a population for up to 49% of the genes. This large variation among individuals within a population is similar to previous studies of Fundulus metabolic gene expression (Oleksiak et al. 2005, Fisher and Oleksiak 2007).
A mixed model also was used to establish variation among all nine populations. This analysis showed that 15-40% of the genes have significant differences in gene expression among any two of the nine populations (p < 0.01). The fewest differences were found between the New Bedford Harbor and Pt. Judith, RI populations, and the most differences were found between the New Bedford Harbor and Elizabeth River populations. These pair-wise comparisons did not consider geography, and geographically, the New Bedford Harbor and Pt. Judith, RI sites are two of the closest populations and the New Bedford Harbor and Elizabeth River sites two of the most distant. Because these Fundulus populations show a significant correlation between genetic and geographical distance (Adams et al., 2006), the fewer differences between New Bedford Harbor and Pt. Judith individuals likely are due to genetic similarities. Similarly, the differences between New Bedford Harbor and Elizabeth River reflect the steep clinal genetic variation between northern and southern populations of Fundulus (Smith et al. 1998, Adams et al. 2006).
In order to obviate problems of neutral genetic drift associated with the steep cline, gene expression in a Superfund population was tested specifically for statistical differences from gene expression in two adjacent reference populations (Table 1). That is, gene expression was tested for statistical differences in each Superfund site (N= 5) as compared to the northern and southern reference population (N= 10). This comparison captures two sources of variation: variation associated with outbred individuals and, most importantly, variation associated with neutral genetic drift due to the steep northern-southern cline. This ensures that differences among Superfund and reference populations are not simply due to genetic distances because the reference populations are geographically more distant from each other than either is to the respective Superfund population.
A linear mixed model was used to test for differences between each Superfund population versus its two surrounding reference sites (Table 2) (Wolfinger et al. 2001). The Elizabeth River, VA population has the smallest number and smallest percentage of differentially expressed genes compared to its two reference populations (20 genes or 8%, p < 0.01). The New Bedford Harbor, MA population has 47 genes that are differently expressed (16%, p < 0.01). The Newark Bay, NJ population has the most differently expressed genes (82 genes or 32%, p < 0.01). These numbers are greater than the 2-3 genes expected by chance (i.e. 1% of analyzed genes). After a Bonferroni correction for multiple comparisons, 2, 8 and 16 genes are significantly different in Elizabeth River, New Bedford Harbor, and Newark Bay populations, respectively.
Table 2. Significant differently expressed genes.
Genes significantly differently expressed at the three Superfund sites, New Bedford Harbor (NBH), Newark Bay (NB) and Elizabeth River (ER). Gene, pathway, function, p-value and relative fold-difference are reported. A gene with a positive fold-difference is more highly expressed in the reference populations, and a gene with a negative fold-difference is more highly expressed in the Superfund population.
| Site | Gene | Pathway | Function | P-value | Fold Difference |
|---|---|---|---|---|---|
| NBH | Cytochrome c oxidase polypeptide VIIA | Oxidative phosphorylation | Energy Metabolism | 7.42E-05 | 1.27 |
| ATP synthase H+ transporting mitochondrial F0 complex subunit f soform 2 | Oxidative phosphorylation | 1.20E-03 | 1.25 | ||
| ATP synthase H+ transporting mitochondrial F0 complex subunit d | Oxidative phosphorylation | 4.13E-03 | 1.15 | ||
| NADH-ubiquinone oxidoreductase MNLL subunit | Oxidative phosphorylation | 8.69E-03 | 1.14 | ||
| NADH-ubiquinone oxidoreductase 19 kDa subunit | Oxidative phosphorylation | 4.82E-03 | 1.14 | ||
| NADH-ubiquinone oxidoreductase chain 3 | Oxidative phosphorylation | 5.14E-03 | -1.18 | ||
| NADH dehydrogenase subunit 3 | Oxidative phosphorylation | 3.92E-04 | -1.31 | ||
| PEP carboxykinase phosphoenolpyruvate carboxykinase | Carbohydrate biosynthesis | 6.16E-03 | 1.19 | ||
| Aldo-keto reductase family 1 member D1 | Carbohydrate metabolism | 2.33E-03 | 1.16 | ||
| Alcohol dehydrogenase [NADP+] | Carbohydrate metabolism | 3.99E-03 | 1.16 | ||
| Aldo-keto reductase family 1 member A1 | Carbohydrate metabolism | 5.84E-03 | 1.13 | ||
| Isocitrate dehydrogenase 2 | Carbohydrate metabolism | 7.82E-04 | -1.28 | ||
| Glutaryl-coenzyme A dehydrogenase | Fatty acid/lipid metabolism | 4.52E-03 | 1.15 | ||
| Long-chain-acyl-CoA dehydrogenase | Fatty acid/lipid metabolism | 4.11E-03 | -1.15 | ||
| Phosphoglycerate kinase | Glycolysis | 7.44E-03 | 1.13 | ||
| Fructose-1 6-bisphosphatase | Glycolysis | 2.96E-04 | -1.23 | ||
| 6-phosphogluconate dehydrogenase | Pentose shunt, electron transport | 1.07E-04 | -1.57 | ||
| 6-phosphogluconolactonase | Pentose-phosphate shunt | 4.89E-03 | 1.15 | ||
| Diacylglycerol synthase | Phospholipid metabolism | 8.81E-05 | -1.53 | ||
| Glutaminase, kidney isoform | Glutamine catabolism | Catabolism | 7.79E-04 | 1.21 | |
| Trypsin precursor | Peptidase | 2.29E-03 | -1.29 | ||
| Isoamyl acetate-hydrolyzing esterase | Probable lipase | 3.41E-03 | 1.13 | ||
| 4-hydroxyphenylpyruvate dioxygenase | Tyrosine catabolic process | 9.78E-04 | 1.22 | ||
| Ubiquitin | Ubiquitin-dependent protein catabolism | 8.58E-03 | -1.14 | ||
| Phenylalanine-4-hydroxylase | Amino acid biosynthetic process | Biosynthesis | 4.03E-03 | 1.16 | |
| Glutamate decarboxylase | Catalyzes the production of GABA | 6.04E-03 | 1.32 | ||
| 3-hydroxy-3-methylglutaryl-CoA reductase | Cholesterol biosynthesis | 3.01E-05 | 1.36 | ||
| Fatty acid synthase | Fatty acid synthesis | 8.50E-04 | 1.46 | ||
| 3-oxoacyl-[acyl-carrier protein] reductase | Fatty acid synthesis | 8.18E-04 | 1.20 | ||
| Isopentenyl-diphosphate delta-isomerase | Isoprenoid biosynthesis | 9.63E-06 | 1.67 | ||
| Ornithine decarboxylase antizyme short isoform | Polyamine biosynthesis | 2.75E-03 | -1.15 | ||
| Cytochrome P450 2N2 | Arachidonic acid metabolism | Regulation/Signaling | 8.22E-03 | 1.18 | |
| Cytochrome P450 2C29 | Arachidonic acid metabolism | 8.29E-04 | -1.27 | ||
| Acyl-CoA-binding protein | Lipid binding /transport | 2.09E-03 | 1.30 | ||
| Fatty acid binding protein H6-isoform | Lipid binding, transporter activity | 9.05E-03 | 1.13 | ||
| Thioredoxin | Signal transduction | 1.32E-03 | 1.18 | ||
| Hepatocyte nuclear factor 4-alpha | Transcription factor | 2.73E-04 | 1.17 | ||
| O-methyltransferase | Inactivation of catecholamine neurotransmitters and catechol hormones | Other | 1.79E-04 | 1.31 | |
| Dimethylaniline monooxygenase [N-oxide forming] 3 | Xenobiotic metabolism | 9.29E-03 | -1.15 | ||
| Protein-glutamine gamma-glutamyltransferase | Coagulation factor XIII A chain | 9.30E-05 | -1.38 | ||
| Superoxide dismutase [Cu-Zn] | Destroys radicals | 5.58E-03 | 1.14 | ||
| GDP-mannose 4 6 dehydratase | Leukocyte adhesion | 6.10E-03 | -1.12 | ||
| Dehydrogenase/reductase SDR family member 2 | May inhibit cell replication | 1.38E-03 | -1.16 | ||
| Glutathione peroxidase 4 (phospholipid hydroperoxidase) | Oxidoreductase Peroxidase | 4.99E-03 | 1.27 | ||
| 5-aminolevulinic acid synthase | Porphyrin metabolism | 4.22E-03 | -1.13 | ||
| Cytokine receptor common gamma chain precursor | Common interleukin receptor subunit | 3.05E-05 | 1.50 | ||
| Zona pellucida binding protein | Sperm binding | 3.47E-04 | -1.37 | ||
|
| |||||
| NB | ATP synthase H+ transporting mitochondrial F0 complex subunit f isoform 2 | Oxidative Phosphorylation | Energy Metabolism | 5.69E-05 | 2.28 |
| ATP synthase H+ transporting mitochondrial F1 complex epsilon subunit | Oxidative Phosphorylation | 1.62E-04 | 1.69 | ||
| Cytochrome c oxidase polypeptide VIIC | Oxidative Phosphorylation | 2.45E-04 | 1.66 | ||
| Cytochrome c oxidase polypeptide VIIA | Oxidative Phosphorylation | 1.90E-05 | 1.66 | ||
| NADH-ubiquinone oxidoreductase MNLL subunit | Oxidative Phosphorylation | 2.98E-04 | 1.57 | ||
| NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 1 | Oxidative Phosphorylation | 7.98E-05 | 1.50 | ||
| Ubiquinol-cytochrome c reductase complex 9.5 kDa protein | Oxidative Phosphorylation | 3.41E-03 | 1.44 | ||
| Cytochrome c oxidase polypeptide VIIb | Oxidative Phosphorylation | 2.56E-05 | 1.37 | ||
| Cytochrome-c oxidase chain VIc | Oxidative Phosphorylation | 2.75E-03 | 1.37 | ||
| Cytochrome c oxidase subunit VIIb | Oxidative Phosphorylation | 9.00E-04 | 1.32 | ||
| NADH-ubiquinone oxidoreductase B17 subunit | Oxidative Phosphorylation | 1.85E-03 | 1.21 | ||
| NADH dehydrogenase (ubiquinone) Fe-S protein 1 | Oxidative Phosphorylation | 6.94E-04 | 1.21 | ||
| Cytochrome c oxidase subunit VIIIb | Oxidative Phosphorylation | 1.04E-03 | 1.19 | ||
| ATP synthase gamma chain | Oxidative Phosphorylation | 3.45E-03 | -1.15 | ||
| ATP synthase alpha chain liver isoform | Oxidative Phosphorylation | 5.37E-03 | -1.19 | ||
| ATP synthase H+ transporting mitochondrial F1 complex gamma polypeptide 1 | Oxidative Phosphorylation | 5.92E-03 | -1.20 | ||
| NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 9 (39kD) | Oxidative Phosphorylation | 3.57E-04 | -1.20 | ||
| Ubiquinol-cytochrome c reductase core protein II | Oxidative Phosphorylation | 1.36E-03 | -1.21 | ||
| NADH-ubiquinone oxidoreductase ASHI subunit | Oxidative Phosphorylation | 1.40E-03 | -1.23 | ||
| NADH dehydrogenase subunit 1 | Oxidative Phosphorylation | 2.88E-03 | -1.25 | ||
| ATP synthase H+ transporting mitochondrial F0 complex subunit b isoform 1 | Oxidative Phosphorylation | 1.22E-04 | -1.30 | ||
| Cytochrome c oxidase subunit VIa precursor polypeptide 2 | Oxidative Phosphorylation | 9.77E-05 | -1.31 | ||
| ATP synthase beta-subunit | Oxidative Phosphorylation | 2.24E-05 | -1.39 | ||
| NADH dehydrogenase (ubiquinone) chain 2 | Oxidative Phosphorylation | 7.41E-04 | -1.42 | ||
| ATP synthase subunit B | Oxidative Phosphorylation | 5.55E-05 | -1.44 | ||
| NADH dehydrogenase subunit 5 | Oxidative Phosphorylation | 1.54E-04 | -1.45 | ||
| NADH-ubiquinone oxidoreductase chain 1 | Oxidative Phosphorylation | 6.36E-05 | -1.51 | ||
| NADH dehydrogenase subunit 4 | Oxidative Phosphorylation | 8.85E-04 | -1.51 | ||
| Cytochrome c oxidase copper chaperone | Copper ion transport | 6.03E-05 | 1.73 | ||
| Electron transfer flavoprotein beta-subunit | Electron transfer flavoprotein | 3.32E-05 | -1.43 | ||
| Acyl-CoA dehydrogenase, medium-chain specific | Fatty acid beta-oxidation | 9.72E-05 | -1.28 | ||
| Alanine aminotransferase | Gluconeogenesis | 1.73E-04 | 1.24 | ||
| Glucose-6-phosphatase | Glycogen metabolic process | 6.28E-06 | -1.88 | ||
| Phosphoglycerate mutase 1 | Glycolysis | 9.53E-03 | 1.19 | ||
| Fructose-1,6-bisphosphatase | Glycolysis | 1.04E-03 | -1.32 | ||
| 6-phosphogluconolactonase | Pentose-phosphate shunt | 5.03E-03 | 1.16 | ||
| ADP, ATP carrier protein, isoform T2 | Transport | 1.31E-05 | -1.56 | ||
| Succinate-CoA ligase (GDP-forming) | Tricarboxylic acid cycle | 2.71E-03 | 1.14 | ||
| Tricarboxylate transport protein | Tricarboxylic acid cycle | 3.50E-03 | -1.27 | ||
| Succinate-CoA ligase (GDP-forming) | Tricarboxylic acid cycle | 2.67E-03 | -1.30 | ||
| Transaldolase | Pentose phosphate pathway | 4.39E-03 | -1.53 | ||
| O-methyltransferase | Inactivation of catecholamine neurotransmitters and catechol hormones | Catabolism | 1.27E-03 | 1.41 | |
| Glutaminase, kidney isoform | Glutamine catabolic process | 9.25E-05 | 1.48 | ||
| Phospholipase A2 precursor | Lipid degradation | 6.51E-04 | 1.96 | ||
| Phospholipase A2 group VII | Lipid degradation | 7.42E-03 | 1.21 | ||
| Trifunctional enzyme beta subunit | Lipid metabolism | 1.18E-03 | 1.28 | ||
| Lipoprotein lipase | Lipid metabolism | 1.49E-04 | -1.67 | ||
| Cytochrome P450 7A1 | Lipid metabolism | 2.31E-04 | -2.11 | ||
| Long-chain-acyl-CoA dehydrogenase | Lipid metabolism; mitochondrial fatty acid beta-oxidation | 1.97E-03 | -1.34 | ||
| Telomerase-binding protein p23 | Lipid metabolism; prostaglandin biosynthesis | 1.46E-04 | 1.35 | ||
| Purine nucleoside phosphorylase | Nucleic acid metabolism | 7.93E-03 | 1.29 | ||
| Dihydropyrimidinase | Nucleotide metabolism | 7.97E-03 | 1.13 | ||
| Ubiquitin-like protein FUBI | Protein ubiquitination during ubiquitin-dependent protein catabolic process | 1.71E-03 | 1.32 | ||
| Ubiquitin conjugation factor E4 B | Protein ubiquitination during ubiquitin-dependent protein catabolic process | 2.52E-03 | -1.21 | ||
| Indoleamine 2,3-dioxygenase | Tryptophan catabolic process | 4.06E-03 | 1.25 | ||
| Tyrosine aminotransferase | Tyrosine catabolic process | 7.20E-04 | -1.34 | ||
| Cytochrome P450 8B1 | Bile acid synthesis | Biosynthesis | 6.52E-03 | -1.51 | |
| Ornithine decarboxylase antizyme, short isoform | Polyamine biosynthesis | 9.97E-03 | -1.24 | ||
| Elongation factor 1-alpha | Protein synthesis | 3.52E-03 | -1.28 | ||
| Elongation factor 1-gamma type 1 | Protein synthesis | 2.12E-03 | -1.33 | ||
| Acyl-CoA-binding protein | Lipid binding/transport | Regulation/Signaling | 8.60E-05 | 1.81 | |
| Glycogen synthase kinase-3 alpha | Protein Kinase | 5.69E-03 | 1.25 | ||
| Troponin I, cardiac muscle | Regulation of muscle contraction | 4.57E-03 | -1.25 | ||
| Troponin I, slow skeletal muscle | Regulation of muscle contraction | 1.44E-05 | -1.72 | ||
| Thioredoxin | Signal transduction | 2.61E-07 | 1.54 | ||
| Forkhead box P2 | Transcription factor | 3.98E-03 | 1.49 | ||
| StAR-related lipid transfer protein 13 | GTPase-activating protein for RhoA | 6.44E-03 | 1.21 | ||
| N-acetyllactosaminide beta-1,6-N-acetyl-glucosaminyltransferase | Acetylglucosaminyltrans ferase activity | Other | 3.97E-03 | 1.25 | |
| 2-oxoisovalerate dehydrogenase alpha subunit | Conversion of alpha-keto acids to acyl-CoA and CO(2) | 3.06E-03 | 1.24 | ||
| Protein disulfide isomerase A3 precursor | Rearrangement of S-S-bonds in proteins | 1.88E-06 | -1.60 | ||
| Dihydrolipoamide dehydrogenase | Dihydrolipoyl dehydrogenase activity | 1.57E-06 | 1.40 | ||
| Triacylglycerol lipase, hepatic precursor | Hydrolysis of phospholipids | 5.48E-06 | 1.76 | ||
| Cytochrome P450 2K2 | Lauric acid hydroxylase | 5.38E-03 | -1.34 | ||
| Cytochrome P450 1A | Xenobiotic metabolism | 2.11E-05 | 1.36 | ||
| Cytochrome P450 1A | Xenobiotic metabolism | 2.11E-03 | 1.32 | ||
| Phosphomannomutase 1 | Mannose metabolic process | 2.18E-03 | 1.52 | ||
| Dehydrogenase/reductase SDR family member 2 | May inhibit cell replication | 1.43E-05 | 1.37 | ||
| Glutathione peroxidase 4 | Oxidoreductase Peroxidase | 4.25E-03 | -1.30 | ||
| Phospholipid hydroperoxide glutathione peroxidase | Protects from radiation and oxidative damage | 9.07E-06 | -1.56 | ||
| Eukaryotic translation initiation factor 1A, X-chromosomal | Protein biosynthesis | 8.43E-03 | -1.19 | ||
| Cold inducible RNA-binding protein | Response to cold | 2.46E-05 | -1.48 | ||
| Triglyceride lipase triacylglycerol | Triglyceride hydrolysis | 5.94E-03 | 1.29 | ||
|
| |||||
| ER | NADH-ubiquinone oxidoreductase MNLL subunit | Oxidative Phosphorylation | Energy Metabolism | 4.98E-03 | -1.15 |
| Cytochrome c oxidase polypeptide VIIC | Oxidative Phosphorylation | 7.78E-03 | -1.16 | ||
| Isocitrate dehydrogenase 1 | Carbohydrate metabolism | 5.29E-03 | -1.23 | ||
| Cystathionine-beta-synthase | Cysteine metabolic process | 6.41E-05 | -1.30 | ||
| Alanine aminotransferase | Gluconeogenesis | 1.62E-03 | -1.15 | ||
| Inositol oxygenase | Polyol metabolism | 5.10E-03 | -1.17 | ||
| Aldo-keto reductase family 1 member A1 | Aldehyde catabolic process | Catabolism | 3.89E-03 | 1.08 | |
| Delta-1-pyrroline-5-carboxylate dehydrogenase mitochondrial precursor | Amino acid catabolism | 6.18E-03 | 1.13 | ||
| Group XIII secreted phospholipase A2 | Lipid degradation | 6.84E-03 | -1.13 | ||
| Methylmalonate-semialdehyde dehydrogenase [acylating] mitochondrial precursor | Metabolism of amino acids | 7.90E-03 | -1.08 | ||
| Trypsin precursor | Peptidase | 3.66E-03 | 1.14 | ||
| Cytochrome P450 8B1 | Bile acid synthesis | Biosynthesis | 2.52E-03 | 1.11 | |
| Myo-inositol 1-phosphate synthase A1 | Myo-inositol biosynthesis | 2.80E-03 | 1.32 | ||
| Cytochrome P450 2N2 | Arachidonic acid metabolism | Regulation/Signaling | 1.99E-05 | -1.44 | |
| Acyl-CoA-binding protein | Lipid transport | 7.68E-04 | -1.23 | ||
| Thioredoxin | Signal transduction | 9.16E-03 | -1.12 | ||
| Forkhead box P2 | Transcription factor | 6.69E-03 | 1.22 | ||
| Superoxide dismutase [Cu-Zn] | Destroys radicals | Other | 1.09E-03 | -1.12 | |
| Fatty acid binding protein H6-isoform | Lipid binding, transporter activity | 2.10E-03 | 1.16 | ||
| Tropomyosin alpha 4 chain | Muscle contraction | 8.98E-03 | 1.14 | ||
Shared Differences in Gene Expression
Significantly differentially expressed genes found in more than one population can suggest both shared mechanisms to cope with or that result from stress. Three genes, acyl-CoA-binding protein, the MNLL subunit of NADH-ubiquinone oxidoreductase, and thioredoxin, are significantly differentially expressed in all three polluted populations (Figure 1), yet none of these genes is consistently expressed in all 3 of the polluted populations. Acyl-CoA-binding protein is more highly expressed in the Elizabeth River population as compared to its respective reference sites but less highly expressed in the New Bedford Harbor and Newark Bay populations as compared to their respective reference sites. Similarly, both the MNLL subunit of NADH-ubiquinone oxidoreductase and thioredoxin are more highly expressed in the Elizabeth River population and less highly expressed in the New Bedford Harbor and Newark Bay populations as compared to their respective reference sites. In total, 8 genes that are significantly differently expressed are shared between the New Bedford Harbor and Elizabeth River populations, 10 genes are shared between the New Bedford Harbor and Newark Bay populations, and 7 genes are shared between the Newark Bay and Elizabeth River populations (Fig. 1).
Figure 1.
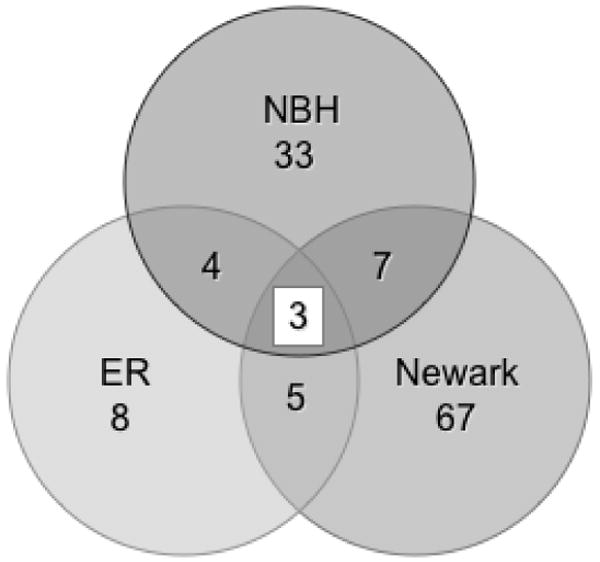
Venn Diagram showing numbers of significant genes for each polluted versus reference sites comparison. Three genes are significantly different in all three polluted Superfund populations compared to their respective reference populations. NBH = New Bedford Harbor comparison, Newark = Newark Bay comparison, ER = Elizabeth River comparison.
All polluted versus all reference site populations
A second approach to discerning patterns of gene expression unique to polluted populations is to compare all of the Superfund populations to all of the reference populations for significant differences in gene expression (Table 3). Again, a mixed model was used for this comparison, and 33 genes are differently expressed in the Superfund populations (15%, p < 0.01). With a Bonferonni correction for multiple testing, this number is reduced to 6.
Table 3. Significant differently expressed genes in all Superfund versus all reference site populations.
A gene with a positive fold-difference is more highly expressed in the Superfund populations, and a gene with a negative fold-difference is more highly expressed in the reference populations. P-values for New Bedford Harbor (NBH), Newark Bay (NB) and Elizabeth River (ER) populations versus their respective reference site populations also are reported. Significant p-values for these three separate comparisons are in bold. Gene names in bold are consistently up or down regulated in the all Superfund versus all reference comparison and in each separate comparison, NBH, NB, and ER.
| Gene | Pathway | Function | P-value | Fold Difference | P-value NBH | P-value NB | P-value ER |
|---|---|---|---|---|---|---|---|
| Cytochrome c oxidase subunit VIIIb | Oxidative Phosphorylation | Energy Metabolism | 8.44E-04 | 1.09 | 4.40E-02 | 1.04E-03 | 5.97E-01 |
| ATP synthase H+ transporting mitochondrial F1 complex delta subunit | Oxidative Phosphorylation | 2.06E-03 | 1.06 | 2.19E-01 | 4.61E-02 | 3.15E-01 | |
| ATP synthase H+ transporting mitochondrial F0 complex subunit b isoform 1 | Oxidative Phosphorylation | 3.81E-05 | -1.09 | 4.28E-01 | 1.22E-04 | 7.06E-02 | |
| 6-phosphogluconolactonase | Pentose-phosphate shunt | 6.66E-04 | 1.07 | 1.07E-04 | 5.03E-03 | 7.76E-01 | |
| PEP carboxykinase phosphoenolpyruvate carboxykinase | Carbohydrate biosynthesis | 7.41E-03 | 1.08 | 6.16E-03 | 9.09E-01 | 1.77E-01 | |
| Isocitrate dehydrogenase 1 (cytosolic or soluble IDH1) | Carbohydrate metabolism | 1.42E-03 | -1.11 | 3.28E-01 | 3.52E-01 | 5.29E-03 | |
| Electron transfer flavoprotein beta-subunit | Electron transfer flavoprotein | 1.64E-04 | -1.10 | 2.31E-01 | 3.32E-05 | 8.17E-01 | |
| Acyl-CoA desaturase | Fatty acid desaturase | 3.47E-03 | 1.09 | 2.55E-01 | 1.07E-01 | 7.07E-01 | |
| Phosphoglycerate mutase type B | Glycolysis | 4.27E-04 | 1.10 | 6.55E-01 | 2.00E-01 | 1.46E-02 | |
| Fructose-1,6-bisphosphatase | Glycolysis | 1.36E-06 | -1.16 | 2.96E-04 | 1.04E-03 | 8.19E-02 | |
| 3-ketoacyl-CoA thiolase, mitochondrial | Lipid metabolism; fatty acid metabolism | 1.20E-03 | -1.07 | 1.40E-01 | 5.36E-01 | 1.23E-02 | |
| ADP, ATP carrier protein, isoform T2 | Transport | 3.79E-04 | -1.10 | 1.85E-01 | 1.31E-05 | 9.45E-01 | |
| Phospholipase A2 precursor | Lipid degradation | Catabolism | 3.36E-04 | 1.19 | 2.89E-01 | 6.51E-04 | 1.08E-02 |
| Group XIII secreted phospholipase A2 | Lipid degradation | 3.30E-04 | -1.11 | 8.80E-01 | 5.73E-02 | 6.84E-03 | |
| Isoamyl acetate-hydrolyzing esterase | Probable lipase | 8.28E-04 | 1.06 | 3.41E-03 | 1.13E-01 | 9.06E-01 | |
| 4-hydroxyphenylpyruvate dioxygenase | Tyrosine catabolic process | 1.88E-03 | 1.06 | 9.78E-04 | 5.75E-01 | 9.22E-02 | |
| Delta-5/delta-6 fatty acid desaturase | Biosynthesis of HUFA | Biosynthesis | 1.99E-03 | 1.10 | 1.32E-02 | 3.14E-02 | 8.20E-01 |
| Cystathionine-beta-synthase | Cysteine Metabolic process | 2.95E-04 | -1.11 | 7.71E-01 | 1.67E-01 | 6.41E-05 | |
| Fatty acid synthase | Fatty acid synthesis | 2.49E-07 | 1.31 | 8.50E-04 | 7.11E-01 | 2.77E-01 | |
| 3-oxoacyl-[acyl-carrier protein] reductase | Fatty acid synthesis | 9.22E-04 | 1.09 | 8.18E-04 | 6.37E-01 | 1.17E-01 | |
| Myo-inositol 1-phosphate synthase A1 | Myo-inositol biosynthesis pathway | 1.72E-04 | 1.22 | 8.44E-01 | 8.91E-01 | 2.80E-03 | |
| Ornithine decarboxylase antizyme, short isoform | Polyamine biosynthesis | 1.91E-05 | -1.11 | 2.75E-03 | 9.97E-03 | 9.15E-02 | |
| Elongation factor 1-alpha | Protein synthesis | 2.03E-03 | -1.08 | 8.12E-02 | 3.52E-03 | 5.81E-01 | |
| Cytochrome P450 2N2 | Arachidonic acid metabolism | Regulation/Signaling | 8.00E-03 | -1.13 | 8.22E-03 | 1.04E-02 | 1.99E-05 |
| Fatty acid binding protein H6-isoform | Lipid binding, transporter activity | 7.75E-05 | 1.12 | 9.05E-03 | 1.16E-01 | 2.10E-03 | |
| Troponin I, slow skeletal muscle | Regulation of muscle contraction | 8.41E-04 | -1.12 | 2.54E-02 | 1.44E-05 | 9.84E-02 | |
| Forkhead box P2 | Transcription factor | 8.35E-05 | 1.22 | 3.61E-01 | 3.98E-03 | 6.69E-03 | |
| Basic transcription factor 3 | Transcription factor | 1.01E-05 | 1.11 | 1.77E-02 | 1.97E-02 | 4.92E-01 | |
| Protein disulfide isomerase A3 precursor | Catalyzes the rearrangement of -S-S- bonds in proteins | Other | 1.09E-04 | -1.13 | 6.81E-01 | 1.88E-06 | 3.53E-02 |
| Cytochrome P450 2K2 | Lauric acid hydroxylase | 4.77E-04 | -1.12 | 1.03E-02 | 5.38E-03 | 5.39E-01 | |
| Phosphomannomutase 1 | Mannose metabolic process | 2.70E-04 | 1.21 | 2.37E-01 | 2.18E-03 | 1.40E-02 | |
| Tropomyosin alpha 4 chain | Muscle contraction | 7.15E-04 | 1.10 | 3.88E-02 | 3.25E-01 | 8.98E-03 | |
| Cytokine receptor common gamma chain precursor | Common interleukin receptor subunit | 8.74E-03 | 1.10 | 3.05E-05 | 5.76E-01 | 6.07E-01 |
Discussion
The goal of this work was to better understand how natural populations cope with chronically polluted habitats. Targeted microarrays were used to gain a snapshot of altered gene expression in natural populations of F. heteroclitus populations inhabiting three different Superfund sites as compared to relatively clean reference populations. A fundamental problem with comparing any two populations is that one expects differences between the populations yet cannot know whether the differences are due to random genetic drift or to other more important factors, in this case, chronic pollution exposure. In order to identify differences due to pollution, one can ask which differences are significant in the polluted population compared to two flanking reference populations. In this joint comparison, the combined genetic drift in between the two reference site populations should be greater than that between each reference and polluted population because the reference populations are geographically more distant from each other than to the polluted population. This comparison assures that identified differences in gene expression are not simply due to genetic drift or clinal variation common to this species. Thus, each of the three Superfund populations was compared with its respective flanking reference sites. Note that although these fish were “common gardened” in the laboratory for four months, these comparisons could identify both physiologically induced changes in gene expression due to any remaining chemical body burden as well as evolved changes in gene expression due to natural selection in the different sites. For instance, while Fundulus eliminate TCDD rapidly (the half-life is < 60 hours (Prince and Cooper 1995)), PCBs have a half-life of ∼4.7 months in Fundulus (Elskus et al. 1999), and methyl mercury has a half-life that ranges from 100 to 1000 days in different fish species (Huckabee et al. 1979). Therefore, these fish likely retain some of the pollutants from their natal environments, and because the polluted versus two reference populations comparison incorporates variation due to random genetic distance, significant differences in gene expression between the polluted population and both reference populations most likely reflect a combination of physiological responses to pollution and evolved differences in gene expression.
In the New Bedford Harbor population, 16% of the genes are significantly differently expressed compared to two flanking reference sites, in Newark Bay, the percentage of significantly differently expressed genes is 32, and in the Elizabeth River population, this percentage is 8. Why are so many more genes significantly differently expressed in the Newark Bay population as compared to the New Bedford Harbor and Elizabeth River populations? One explanation is that the Newark Bay population may be exposed to a broader diversity of pollutants. The New Bedford Harbor site is predominantly contaminated with PCBs and metals, the Elizabeth River site is predominantly contaminated with PAHs and metals, and the Newark Bay site is contaminated with PAHs, PCBs, pesticides, and metals. Additionally, compared to both New Bedford Harbor and the Elizabeth River, Newark Bay is part of a much larger and more highly urbanized environment with greater potential for urban runoff, illegal dumping, and accidental spills. Finally, there will be differential metabolism and mobilization of the chemicals during four months of depuration. Many of the less lipophilic compounds may be completely eliminated while other compounds are only partially mobilized, if at all. Thus, the greater number of changes in Newark Bay may reflect a greater number of induced changes in gene expression due to mobilized but not eliminated pollutants. This idea is strengthened by the fact that the Newark Bay fish were larger than the fish in the surrounding reference sites (t-test, p < 0.01), and although ANCOVA was used to account for body weight, bigger fish will have a greater body burden of pollutants and will depurate more slowly. Interestingly, previous studies have not found a significant effect of body weight on gene expression (Oleksiak et al. 2002, Oleksiak et al. 2005). Thus, the effect of weight seen in these analyses might be a reflection of physiological induction due to incomplete depuration. Whether the differences among superfund sites are due to physiologically induced changes or evolved changes in gene expression due to chronic pollutant exposure can only be determined by comparing fish that have been completely depurated or by comparing laboratory raised progeny.
A striking feature of the genes that are significantly differently expressed in the Newark Bay comparison is the number that are involved in the oxidative phosphorylation pathway (Table 2). 34% of the significantly differentially expressed genes are involved in this pathway. In comparison, only 2% of the significantly differently expressed genes in the New Bedford Harbor comparison and 1% in the Elizabeth River comparison are involved in the oxidative phosphorylation pathway. This suggests that pollution may have a significant effect on energy metabolism in these fish.
In New Bedford Harbor, the most significantly differently expressed gene is the down regulation of isopentenyl-diphosphate delta-isomerase. Isopentenyl-diphosphate delta-isomerase catalyzes a central reaction in the biosynthesis of isoprenoids and is necessary for the synthesis of a wide variety of essential cellular metabolites, including dolichols, vitamins A, D, E, and K, sterols, steroid hormones and bile acids (Ramos-Valdivia et al. 1997). It also is involved in cholesterol biosynthesis. Interestingly, the second most significant gene in the New Bedford Harbor population is the down regulation of 3-hydroxy-3-methylglutaryl-CoA reductase which is also involed in cholesterol biosynthesis (Goldstein and Brown 1990).
In Newark Bay, the most significantly differently expressed gene is thioredoxin. Thioredoxin is a ubiquitous and evolutionarily conserved protein that modulates the structure and activity of proteins involved in a spectrum of processes, such as gene expression, apoptosis, and the oxidative stress response (Kumar et al. 2004). Thioredoxin is significantly less expressed in the Newark Bay population and also in the New Bedford Harbor population but is significantly more highly expressed in the Elizabeth River population. Its altered regulation in these polluted populations may reflect response to oxidative stress previously measured in some of these populations (Meyer et al. 2003). This difference between populations for thioredoxin expression also was seen in a study of brain gene expression using the same arrays and same populations (Fisher and Oleksiak 2007). In this study, thioredoxin was significantly less highly expressed in the brain in the New Bedford Harbor and Elizabeth River populations but significantly more highly expressed in the Newark Bay population suggesting tissue specific differences in regulation.
Cytochrome P4502N2 (CYP2N2) shows both the smallest p-value and greatest magnitude of change in the Elizabeth River population comparison; it is significantly more highly expressed in the Elizabeth River population. It also is significantly less highly expressed in the New Bedford Harbor population. CYP2N2 is an arachidonic acid epoxygenase and hydroxylase, and is potentially involved in xenobiotic metabolism (Oleksiak et al. 2000). It has been shown to be a good indicator of exposure to anthracene at a range of concentrations in laboratory exposures as well as in field collected F. heteroclitus collected from a PAH contaminated site (Peterson and Bain 2004), showing statistically significant up-regulation. This up-regulation is similar to what was found for the Elizabeth River population; PAHs, including anthracene, are predominant pollutants in the Elizabeth River.
Significantly differentially expressed genes are found in more than one population suggest shared mechanisms to cope with pollutants or genes whose expression may be more sensitive to stress and thus more labile. The latter is suggested when a gene is significantly more highly expressed in one polluted population compared to both reference populations and significantly less expressed in a different polluted population compared to its reference populations. In total, 8 significantly differently expressed genes are shared between the New Bedford Harbor and Elizabeth River populations, 10 genes are shared between the New Bedford Harbor and Newark Bay populations, and 7 genes are shared between the Newark Bay and Elizabeth River populations (Fig. 1, Table 2). However, six of the shared genes between the New Bedford Harbor and Elizabeth River populations are not similarly expressed in the two populations: thus, a gene more highly expressed in the New Bedford Harbor population compared to its reference populations is less highly expressed in the Elizabeth River population or vice versa. The two genes with a conserved response in the New Bedford Harbor and Elizabeth River populations, aldo-keto reductase (family 1 member A1) and fatty acid binding protein (H6-isoform), both are less expressed in these populations compared to the reference populations. Similarly, between the Elizabeth River and Newark Bay populations, only one of the seven shared genes is significantly altered in the same direction; the transcription factor forkhead box P2 is significantly less expressed in the Superfund populations. In contrast, the New Bedford Harbor population has more similarities with the Newark Bay population with nine of the ten shared, significantly differently expressed genes being in the same direction (acyl-CoA-binding protein, 6-phosphogluconolactonase, ATP synthase H+ transporting mitochondrial F0 complex subunit f isoform 2, cytochrome c oxidase polypeptide VIIA, Fructose-1,6-bisphosphatase, the MNLL subunit of NADH-ubiquinone oxidoreductase, O-methyltransferase, ornithine decarboxylase, and thioredoxin).
Only three genes are significantly differently expressed in all three polluted populations (Figure 1). Yet, the pattern of expression (whether relatively up or down) differs among the three Superfund sites. All three of these genes, acyl-CoA-binding protein, the MNLL subunit of NADH-ubiquinone oxidoreductase and thioredoxin, are more highly expressed in the Elizabeth River population and less highly expressed in the New Bedford Harbor and Newark Bay populations. The different patterns of gene expression among the three Superfund populations suggest that there are multiple ways to adapt to toxicants; differences seen among these populations may be due to both individual variation and different pollutant exposures.
The comparison of all Superfund individuals versus all reference individuals is more powerful than the individual Superfund population comparisons because the sample size is increased 3-fold. As in each individual Superfund comparison, this comparison controls for genetic drift because the reference site populations are geographically more distant from each other than they are to the Superfund site populations. In this comparison, 33 genes (15%) are significantly differently expressed and include 6 genes not found in any of the paired analyses (Table 3). However, the results need to be considered carefully because significant changes in gene expression in only one or two populations can drive the results (see Table 3). Yet, for 58% (19 genes) of the significantly differentially expressed genes, the pattern of change is shared among all three Superfund sites. Fatty acid synthase is the most significantly differently expressed of this 58%. Fatty-acid synthase is down-regulated in all the polluted populations as compared to the reference populations. A primary function of the fatty acid synthesis pathway is to store excess energy as fat (Kuhajda 2000). The down-regulation of fatty acid synthase in the polluted populations suggests that fish from these populations may have less stored energy. Since the fish had been on the same diet for four months, this is not due to diet, but instead may be due to an extra energy cost due to coping with pollution. For instance. during short-term ethanol stress in Saccharomyces cerevisiae, global gene expression studies show that a large number (17%) of the up-regulated genes are involved in energy metabolism (Alexandre et al. 2001). Thus, the down-regulation of fatty acid synthase in polluted populations might reflect a generalized result of stress: a decrease in available energy.
Conclusions
Between any two Superfund populations, up to 9 genes show a consistent difference in gene expression. The most parsimonious explanation for shared differences is that they are due to similar pollutant exposures. Shared pollutant exposures can result in both similar induced responses and similar adaptive responses. Due to the lipophilic and persistent nature of many of the pollutants at the Superfund sites and the short depuration period of four months, the altered gene expression patterns in the Superfund populations most likely represent both induced and adaptive strategies for responding to pollution.
Acknowledgments
Funding for this work was received from NIH 5 RO1 ES011588, 2P42 ES010356, and 2 P42 ES007381. The Author thanks Douglas L. Crawford and Jeffrey D. VanWye for help with production of the metabolic arrays and DLC for review of the manuscript.
References
- Adams SM, Lindmeier JB, Duvernell DD. Microsatellite analysis of the phylogeography, Pleistocene history and secondary contact hypotheses for the killifish, Fundulus heteroclitus. Mol Ecol. 2006;15:1109–1123. doi: 10.1111/j.1365-294X.2006.02859.x. [DOI] [PubMed] [Google Scholar]
- Alden RWI, Winfield JG. Regional Action Plan for the Elizabeth River. 1995. Defining the Problem: The Elizabeth River, A Region of Concern. Report Submitted to Virginia Department of Environmental Quality. [Google Scholar]
- Alexandre H, Ansanay-Galeote V, Dequin S, Blondin B. Global gene expression during short-term ethanol stress in Saccharomyces cerevisiae. FEBS Lett. 2001;498:98–103. doi: 10.1016/s0014-5793(01)02503-0. [DOI] [PubMed] [Google Scholar]
- Bello SM, Franks DG, Stegeman JJ, Hahn ME. Acquired resistance to Ah receptor agonists in a population of Atlantic killifish (Fundulus heteroclitus) inhabiting a marine superfund site: in vivo and in vitro studies on the inducibility of xenobiotic metabolizing enzymes. Toxicol Sci. 2001;60:77–91. doi: 10.1093/toxsci/60.1.77. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:155–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Cooper PS, Vogelbein WK, Van Veld PA. Altered expression of the xenobiotic transporter P-glycoprotein in liver and liver tumours of mummichog (Fundulus heteroclitus) from a creosote-contaminated environment. Biomarkers. 1999;4:48–58. doi: 10.1080/135475099230994. [DOI] [PubMed] [Google Scholar]
- Elskus AA, Monosson E, McElroy AE, Stegeman JJ, Woltering DS. Altered CYP1A expression in Fundulus heteroclitus adults and larvae: a sign of pollutant resistance? Aquatic Toxicology. 1999;45:99–113. [Google Scholar]
- Fisher MA, Oleksiak MF. Convergence and divergence in gene expression among natural populations exposed to pollution. BMC Genomics. 2007;8:108. doi: 10.1186/1471-2164-8-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- Greaves J. Analysis of Organic Pollutants in Sediments and Blue Crab (Callinectes sapidus) Tissues. 1990. Elizabeth River Long-Term Monitoring Program - Phase I: 1989. Final Report to the Virginia Water Control Board:222. [Google Scholar]
- Hamadeh HK, Bushel PR, Jayadev S, Martin K, DiSorbo O, Sieber S, Bennett L, Tannant R, Stoll R, Barrett JC, Blanchard K, Paules RS, Afshari CA. Gene expression analysis reveals chemical-specific profiles. Toxicological Sciences. 2002;67:219–231. doi: 10.1093/toxsci/67.2.219. [DOI] [PubMed] [Google Scholar]
- Huckabee JW, Elwood JW, Hildebrand SG. Accumulation of Mercury in Freshwater Biota. In: Nriagu JO, editor. The Biogeochemistry of Mercury in Environment. Elsevier/North-Holland Biomedical Press; N.Y., New York: 1979. pp. 277–301. [Google Scholar]
- Kerr M, Churchill G. Experimental design for gene expression microarrays. Biostatistics. 2001;2:183–201. doi: 10.1093/biostatistics/2.2.183. [DOI] [PubMed] [Google Scholar]
- Kuhajda FP. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition. 2000;16:202–208. doi: 10.1016/s0899-9007(99)00266-x. [DOI] [PubMed] [Google Scholar]
- Kumar JK, Tabor S, Richardson CC. Proteomic analysis of thioredoxin-targeted proteins in Escherichia coli. Proc Natl Acad Sci U S A. 2004;101:3759–3764. doi: 10.1073/pnas.0308701101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake JL, McKinney R, Lake CA, Osterman FA, Heltshe J. Comparisons of patterns of polychlorinated biphenyl congeners in water, sediment, and indigenous organisms from New Bedford Harbor, Massachusetts. Archives of Environmental Contamination & Toxicology. 1995;29:207–220. [Google Scholar]
- Long ER, Wolfe DA, Scott KJ, Thursby GB, Stern EA, Peven C, Schwartz T. Magnitude and Extent of Sediment Toxicity in the Hudson-Raritan Estuary 1995 [Google Scholar]
- Meyer JN, Smith JD, Winston GW, Di Giulio RT. Antioxidant defenses in killifish (Fundulus heteroclitus) exposed to contaminated sediments and model prooxidants: short-term and heritable responses. Aquat Toxicol. 2003;65:377–395. doi: 10.1016/j.aquatox.2003.06.001. [DOI] [PubMed] [Google Scholar]
- Oleksiak MF, Churchill GA, Crawford DL. Variation in gene expression within and among natural populations. Nat Genet. 2002;32:261–266. doi: 10.1038/ng983. [DOI] [PubMed] [Google Scholar]
- Oleksiak MF, Roach JL, Crawford DL. Natural variation in cardiac metabolism and gene expression in Fundulus heteroclitus. Nature Genet. 2005;37:67–72. doi: 10.1038/ng1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksiak MF, Wu S, Parker C, Karchner SI, Stegeman JJ, Zeldin DC. Identification, functional characterization, and regulation of a new cytochrome P450 subfamily, the CYP2Ns. J Biol Chem. 2000;275:2312–2321. doi: 10.1074/jbc.275.4.2312. [DOI] [PubMed] [Google Scholar]
- Ownby DR, Newman MC, Mulvey M, Vogelbein WK, Unger MA, Arzayus LF. Fish (Fndulus hereroclitus) populations with different exposure histories differ in tolerance of creosote-contaminated sediments. Environmental Toxicology and Chemistry. 2002;21:1897–1902. [PubMed] [Google Scholar]
- Peterson JS, Bain LJ. Differential gene expression in anthracene-exposed mummichogs (Fundulus heteroclitus) Aquat Toxicol. 2004;66:345–355. doi: 10.1016/j.aquatox.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Prince R, Cooper KR. Comparisons of the effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on chemically impacted and nonimpacted subpopulations of Fundulus heteroclitus: I. TCDD Toxicity. Environ Toxicol Chem. 1995;14:579–587. [Google Scholar]
- Pruell RJ, Norwood CB, Bowen RD, Boothman WS, Rogerson PF, Hackett M, Butterworth BC. Geochemical study of sediment contamination in New Bedford Harbor, Massachusetts. Mar Environ Res. 1990;29:77–102. [Google Scholar]
- Raghavachari N, Bao Y, Li G, Xie X, Muller U. Reduction of autofluorescence on DNA microarrays and slide surfaces by treatment with sodium borohydride. Anal Biochem. 2003;312:101–105. doi: 10.1016/s0003-2697(02)00440-2. [DOI] [PubMed] [Google Scholar]
- Ramos-Valdivia AC, van der Heijden R, Verpoorte R. Isopentenyl diphosphate isomerase: a core enzyme in isoprenoid biosynthesis. A review of its biochemistry and function. Nat Prod Rep. 1997;14:591–603. doi: 10.1039/np9971400591. [DOI] [PubMed] [Google Scholar]
- Smith M, Chapman R, Powers D. Mitochondrial DNA analysis of Atlantic Coast, Chesapeake Bay, and Delaware Bay populations of the teleost Fundulus heteroclitus indicates temporally unstable distributions over geologic time. Molecular Marine Biology and Biotechnology. 1998;7:79–87. [Google Scholar]
- Thomas RS, Rank DR, P SG, Zastrow GM, Hayes KR, Pande K, Glover E, Silander T, Craven MW, Reddy JK, Jovanovich SB, Bradfield CA. Identification of toxicologically predictive gene sets using cDNA microarrays. Molecular Pharmacology. 2001;60:1189–1194. doi: 10.1124/mol.60.6.1189. [DOI] [PubMed] [Google Scholar]
- Vogelbein WK, Fournie JW, Veld PAV, Huggett RJ. Hepatic neoplasms in the mummichog Fundulus heteroclitus from a creosote-contaminated site. Cancer Res. 1990;50:5978–5986. [PubMed] [Google Scholar]
- Vogelbein WK, Williams CA, Van Veld PA, Unger MA. Acute toxicity resistance in a fish populations with a high prevalence of cancer. Society for Environmental Toxicology and Chemistry; Washington, D. C.: 1996. [Google Scholar]
- Wirgin I, Waldman JR. Resistance to contaminants in North American fish populations. Mutat Res. 2004;552:73–100. doi: 10.1016/j.mrfmmm.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Wolfe DA, Long ER, Thursby GB. Sediment toxicity in the Hudson-Raritan Estuary: Distribution and correlations with chemical contamination. Estuaries. 1996;19:901–912. [Google Scholar]
- Wolfinger RD, Gibson G, Wolfinger ED, Bennett L, Hamadeh H, Bushel P, Afshari C, Paules RS. Assessing gene significance from cDNA microarray expression data via mixed models. J Comput Biol. 2001;8:625–637. doi: 10.1089/106652701753307520. [DOI] [PubMed] [Google Scholar]
- Wu H, Kerr K, Cui X, Churchill G. MAANOVA: a software package for the analysis of spotted cDNA microarray experiments. The Analysis of Gene Expression Data: Methods and Software 2003 [Google Scholar]