

Abstract
Prostate cancer (PCa) is the second leading cause of cancer-related deaths in men. Hormone refractory invasive PCa is the end stage and accounts for the majority of PCa patient deaths. We present here that plumbagin (PL), a quinoid constituent isolated from the root of the medicinal plant Plumbago zeylanica L, may be a potential novel agent in the control of hormone refractory PCa. Specific observations are the findings that PL inhibited PCa cell invasion and selectively induced apoptosis in PCa cells but not in immortalized non-tumorigenic prostate epithelial RWPE-1 cells. Also, intraperitoneal administration of PL (2mg/kg body weight), beginning 3 days after ectopic implantation of hormone refractory DU145 PCa cells, delayed tumor growth by 3 weeks and reduced both tumor weight and volume by 90%. Discontinuation of PL treatment in PL- treated mice, for as long as 4 weeks did not result in progression of tumor growth. PL, at concentrations as low as 5 μM, inhibited both in cultured PCa cells and DU145 xenografts the expression of: 1) PKCε, PI3K, pAKT, pJAK-2 and pStat3; 2) the DNA-binding activity of transcription factors AP-1, NFkB, and Stat3 and 3) Bcl-xL, cdc25A and COX-2 expression. The results indicate for the first time, using both in vitro and in vivo preclinical models, that PL inhibits the growth and invasion of PCa. PL inhibits multiple molecular targets including PKCε, a predictive biomarker of PCa aggressiveness. PL may be a novel agent for therapy of hormone refractory PCa.
Keywords: Plumbagin, PKCε, Stat3, prostate cancer
Introduction
Prostate cancer (PCa) is the most frequently diagnosed cancer among men and is the second leading cause of cancer-related deaths (1). The risk of PCa increases rapidly after age 50, with two-thirds of all PCa cases found in men after age 50. PCa first manifests as an androgen-dependent (AD) disease and can be treated with androgen-deprivation therapy. Despite the initial success of androgen ablation therapy, PCa progresses from AD to androgen-independent (AI). The hormone refractory invasive PCa is the end stage and accounts for the majority of PCa patient deaths (2-6). At present, there is no effective treatment for AI metastatic PCa. There is an urgent need for novel agents which can be effective and selective in the prevention and treatment of hormone refractory PCa. Plumbagin (PL), a medicinal plant-derived naphthoquinone (7), appears to possess such properties.
PL (5-hydroxy-2-methyl-1,4-napthoquinone) (Fig. 1A) was isolated from the roots of the medicinal plant Plumbago zeylanica L (also known as Chitrak) (7). The roots of Plumbago zeylanic have been used in Indian medicine for more than 2,500 years for treatments of various ailments. PL is also present in black walnut and other various medicinal plants (7). PL has been shown to exert anticancer and antiproliferative activities in animal models and in cell culture (7). PL, fed in the diet (200ppm), inhibits azoxymethane-induced intestinal tumors in rats (8). PL inhibits ectopic growth of breast cancer MDA-MB-231 cells (9), non-small cell lung cancer A549 cells (10), and melanoma A375-S2 cells in athymic nude mice (11). PL has also been shown induce apoptosis in human PCa cell lines (12). However, no study exists about the effects of PL in the prevention and/or treatment of PCa progression.
Figure 1. Plumbagin induces apoptosis and inhibits cells invasion in prostate cancer cells.

PCa cell lines (DU145, CWR22rv1, LNCaP and RWPE-1) at 70-80% confluency were serum starved for 24 hrs and then treated with PL at various (0, 5, 10, and 15 μM) concentrations in DMSO (final concentration 0.1%). At 24hrs after treatment, cells were collected for apoptosis analysis. CWR22rv1, DU145, and PC-3 cells were treated with 5μM or 20μM PL in DMSO (final concentration 0.1%) for 48hr and assayed cell invasion as described before (14). A: Structure of PL; B: Induction of apoptosis. Each value in the graph is the mean ± S.E. from three separate dishes. C: PCa cell invasion. Cells were stained with crystal violet and photographed at 40X magnification. D: Number of invading cells was estimated by colorimetric measurements at 560 nm according to assay instructions (Chemicon International, Temecula, CA). Each value in the graph is the mean ± S.E. from three separate wells. Similar results were observed in a repeat experiment.
We present in this communication for the first time that PL is a novel inhibitor of the growth and invasion of hormone refractory PCa cells. Intra-peritoneal administration of PL reduced both the weight and volume of ectopically xenografted DU145 cells by 90%. PL inhibited PCa cell invasion and selectively induced apoptosis in PCa cells. PL inhibited constitutive expression of multiple molecular targets including PKCε, PI3K, AKT, and activation of transcription factors AP-1, NFkB, and Stat3 in PCa cells. PL may be a novel agent for therapy of hormone refractory PCa.
Materials and Methods
Chemicals, antibodies and assay kits: Plumbagin (practical grade, purity >95%) was purchased from Sigma-Aldrich (St Louis, MO)
The sources of antibodies used in this study were: PKCε, other PKC isoforms, Stat3, pStat3Tyr705, PI3K (p85), PI3K (p110), p21, p27, VEGF, MMP-9, Bcl-xL, COX-2, Cdc25A, β-actin, (Santa Cruz Biotechnology, Santa Cruz, CA); pJAK-1 (Tyr1022/1023), pJAK-2 (Tyr1007/1008), pAkt (Ser473), pAkt (Thr308), AKT (Cell Signaling Tech., Danvers, MA); pStat3Ser727 (BD Biosciences, San Jose, CA); and PCNA (Dako North America, Inc., Carpinteria, CA). The oligonucleotides for AP-1 (5′—CGC TTG ATG ACT CAG CCG GAA—3′), NFkB (5′—AGT TGA GGG GAC TTT CCC AGG C –3′) and Stat3 (5′—GAT CCT TCT GGG AAT TCC TAG ATC –3′) were obtained from Santa Cruz Biotechnology, Santa Cruz, CA. Collagen-Based Cell Invasion Assay kit was from Millipore, Temecula, CA.
Cell lines
Cell lines (RWPE-1, CWR22rv1, LNCaP, PC-3 and DU145) were obtained from ATCC (Manassas, VA).
Apoptosis
Percent of cells undergoing apoptosis was determined by flow cytometric analysis of propidium iodide stained cells (13).
Cell invasion assay
Cell invasion was assayed using a Collagen-Based Cell Invasion Assay kit as per the manufacturer's instructions (14). Briefly, prostate cancer cell lines at 80% confluency were serum starved for 18 to 24 h before the assay. The cells were harvested and the pellet was gently resuspended in serum-free medium. In the upper chamber, 0.5 × 106 cells per well were plated in triplicates and incubated for 2 h at 37°C in a humidified incubator with 5% CO2 before PL treatment. Both the insert and the holding well were subjected to the same medium composition with the exception of serum. The insert contained no serum, whereas the lower well contained 10% fetal bovine serum that served as a chemoattractant. The untreated groups were used as a control. Forty-eight hours after PL treatment, the cell invasion assay was performed as per the manufacturer's instructions. The cells in the insert were removed by wiping gently with a cotton swab. Migrated cells sticking to the bottom side of the insert were stained with Cell Stain. Invading cells on the bottom side of the membrane were photographed using light inverted microscopy (Nikon Eclipse TS 100) at 40X magnification. In addition, the number of cells migrating to the bottom side was estimated by colorimetric measurements at 560 nm according to assay instructions. Mean ± SE was calculated from three independent experiments.
Ectopic DU145 tumor xenografts
Male athymic nude mice were purchased from Jackson Laboratory and raised in a pathogen-free environment. Mice were used for experimentation 2 weeks after acclimatization. DU145 cells (2.5 × 106 cells in Matrigel) were implanted on both flanks of nude mice. The animals (n=10) were treated with PL (2mg/kg body weight in 0.1 ml PBS, five days a week) by intra-peritoneal injection after 3 days post cell implantation. The untreated animals (n=10) were used as a control. Mice were weighed and examined for twice weekly for the presence of palpable tumors. Tumor size was measured by calipers and recorded. Tumor volume (V) was determined by the following equation: V = (L × W × H × 0.5236), where L is length, W is the width, and H is the height of the xenograft tumor. At the end of study, mice were euthanized and digital photographs were taken of their tumors. The mean calculated tumor volume was plotted as a function of time. After 11 weeks, PL treatment was stopped and tumor's growth was measured through 16 weeks post cell implantation.
Statistical Analysis
Statistical differences between the tumor volume means of control and PL treated mice were analyzed by Student's t-test.
Western Blot Analysis
Human prostate cancer cells and xenograft samples were lysed in immunoprecipitation (IP) lysis buffer (50 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [HEPES, pH 7.5], 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride [PMSF], 200 mM Na3VO4, 200 mM NaF and 1 mM EGTA). The homogenate was centrifuged at 14, 000×g for 30 min at 4°C. 25 μg of whole cell lysate was fractionated on 10-15% SDS-polyacrylamide gels. The proteins were transferred to Hybond-P polyvinylidene difluoride (PVDF) transfer membrane (Amersham). The membranes were then incubated with the indicated primary antibodies followed by a horseradish peroxidase secondary antibody and developed with Amersham's enhanced chemiluminescence reagent and autoradiography using BioMax film (Kodak Co., Rochester, NY). The Western blots signals were quantitated by densitometric analysis using Totallab Nonlinear Dynamic Image analysis software (Nonlinear USA Inc., Durham, NC).
Histology
Xenograft samples were fixed for 24 h in 10% neutral buffered formalin, transferred to phosphate buffered saline (PBS, pH 7.4) and then embedded in paraffin. Sections (4 μm thickness) of each specimen were cut for histologic and immunohistochemical examination.
Immunohistochemical Analysis
Immunohistochemistry (IHC) was carried out with rabbit anti-PKCε (1:200 dilution), rabbit anti-Stat3 (1:150 dilution) or mouse anti-PCNA (1:150) antibody in a Lab Vision autostainer 3600, and PT module (Lab Vision, Fremont, CA) with a standard protocol for IHC (14). Briefly, the samples of xenograft tumor were deparaffinized and antigen retrieval was done by heating in citrate buffer, pH 6.0 buffer (Lab Vision, Fremomt, CA) at 98°C for 20 min, and then incubated in peroxidase for 5 min to block endogenous peroxidase. Non-specific proteins were blocked with Biocare Medical Terminator (Biocare Medical, Concord, CA) for 10 min, then samples were incubated with appropriate primary antibody at room temperature for 60 min, followed by horseradish peroxidase-labeled IgG secondary antibody (Biocare Medical, Concord, CA) for 40 min. Color was developed by incubating samples with diaminobenzidine (DAB)+ (Dako North America, Inc., Carpinteria, CA) for 1 min. CAT-Hematoxylin (Biocare Medical, Concord, CA) was used for 1 min as counter stain. The specific staining of PKCε, Stat3 or PCNA in the sections was examined using Olympus BX 51 microscope. Negative controls (without primary antibody) were included for each study. For the quantitation of Stat3 and PCNA-positive staining cells, 10 random areas were selected for each mouse at each time point. The number of cells showing positive labeling and the total number of cells counted were recorded. An average percentage was then calculated based on the total number of cells and the number of positive staining cells from each set of 10 fields counted. Results are expressed as mean of percentages ± SE.
Electrophoretic mobility shift assay (EMSA)
PCa cells (DU145, PC-3, CWR22rv1 and LNCaP) at 70-80% confluency were serum starved for 24 hrs. Cells were treated with 0, 5, 10, 15, or 20 μM of PL for 3hrs. Nuclear protein extracts were prepared by lysing cells in a hypotonic solution (10 mM HEPES, pH 7.5; 10 mM KCl; 0.1 mM EDTA, pH 8.0; 0.1 mM EGTA pH 8.0; 1 mM DTT; 0.5 mM PMSF; 0.5 mg/ml benzamide; 2 μg/ml aprotinin; 2 μg/ml leupeptin), with detergent (NP-40 at 6.25% (v/v)) followed by low speed (1500 × g for 30 secs) to collect nuclei. Nuclear proteins were extracted in a high-salt buffer (20 mM HEPES, pH 7.5; .4 M NaCl; 1 mM EDTA, pH 8.0; 1 mM EGTA pH 8.0; 1 mM DTT; 1 mM PMSF; 0.5 mg/ml benzamide; 2 μg/ml aprotinin; 2 μg/ml leupeptin) and nuclear membranes and genomic DNA removed by high-speed (16,000×g) centrifugation for 5 minutes. Nuclear protein extracts were stored at −70°C until used. The nuclear protein extract was incubated in a final volume of 20 μl of 10 mM HEPES (pH 7.9), 80 mM NaCl, 10% glycerol, 1 mM DTT, 1 mM EDTA, and 100 μg/ml poly(deoxyinosinic-deoxycytidylic acid) for 15 minutes. γ-p32-radiolabeled double-stranded oligonucleotides of the consensus binding sequences of AP-1, NFkB or Stat3, were then added and the complexes were incubated for 20 minutes at room temperature. The protein-DNA complexes were resolved on a 4.5% acrylamide gel containing, 2.5% glycerol and 0.5X Tris-borate EDTA at room temperature. Gels were dried and autoradiographed to determine binding activity (14)
Results
A. Plumbagin inhibits invasion and induces apoptosis in PCa cells
Cell invasion requires cells to migrate through an extra cellular matrix or basement membrane barrier, by first enzymatically degrading the barrier, then becoming established in a new location. Cell invasion is exhibited by tumor cells during metastasis. The effects of PL on the invasive ability of AI human PCa cell lines were determined. In this experiment (Fig.1C), PCa cells (DU145, PC3, and CWR22rv1) were treated with 5 or 20 μM PL for 48 hours and cell invasion was assayed using a collagen-based cell invasion assay kit (14). PL, at both 5μM and 20μM concentration, significantly (p value<0.001) inhibited the invasion of DU145, PC3, and CWR22rv1. The inhibitory effect of PL on cell invasion did not differ among these cell lines (DU145, PC3, and CWR22rv1) (p value > 0.1) (Fig. 1C and D). The effect of PL on the induction of apoptosis in human PCa has recently been reported (12). PL induced apoptosis in human PCa cells (PC3, LNCaP, and C4-2) irrespective of androgen responsiveness and p53 status. PL-induced apoptosis in human PCa cells was associated with modulation of cellular redox status and generation of reactive oxygen species (12). We also determined the effects of PL on the induction of apoptosis in PCa cell lines (DU145, CWR22rv1 and LNCaP) and non-tumorigenic immortalized prostate epithelial RWPE-1 cells. PL at concentration as high as 20 μM did not significantly (p value 0.42) induce apoptosis in RWPE-1 cells (Fig. 1B). PL at all concentrations significantly (p value< 0.009) induced apoptosis in PCa cell lines (DU145, CWR22rv1 and LNCaP). AI PCa cells (DU145, CWR22rv1) appear to be more sensitive than AD PCa cells (LNCaP) to the induction of apoptosis by PL (Fig.1B).
B. Plumbagin inhibits growth of DU145 cells in athymic nude mice
In this experiment (Fig. 2A and B), PL (2mg/kg body weight) was administered i.p. 3 days post ectopic implantation of hormone refractory DU145 cells. PL treatment delayed tumor growth by three weeks and significantly (p value < 0.05) reduced both the tumor weight and volume throughout the experimental period (Fig. 2A and B). Discontinuation of PL treatment in PL- treated mice, for as long as 4 weeks, did not result in an increase in tumor growth (Fig. 2B). PL treatment significantly (p value =0.000) inhibited PCNA expression and constitutive expression of Stat3 and PKCε (Fig. 1C). Also, PL treatment inhibited the expression of VEGF and MMP-9 (Fig. 2D). The PL treated mice gained weight and exhibited no obvious toxic effects.
Figure 2. Plumbagin inhibits expression of PKCε, activated Stat3, PCNA, VEGF and MMP-9 in ectopically xenografted DU145 cells.
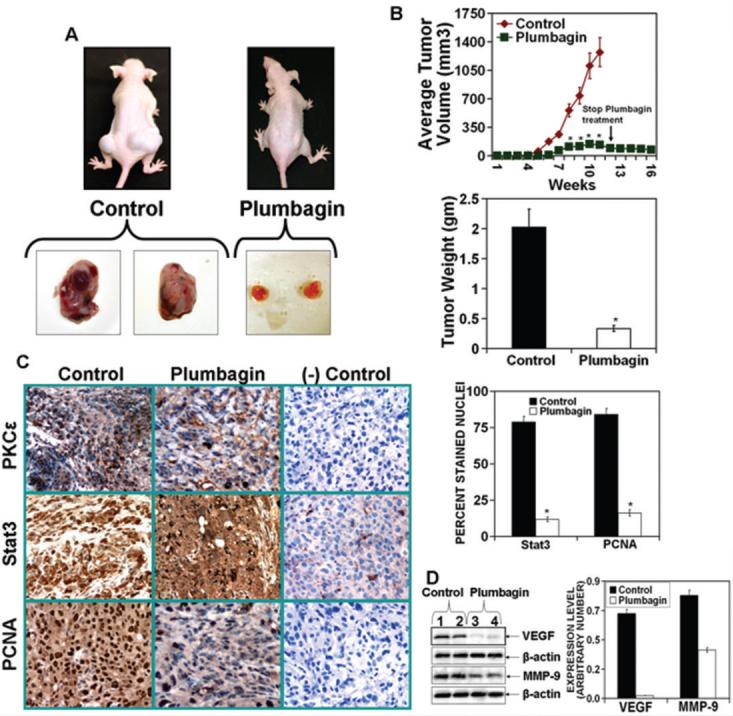
The DU145 cells (2.5 × 106 cells in 100 μl of a 1:1 mixture of media:Matrigel) were implanted on both flanks of athymic nude mice (N=10 mice per group). The animals were treated with PL (2mg/kg body weight in PBS or PBS only, five days a week) by intra-peritoneal injection beginning at three days post cell implantation. At the end of the study, mice were sacrificed and digital photographs were taken. A (top): photographs of representative mice A (bottom): photographs of excised tumors. B (top): tumor growth kinetics. Tumor growth was measured weekly using digital calipers and the average tumor volume was graphed as a function of time. After 11 weeks, PL treatment was stopped and tumor growth was measured through 16 weeks post cell implantation. *, p<0.05 from the control group. B (bottom): Tumor weight at 11 weeks post cell implantation. C (left): Immunohistochemistry of tumor tissue for PKCε, Stat3 and PCNA with negative controls for specificity. Magnification, x40 (left). C (right): Quantitation of Stat3 and PCNA-positive stained nuclei (right). Columns: mean from ten different views; bars: SE. *, p<0.000 from the control group. D (left): Expression of VEGF and MMP-9 in tumors from PL treated and control mice. D (right): Quantitation of VEGF and MMP-9 expression.
C. Plumbagin induced-inhibition of PCa cell growth accompanies inhibition of the expression of multiple molecular targets including PKCε
To obtain clues about the mechanism by which PL may inhibit growth and invasion of PCa, we used both DU145 cells cultured in vitro and DU145 tumor xenografts from vehicle treated and PL treated mice. The results are illustrated in Fig. 3. PKCε expression and constitutive activation of Stat3 have been shown to play a role in the progression of human PCa (14). Stat3 activation, which involves dimerization, nuclear translocation, DNA-binding and transactivation of transcription, requires phosphorylation of both tyrosine 705 and serine 727 (14). Stat3Tyr705 phosphorylation is mediated by a wide variety of growth factors (e.g., IL-6). IL-6 signaling is mediated through Janus kinase (JAK). JAK-STAT is the classical pathway that has been shown to mediate cellular responses to a variety of cytokines including IL-6. In response to IL-6, Stat3 is transiently associated with gp130 and subsequently phosphorylated by JAKs on tyrosine 705 of Stat3. PKCε-mediated Stat3 Ser727 phosphorylation is also essential for both optimal DNA-binding and transcriptional activities of Stat3 (14). A shown in Fig. 3A-D, PL treatment inhibited the expression of pJAK-2 and PKCε. PL-mediated inhibition of pJAK-2 and PKCε expression accompanied inhibition of both Stat3Ser727 and Stat3Tyr705 phosphorylation (Fig.3 A-D) and Stat3 DNA-binding activity (Fig. 6A). The effects of PL on the expression of other PKC isoforms was also determined (Fig. 4). PL inhibited the expression of PKCε and PKC β1. PKCα expression was slightly increased while expression levels of other PKC isoforms (PKCβ,γ,δ,η,ζ,μ) were unaffected (Fig. 4). Constitutively activated PKCε is linked to cell survival essential for maintenance of PCa. We observed in PCa from TRAMP mice that PKCε expression accompanied upregulation of phosphorylated PI3K and AKT, major components of the cell survival pathway (14). These results prompted us to analyze the effects of PL on the expression of PI3K and AKT in DU145 cells and tumors. The results are shown in Fig. 5. PL treatment inhibited the expression of the PI3K (p85) and PI3K (p110) regulatory subunits and pAKT (Ser473 and Thr308) (Fig. 5 A and B). We also observed PL treatment induced the expression of p21 and p27 (Fig. 5 C and D).
Figure 3. Plumbagin inhibits PKCε expression as well as JAK-2 and Stat3 phosphorylation in DU145 cells in vitro and in vivo.
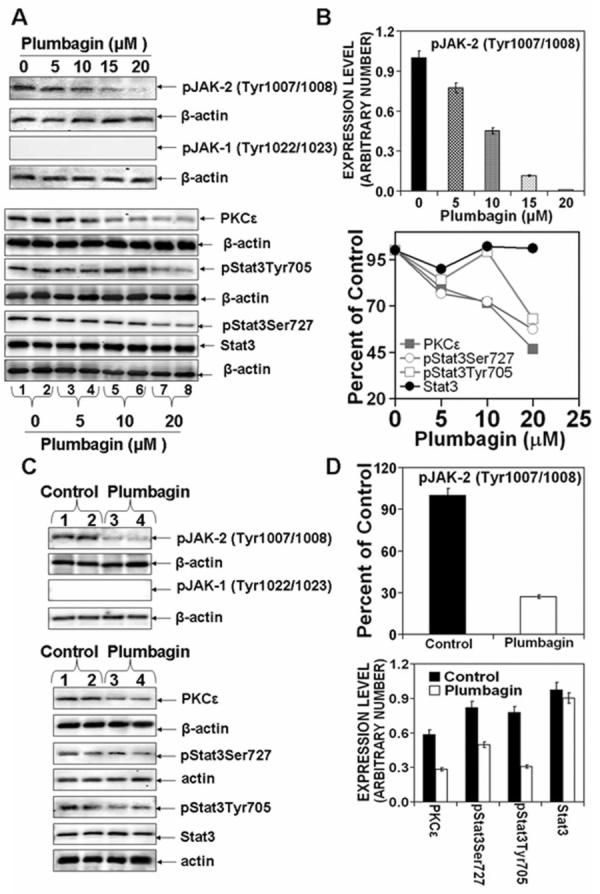
A and B: DU145 cells at 70-80% confluency were serum starved for 24 hrs. Cells were treated with 0, 5, 10, 15 or 20 μM PL in DMSO (final concentration 0.1%) for 6hrs. Whole cell lysates were prepared and used for Western blot analysis of the indicated proteins. C and D: DU145 cells (2.5 × 106 cells in 100 μl in a 1:1 of media:Matrigel) were implanted on both flanks of nude mice. Animals were treated with PL (2 mg/kg body weight in PBS or PBS only, five days a week) by intra-peritoneal injection beginning three days post implantation. At the end of the study, tumors from PL treated or control mice were excised and whole-cell lysates were prepared. Protein extracts (25 μg protein) were immunoblotted and indicated proteins detected with the appropriate antibodies. Protein levels were normalized to β-actin. Western blots (A and C) were quantitated (B and D) by densitometric analysis using Total lab Nonlinear Dynamic Image analysis software (Nonlinear USA Inc., Durham, NC).
Figure 6. Plumbagin inhibits DNA binding of transcription factors (TF) Stat3, NFkB and AP-1 and TF-regulated gene expression.
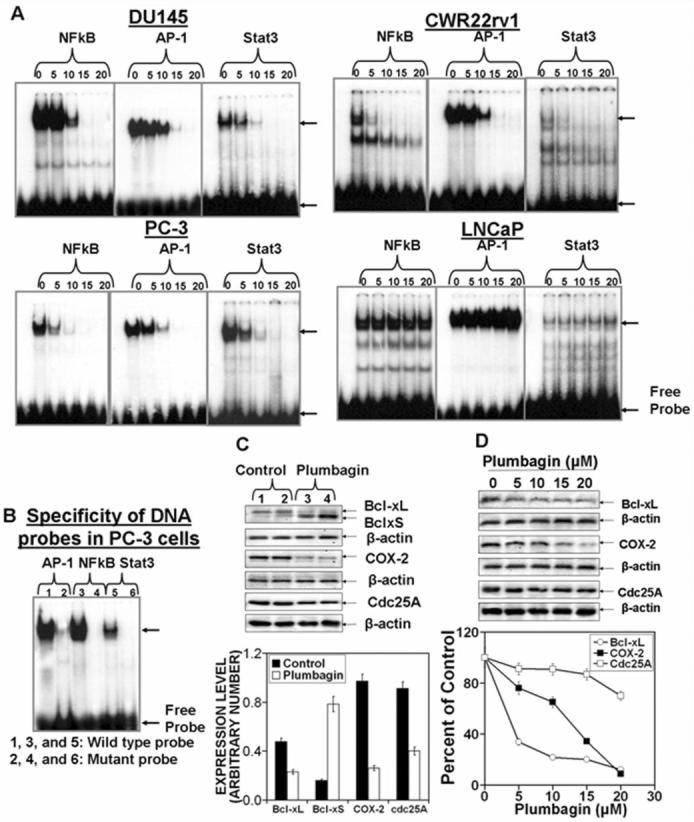
PCa cells (DU145, PC-3, CWR22rv1 and LNCaP) at 70-80% confluency were serum starved for 24 hrs. Cells were treated with 0, 5, 10, 15, or 20 μM PL in DMSO (final concentration 0.1%) for 3hrs. Nuclear protein extracts were prepared by lysing cells in a hypotonic solution (10 mM HEPES, pH 7.5; 10 mM KCl; 0.1 mM EDTA, pH 8.0; 0.1 mM EGTA pH 8.0; 1 mM DTT; 0.5 mM PMSF; 0.5 mg/ml benzamide; 2 μg/ml aprotinin; 2 μg/ml leupeptin), with detergent (NP-40 at 6.25% (v/v)) followed by low speed (1500 × g for 30 secs) to collect nuclei. Nuclear proteins were extracted in a high-salt buffer (20 mM HEPES, pH 7.5; .4 M NaCl; 1 mM EDTA, pH 8.0; 1 mM EGTA pH 8.0; 1 mM DTT; 1 mM PMSF; 0.5 mg/ml benzamide; 2 μg/ml aprotinin; 2 μg/ml leupeptin) and nuclear membranes and genomic DNA removed by high-speed centrifugation. Nuclear protein extracts were stored at −70°C until used. A: EMSA of NFkB, AP-1 and Stat3 DNA binding; B: Specificity of AP-1, NFkB and Stat3 DNA binding. C and D: TF-regulated gene expression. C: Tumors from PL treated or control mice were excised and whole-cell lysates were prepared to analyze the expression of indicated proteins. D: DU145 cells at 70-80% confluency were serum starved for 24 hrs. Cells were treated with 0, 5, 10, 15 or 20 μM PL for 6hrs. Whole cell lysates were prepared and used for Western blot analysis of indicated proteins. Quantitation of Western blots C and D (Bottom).
Figure 4. Effects of plumbagin on the expression of PKC isoforms.
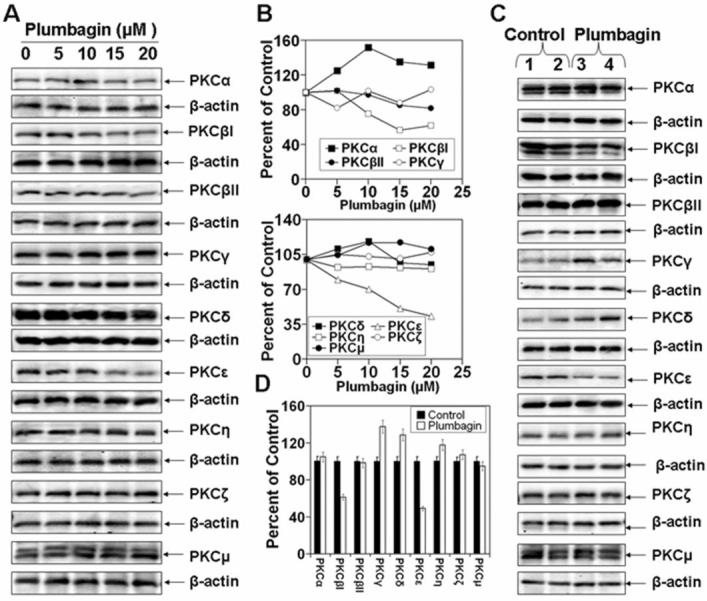
DU145 cells at 70-80% confluency were serum starved for 24 hrs. Cells were treated with 0, 5, 10, 15 or 20 μM PL in DMSO (final concentration 0.1%) for 6hrs. Whole cell lysates were prepared and used for Western blot analysis of PKC isoforms (A). Tumors from PL treated or control mice were excised and whole-cell lysates were prepared to analyze the expression of PKC isoforms (C). Quantitation of Western blots (B and D).
Figure 5. Effects of plumbagin on the expression PI3K, AKT, p21 and p27.

DU145 cells at 70-80% confluency were serum starved for 24 hrs. Cells were treated with 0, 5, 10, 15 or 20 μM PL DMSO (final concentration 0.1%) for 6hrs. Whole cell lysates were prepared and used for Western blot analysis of PI3K, AKT, p21 and p27 (A and C). Tumors from PL treated or control mice were excised and whole-cell lysates were prepared to analyze the expression PI3K, AKT, p21 and p27 (B and D).
D. Plumbagin treatment indiscriminately inhibits the DNA binding activity of transcriptional factors AP-1, NFkB, and Stat3 in PCa cell lines
Activation of PKCε and PI3K/AKT pathways culminate in the activation of transcription factors (AP-1, NFkB, and Stat3), which drive the expression of cell survival genes (14). Sandur et. al. (1) have reported that PL-modulated cell proliferation, carcinogenesis, and radioresistance may be due to inhibition of NFkB pathway. We found that PL not only inhibited the DNA-binding of NFkB, but also of AP-1 and Stat3, in PCa cell lines DU145, PC3, and CWR22rv1 (Fig. 6). Inhibition of the DNA-binding activity was observed at PL concentrations as low as 5 μM (Fig. 6). Fig. 6 also shows that PL inhibited the expression of several cell survival genes (COX-2, cdc25A, Bcl-xL) (Fig. 6 C and D).
Discussion
Prostate cancer (PCa) is the most common type of cancer in American men and ranks second to lung cancer in cancer-related deaths (1). Hormone refractory invasive PCa is the end stage and accounts for the majority of PCa patient deaths (2-7). Men with hormone refractory cancer are at high risk for developing bone metastasis which result in clinically significant skeletal morbidity (15-18). The management of locally advanced PCa is difficult and complex because the cancer often becomes unresponsive to current chemotherapeutic agents. Several agents such as selenium, lycopene, soy products, green tea, pomegranate phenolics, apigenin and vitamins D and E have been shown to be effective in the prevention of the induction of PCa (19-22). However, there is no agent which is in fact effective and selective in the prevention and/or treatment of late stage hormone refractory PCa. We present here that PL, a quinoid constituent isolated from the roots of medicinal plant Plumbago zeylanica L (also known as Chitrak) (7), induces apoptosis and inhibits invasion of AI PCa cells (Fig. 1). Administration of PL (2mg/kg body weight), beginning 3 days after ectopic implantation of hormone refractory DU145 PCa cells, delays tumor growth by 3 weeks and reduces both tumor weight and volume by 90% (Fig. 2). Also, PL abrogates the expression of PKCε (Fig. 3), which plays a role in the development and maintenance of AI PCa (14).
The results (Fig. 1) involving the induction of apoptosis in PCa cells by PL are consistent with findings using other cancer cell lines such as ovarian cancer BG1 cells (23), cervical cancer cells (24) and breast cancer cells (9). PL-induced apoptosis involves G2/M arrest and generation of reactive oxygen species (ROS) (10). ROS-mediated inhibition of topoisomerase II has been suggested to be a mechanism contributing to the apoptosis-inducing properties of PL (25). It is also noteworthy that PL in breast cancer cell lines has been reported to trigger autophagic cell death but not predominantly apoptosis (9).
We provide direct experimental evidence that PL has efficacy in preclinical model of ectopic growth of PCa cells in nude mice (Fig. 2). Inhibition of tumor growth may be the result of inhibition of the expression of cell proliferative marker PCNA as well as inhibition of the constitutive activation of cell survival markers PKCε and Stat3 (Fig. 3).
Metastasis is the primary cause of mortality from cancer (15-18). Cell migration and invasion play critical roles in cancer metastasis (15-18). PL was observed to be a potent inhibitor of PCa cell invasion (Fig. 1). The molecular mechanism linked to PL-induced inhibition of PCa cell invasion may involve inhibition of the expression of MMP-9 and VEGF (Fig. 1), the components in cell invasion and metastasis (26-28).
PL inhibits PKCε expression and Stat3 activation (Fig. 3 and 4). PKCε is a member of the novel PKC subfamily (29-33). PKCε is an important component of the mechanism of induction and progression of PCa (14). PKCε is overexpressed in human PCa and PCa developed either in C57BL/6 or [C57BL/6 × FVB] F1 TRAMP mice (14). The fact that PKCε expression is significantly elevated in PCa and correlates with PCa aggressiveness (14, 34) implies that PKCε is probably linked to the maintenance of AI PCa. In this context, the pioneering work of Terrian and his associates on the role of PKCε in prostate carcinogenesis, using PCa derived cell lines, is noteworthy (34-37). In their reports, PKCε overexpression transformed AD LNCaP tumor cells to AI cells (35). The transformation of AD LNCaP cells to an AI variant was associated with increased cell proliferation and resistance to apoptosis. Antisense experiments established that endogenous PKCε plays an important role in regulating the growth and survival of AI PCa cells, suggesting that PKCε expression may be sufficient to maintain PCa growth and survival after androgen ablation (35). PKC has been shown to be a transforming oncogene, and a predictive biomarker of breast cancer and PCa (14).
PKCε associates with Stat3 and regulates Stat3 activation. Stat3 is activated by phosphorylation at both tyrosine705 and serine727 residues. Constitutively activated STATs, in particular Stat3 have been found in a number of human cancers (e.g., SCCs, head and neck, breast, ovary, prostate, lung) (38-45). PKCε activation transduces multiple signals involving inhibition of apoptotic pathways and promotion of cell survival pathways. PKCε-mediated cell survival pathway involves constitutive activation of Stat3. PKCε is an initial signal that regulates activation of Stat3. PKCε may be a primary target of PL for prevention of AI PCa progression.
PL inhibits the activation of PI3K/AKT. As observed in PCa from TRAMP mice, PKCε expression accompanied up-regulation of phosphorylated PI3K and AKT, major components of the cell survival pathway (14). Consistent with these findings, using CWR22 xenografts, it was shown by proteomic analysis that the association of PKCε with Bax may neutralize apoptotic signals propagated through the mitochondrial deathsignaling pathway (46). Also, integrin signaling links PKCε to the PKB/Akt survival pathway in recurrent PCa cells (34). PL inhibits PKCε overexpression, which correlates with PCa aggressiveness and accompanies an increase in proteins that modulate apoptosis (survivin, Bcl-2, Bcl-xL), cell cycle progression (p21, p27) and cell survival (Stat3).
It is notable that Sandur et. al. (7) reported that PL is a specific inhibitor of NFkB and does not suppress activation of other transcription factors AP-1 and Stat3 in KBM-5 (human chronic myeloid leukemia) and U266 (human multiple myeloma) cells. The discrepancy between our results with the PCa cell lines (PC-3, DU145 and CWR22rv1) and their results with KBM-5 and U266 cells may be due to cellular context.
In several repeat experiments, PL inhibited the constitutive activation of AP-1, NFkB and Stat3 in AI PCa cell lines PC-3, DU145 and CWR22rv1 but not in AD PCa cell line LNCaP. These results indicate that androgen receptor (AR) status may determine PL-induced suppression of transcription factors AP-1, NFkB and Stat3. The mechanism by which PL suppresses the constitutive activation of AP-1, NFkB and Stat3 in AI PCa cells is unclear. However PL inhibits constitutive expression of PKCε, which may play role in the activation of AP-1, NFkB and Stat3.
The role of PKCε in PL-induced inhibition of growth and invasion of AI PCa is speculative. Most AI PCa continue to express AR as well as the AD gene PSA, which indicates that these cells maintain a functional AR signaling pathway despite castrate levels of testosterone. Gene amplification and mutations in AR are frequently observed in recurrent PCa, which may account for hypersensitivity of the AR to low castrate level of androgens, and altered ligand specificity (47). Increased AR activity in AI PCa is perhaps caused by cross talk of AR with multiple intacellular signaling cascades including peptide growth factors (EGF, TGFβ, IGF-1) (48). In this context, it is noteworthy that HER-2/neu, a member of the EGF family of receptor tyrosine kinases, activates the androgen receptor pathway in the absence of ligand (49). It remains to be determined the whether there is cross talk between AR and PKCε signal transduction pathway in the progression of AI PCa,
PL has also been extensively evaluated for toxic side effects in rodents. Toxic side effects included diarrhea, skin rashes, hepatic and reproductive toxicity. These toxic side effects were dose-related. The LD50 for these side effects in mice was 8-65 mg/kg body weight for oral administration, and 16 mg/kg body weight for i.p. (7). PL has been reported to be nontoxic at doses (2 mg/kg body weight i.p. or 200 ppm in diet) shown to elicit chemopreventive and therapeutic effects (7). Also, the mutagenic activity of PL in Escherichia coli has been examined and was negative in the Ames test (7).
In summary, PL, a plant-derived naphthoquinone, inhibits the growth and invasion of AI PCa cells (Figs. 1 and 2). PL-induced inhibition of PCa cell growth and invasion accompany inhibition of multiple targets including PKCε and transcription factors AP-1, NFkB and Stat3 (Figs. 3, 4 and 6). The results (Figs. 1-6) presented have led us to propose that PKCε is a master switch in the progression and invasion of hormone refractory PCa. PKCε directly or indirectly via association with other protein kinases (e.g. Raf-1, MEK1/2, ERK1/2, p38MAPK) phosphorylates Stat3Ser727. Constitutive activation of PKCε and Stat3 are correlated with the aggressiveness of PCa (14). PI3K/PKD3/Akt may phosphorylate AR, enabling to form dimmers, thus enhancing AR-DNA binding and gene expression (50). We hypothesize that PL inhibits the expression of PKCε, an initial signal in the development of AI PCa.
Acknowledgements
This work was supported in parts by DOD Grant W81XWH and NIH Grant CA35368 to AKV.
Abbreviations
- PL
Plumbagin
- PCa
Prostate cancer
- AD
androgen dependent
- AI
androgen independent
- STAT
signal transducers and activators of transcription
- PKC
Protein Kinase C
Footnotes
Disclosure of Potential Conflicts of Interest
None
References
- 1.Jemal A, Murray T, Ward E, et al. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.Edwards J, Bartlett JM. The androgen receptor and signal-transduction pathways in hormone-refractory prostate cancer. Part 2: Androgen-receptor cofactors and bypass pathways. BJU Int. 2005;95:1327–35. doi: 10.1111/j.1464-410X.2005.05527.x. [DOI] [PubMed] [Google Scholar]
- 3.Zhou J, Scholes J, Hsieh JT. Signal transduction targets in androgen-independent prostate cancer. Cancer Metastasis Rev. 2001;20:351–62. doi: 10.1023/a:1015504015302. [DOI] [PubMed] [Google Scholar]
- 4.Silvestris N, Leone B, Numico G, et al. Present status and perspectives in the treatment of hormone-refractory prostate cancer. Oncology. 2005;69:273–82. doi: 10.1159/000089676. [DOI] [PubMed] [Google Scholar]
- 5.Chau CH, Figg WD. Molecular and phenotypic heterogeneity of metastatic prostate cancer. Cancer Biol Ther. 2005;4:166–7. doi: 10.4161/cbt.4.2.1571. [DOI] [PubMed] [Google Scholar]
- 6.Quinn DI, Henshall SM, Sutherland RL. Molecular markers of prostate cancer outcome. Eur J Cancer. 2005;41:858–87. doi: 10.1016/j.ejca.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 7.Sandur SK, Ichikawa H, Sethi G, et al. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) suppresses NF-kappaB activation and NF-kappaB-regulated gene products through modulation of p65 and IkappaBalpha kinase activation, leading to potentiation of apoptosis induced by cytokine and chemotherapeutic agents. J Biol Chem. 2006;281:17023–33. doi: 10.1074/jbc.M601595200. [DOI] [PubMed] [Google Scholar]
- 8.Sugie S, Okamoto K, Rahman KM, et al. Inhibitory effects of plumbagin and juglone on azoxymethane-induced intestinal carcinogenesis in rats. Cancer Lett. 1998;127:177–83. doi: 10.1016/s0304-3835(98)00035-4. [DOI] [PubMed] [Google Scholar]
- 9.Kuo PL, Hsu YL, Cho CY. Plumbagin induces G2-M arrest and autophagy by inhibiting the AKT/mammalian target of rapamycin pathway in breast cancer cells. Mol Cancer Ther. 2006;5:3209–21. doi: 10.1158/1535-7163.MCT-06-0478. [DOI] [PubMed] [Google Scholar]
- 10.Hsu YL, Cho CY, Kuo PL, et al. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) induces apoptosis and cell cycle arrest in A549 cells through p53 accumulation via c-Jun NH2-terminal kinase-mediated phosphorylation at serine 15 in vitro and in vivo. J Pharmacol Exp Ther. 2006;318:484–94. doi: 10.1124/jpet.105.098863. [DOI] [PubMed] [Google Scholar]
- 11.Wang CC, Chiang YM, Sung SC, et al. Plumbagin induces cell cycle arrest and apoptosis through reactive oxygen species/c-Jun N-terminal kinase pathways in human melanoma A375.S2 cells. Cancer Lett. 2008;259:82–98. doi: 10.1016/j.canlet.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 12.Powolny AA, Singh SV. Plumbagin-induced Apoptosis in Human Prostate Cancer Cells is Associated with Modulation of Cellular Redox Status and Generation of Reactive Oxygen Species. Pharm Res. 2008 doi: 10.1007/s11095-008-9533-3. [DOI] [PubMed] [Google Scholar]
- 13.Rajesh D, Schell K, Verma AK. Ras mutation, irrespective of cell type and p53 status, determines a cell's destiny to undergo apoptosis by okadaic acid, an inhibitor of protein phosphatase 1 and 2A. Mol Pharmacol. 1999;56:515–25. doi: 10.1124/mol.56.3.515. [DOI] [PubMed] [Google Scholar]
- 14.Aziz MH, Manoharan HT, Church DR, et al. Protein kinase Cepsilon interacts with signal transducers and activators of transcription 3 (Stat3), phosphorylates Stat3Ser727, and regulates its constitutive activation in prostate cancer. Cancer Res. 2007;67:8828–38. doi: 10.1158/0008-5472.CAN-07-1604. [DOI] [PubMed] [Google Scholar]
- 15.Bussard KM, Gay CV, Mastro AM. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. 2008;27:41–55. doi: 10.1007/s10555-007-9109-4. [DOI] [PubMed] [Google Scholar]
- 16.Keller ET, Dai J, Escara-Wilke J, et al. New trends in the treatment of bone metastasis. J Cell Biochem. 2007;102:1095–102. doi: 10.1002/jcb.21540. [DOI] [PubMed] [Google Scholar]
- 17.Kingsley LA, Fournier PG, Chirgwin JM, et al. Molecular biology of bone metastasis. Mol Cancer Ther. 2007;6:2609–17. doi: 10.1158/1535-7163.MCT-07-0234. [DOI] [PubMed] [Google Scholar]
- 18.Valdespino V, Tsagozis P, Pisa P. Current perspectives in the treatment of advanced prostate cancer. Med Oncol. 2007;24:273–86. doi: 10.1007/s12032-007-0017-9. [DOI] [PubMed] [Google Scholar]
- 19.Gupta S. Prostate cancer chemoprevention: current status and future prospects. Toxicol Appl Pharmacol. 2007;224:369–76. doi: 10.1016/j.taap.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Patel D, Shukla S, Gupta S. Apigenin and cancer chemoprevention: progress, potential and promise (review) Int J Oncol. 2007;30:233–45. [PubMed] [Google Scholar]
- 21.Adhami VM, Mukhtar H. Polyphenols from green tea and pomegranate for prevention of prostate cancer. Free Radic Res. 2006;40:1095–104. doi: 10.1080/10715760600796498. [DOI] [PubMed] [Google Scholar]
- 22.Bemis DL, Katz AE, Buttyan R. Clinical trials of natural products as chemopreventive agents for prostate cancer. Expert Opin Investig Drugs. 2006;15:1191–200. doi: 10.1517/13543784.15.10.1191. [DOI] [PubMed] [Google Scholar]
- 23.Srinivas G, Annab LA, Gopinath G, et al. Antisense blocking of BRCA1 enhances sensitivity to plumbagin but not tamoxifen in BG-1 ovarian cancer cells. Mol Carcinog. 2004;39:15–25. doi: 10.1002/mc.10164. [DOI] [PubMed] [Google Scholar]
- 24.Srinivas P, Gopinath G, Banerji A, et al. Plumbagin induces reactive oxygen species, which mediate apoptosis in human cervical cancer cells. Mol Carcinog. 2004;40:201–11. doi: 10.1002/mc.20031. [DOI] [PubMed] [Google Scholar]
- 25.Kawiak A, Piosik J, Stasilojc G, et al. Induction of apoptosis by plumbagin through reactive oxygen species-mediated inhibition of topoisomerase II. Toxicol Appl Pharmacol. 2007;223:267–76. doi: 10.1016/j.taap.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 26.Shankar S, Ganapathy S, Chen Q, et al. Curcumin sensitizes TRAIL-resistant xenografts: molecular mechanisms of apoptosis, metastasis and angiogenesis. Mol Cancer. 2008;7:16. doi: 10.1186/1476-4598-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kong D, Li Y, Wang Z, et al. Inhibition of angiogenesis and invasion by 3,3′-diindolylmethane is mediated by the nuclear factor-kappaB downstream target genes MMP-9 and uPA that regulated bioavailability of vascular endothelial growth factor in prostate cancer. Cancer Res. 2007;67:3310–9. doi: 10.1158/0008-5472.CAN-06-4277. [DOI] [PubMed] [Google Scholar]
- 28.Adhami VM, Ahmad N, Mukhtar H. Molecular targets for green tea in prostate cancer prevention. J Nutr. 2003;133:2417S–24S. doi: 10.1093/jn/133.7.2417S. [DOI] [PubMed] [Google Scholar]
- 29.Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007;7:281–94. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 30.Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J. 1998;332(Pt 2):281–92. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newton AC. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev. 2001;101:2353–64. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- 32.Mochly-Rosen D, Kauvar LM. Modulating protein kinase C signal transduction. Adv Pharmacol. 1998;44:91–145. doi: 10.1016/s1054-3589(08)60126-x. [DOI] [PubMed] [Google Scholar]
- 33.Basu A, Sivaprasad U. Protein kinase Cepsilon makes the life and death decision. Cell Signal. 2007;19:1633–42. doi: 10.1016/j.cellsig.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cornford P, Evans J, Dodson A, et al. Protein kinase C isoenzyme patterns characteristically modulated in early prostate cancer. Am J Pathol. 1999;154:137–44. doi: 10.1016/S0002-9440(10)65260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu D, Foreman TL, Gregory CW, et al. Protein kinase cepsilon has the potential to advance the recurrence of human prostate cancer. Cancer Res. 2002;62:2423–9. [PubMed] [Google Scholar]
- 36.Wu D, Thakore CU, Wescott GG, et al. Integrin signaling links protein kinase Cepsilon to the protein kinase B/Akt survival pathway in recurrent prostate cancer cells. Oncogene. 2004;23:8659–72. doi: 10.1038/sj.onc.1207900. [DOI] [PubMed] [Google Scholar]
- 37.Wu D, Terrian DM. Regulation of caveolin-1 expression and secretion by a protein kinase cepsilon signaling pathway in human prostate cancer cells. J Biol Chem. 2002;277:40449–55. doi: 10.1074/jbc.M206270200. [DOI] [PubMed] [Google Scholar]
- 38.Huang HF, Murphy TF, Shu P, et al. Stable expression of constitutively-activated STAT3 in benign prostatic epithelial cells changes their phenotype to that resembling malignant cells. Mol Cancer. 2005;4:2. doi: 10.1186/1476-4598-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alvarez JV, Febbo PG, Ramaswamy S, et al. Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors. Cancer Res. 2005;65:5054–62. doi: 10.1158/0008-5472.CAN-04-4281. [DOI] [PubMed] [Google Scholar]
- 40.Burke WM, Jin X, Lin HJ, et al. Inhibition of constitutively active Stat3 suppresses growth of human ovarian and breast cancer cells. Oncogene. 2001;20:7925–34. doi: 10.1038/sj.onc.1204990. [DOI] [PubMed] [Google Scholar]
- 41.Ni Z, Lou W, Leman ES, et al. Inhibition of constitutively activated Stat3 signaling pathway suppresses growth of prostate cancer cells. Cancer Res. 2000;60:1225–8. [PubMed] [Google Scholar]
- 42.Fernandes A, Hamburger AW, Gerwin BI. ErbB-2 kinase is required for constitutive stat 3 activation in malignant human lung epithelial cells. Int J Cancer. 1999;83:564–70. doi: 10.1002/(sici)1097-0215(19991112)83:4<564::aid-ijc20>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 43.Kobielak A, Fuchs E. Links between alpha-catenin, NF-kappaB, and squamous cell carcinoma in skin. Proc Natl Acad Sci U S A. 2006;103:2322–7. doi: 10.1073/pnas.0510422103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levy DE, Darnell JE., Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 45.Chan KS, Sano S, Kiguchi K, et al. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J Clin Invest. 2004;114:720–8. doi: 10.1172/JCI21032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McJilton MA, Van Sikes C, Wescott GG, et al. Protein kinase Cepsilon interacts with Bax and promotes survival of human prostate cancer cells. Oncogene. 2003;22:7958–68. doi: 10.1038/sj.onc.1206795. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki H, Ueda T, Ichikawa T, et al. Androgen receptor involvement in the progression of prostate cancer. Endocr Relat Cancer. 2003;10:209–16. doi: 10.1677/erc.0.0100209. [DOI] [PubMed] [Google Scholar]
- 48.Danielpour D. Functions and regulation of transforming growth factor-beta (TGF-beta) in the prostate. Eur J Cancer. 2005;41:846–57. doi: 10.1016/j.ejca.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 49.Craft N, Shostak Y, Carey M, et al. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med. 1999;5:280–5. doi: 10.1038/6495. [DOI] [PubMed] [Google Scholar]
- 50.Chen J, Deng F, Singh SV, et al. Protein kinase D3 (PKD3) contributes to prostate cancer cell growth and survival through a PKCepsilon/PKD3 pathway downstream of Akt and ERK 1/2. Cancer Res. 2008;68:3844–53. doi: 10.1158/0008-5472.CAN-07-5156. [DOI] [PubMed] [Google Scholar]