

Abstract
In mammals, dosage compensation is achieved by X-chromosome inactivation (XCI) in the female. The noncoding Xist gene initiates silencing of the X, while its antisense partner Tsix blocks silencing. The complementarity of Xist and Tsix RNAs has long suggested a role for RNAi. Here, we report that murine Xist and Tsix form duplexes in vivo. During XCI, the duplexes are processed to small RNAs, most likely on the active X (Xa) in a Dicer-dependent manner. Deleting Dicer compromises small RNA production and de-represses Xist. Furthermore, without Dicer, Xist RNA cannot accumulate and H3-K27 trimethylation is blocked on the inactive X (Xi). Intriguingly, the defects are partially rescued by truncating Tsix. Thus, XCI and RNAi intersect, downregulating Xist on Xa and spreading silencing on Xi.
X-chromosome inactivation (XCI) (1) balances X-chromosome dosages between XX and XY individuals. XCI is initiated by Xist (2, 3) and opposed by Tsix (4). How Xist induces XCI on Xi and how Tsix stably silences Xist on Xa remain two unanswered questions. A role for RNA-interference (RNAi) has long been speculated. RNAi refers to the repressive influence of double-stranded RNA (dsRNA) on gene transcription and transcript stability (5, 6). Numerous similarities, including the involvement of noncoding RNAs, can be found between XCI and RNAi-silencing of constitutive heterochromatin. However, a Dicer (Dcr) efficiency has no obvious effect on maintaining Xi in T cells (7) and, although Xist and Tsix RNAs are perfectly complementary, dsRNAs had never been observed in vivo.
Here, we formally explore a role for RNAi in XCI. To search for small RNAs within Xist/Tsix, we performed Northern analysis in mouse embryonic stem (ES) cells, a model that recapitulates XCI during cell differentiation ex vivo, and in mouse embryonic fibroblasts (MEFs), post-XCI cells which faithfully maintain one Xi. At repeat A, a region of Xist required for silencing (8), we observed small RNAs at ∼30 nt and ∼37 nt in the Tsix orientation and at ∼25 nt and ∼35 nt in the Xist orientation (Fig. 1A). At Xist exon 7, small RNAs occurred between 24-42 nt on the Tsix strand and at ∼25 and ∼35 nt on the Xist strand (Fig. 1B). At the promoter, robust quantities of Tsix-strand small RNAs were observed (Fig. 1C). Small RNAs were also seen on the Xist strand, implying low level sense transcription must occur at the promoter. The integrity of all Northern blots were confirmed by miRNA292-as and tRNA controls (Fig. 1, S1). Intriguingly, the small RNAs were developmentally regulated, being unmeasurable in the pre-XCI (day 0, d0) and post-XCI (MEF) states and detectable only during XCI (d4, d10). Furthermore, small RNAs occurred in both XX and XY cells. For discussion purposes, we tentatively call them ‘xiRNA’ for their X-inactivation center origin, distinct from the smaller siRNAs and miRNAs.
Figure 1. Small RNAs derived from Tsix/Xist.

(A) xiRNAs from Xist repeat A (XA) region (map) detected by Northern analysis. Sense (s) and antisense (as) riboprobes detect Tsix and Xist, respectively. miR292-as controls are shown on same blots. M, male. F, female.
(B) Northern analysis of xiRNAs from Xist exon 7.
(C) Northern analysis of Xist promoter region.
(D) Northern analysis of mutant cells. WT lanes identical to those in panel A (concurrent analysis).
To determine if xiRNA production depends on antisense expression, we investigated ES cells deleted for Tsix (TsixΔCpG) (4) and the Tsix regulator, Xite (XiteΔL) (9). Deleting Tsix resulted in a dramatic reduction in antisense-strand xiRNA (Fig. 1D). A residual level of xiRNAs was still detectable, consistent with cryptic promoter activity in TsixΔCpG (4). Deleting Xite likewise reduced antisense xiRNA levels, consistent with a requirement for Xite in transactivating Tsix (9). In the sense orientation, both deletions also compromised xiRNA production. Thus, small RNAs are indeed generated from Xist/Tsix and depend on Tsix and Xite expression.
The presence of xiRNAs implied that Tsix and Xist must exist as long duplex precursors. However, the developmental timing of xiRNA appearance is paradoxical: Although Tsix and Xist are biallelically expressed on d0, they become monoallelically expressed on opposite Xs during XCI (4). On d0, 3-5 copies/per chromosome of Xist RNA are present, while Tsix occurs at >10-fold molar excess (10-12). Upon XCI, Tsix is downregulated on Xi as Xist upregulates >30-fold. On Xa, Tsix persists as Xist is downregulated. How would dsRNA form when Tsix and Xist – both cis-limited – are on opposite chromosomes during XCI?
To determine if Tsix and Xist formed dsRNA, we devised an in vivo RNase protection assay based on differential susceptibility of ssRNA and dsRNAs to RNase A/T1. We permeabilized replicate preparations of d0 ES cells in a nondenaturing buffer containing DNAse I and RNAse, treated with RNase, and performed strand-specific RT-PCR on the protected RNAs. To confirm assay sensitivity, a positive control -- into which 1 copy/cell of in vitro-transcribed and annealed Tsix:Xist dsRNA is ‘spiked’ -- could readily be detected using this protocol (Fig. 2A,B). The abundant single-stranded Rrm2 and Gapdh transcripts were not amplified, indicating that our assay was specific for dsRNA. We consistently observed RNase-protected Xist and Tsix RNA strands in XX and XY ES cells, suggesting the presence of dsRNA (Fig. 2B). Real-time RT-PCR quantitation showed that ∼16% of Xist and ∼13% of Tsix were protected (Fig. 2C). As expected of duplexes, approximately equal stoichiometric ratios of the two strands were present in the RNase-protected fraction. Kinetic analysis revealed decreasing amounts of dsRNA during differentiation in XX and XY cells (Fig. 2D). Thus, steady state quantities of both dsRNA and xiRNA are developmentally regulated, but in an opposite manner.
Figure 2. Tsix and Xist RNA form long duplexes in vivo.
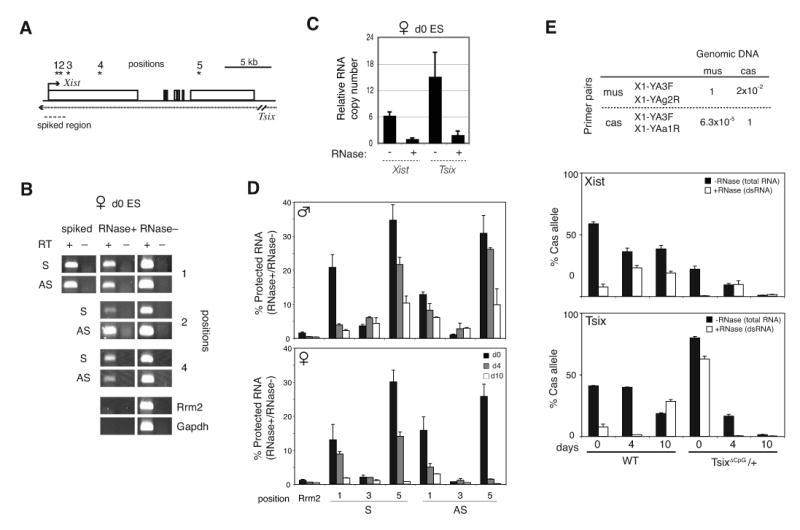
(A) Map of Tsix/Xist and primer pairs (*).
(B) In vivo RNAse protection assay. S, sense. AS, antisense.
(C) Relative quantities of Xist and Tsix in duplexes measured at position 2 (bp 1206-1337 of Xist) by strand-specific real-time RT-PCR of protected RNA (RNAse+) as compared to total levels (RNAse–). Quantities are standardized to Xist in the Xist-Tsix duplex (Xist, RNAse+ = 1). Error bars=one standard deviation (SD) in triplicate reactions.
(D) Quantities of protected Tsix or Xist RNAs (RNase+) relative to total Tsix or Xist (RNase-) for in vivo RNAse protection assays. Error bar=1SD, triplicate reactions.
(E) In vivo RNAse protection assays to test allelic origin of dsRNA using strand-specific, allele-specific realtime RT-PCR with SNP-based primers for Xcas or Xmus alleles (Table). PCR of control genomic DNA shows high specificity (98% for mus, >99.99% for cas). Error bar=1SD, triplicate reactions. For test samples, the mus and cas fractions were amplified separately, normalized to genomic DNA (Xcas: Xmus =1:1), and plotted as a function of time. %cas = [Xcas RNA/ (Xcas RNA + Xmus RNA)] × 100. Because the bars show relative allelic fractions, quantities are only comparable within a single timepoint.
The inverse correlation over time raised the possibility that dsRNA is processed to xiRNA. To address potential allelic differences, we performed allele-specific RNase protection assays using two genetically marked female ES cell lines – wildtype 16.7, which carries Xs from Mus castaneus (Xcas) and M. musculus (Xmus) and undergoes random XCI (with a natural 30:70 specific-specific bias (13)); and TsixΔCpG/+ mutants, which harbors a Tsix deletion on Xmus (4) and therefore always inactivates Xmus in the 16.7 background. Total Tsix RNA (no RNase treatment) decreased >10-fold over time, but a low residual level could still be detected at d4 and d10 as expected (data not shown) (4, 9, 14). From this residual pool, using SNP-based allele-specific primers at position 3, we unexpectedly found that duplexed Tsix (RNase-protected) in TsixΔCpG/+ cells predominantly originated from Xi (Xmus) (Fig. 2E) – the X on which the major Tsix promoter is deleted. Likewise, the Xist strand found in duplex form originated from Xi. Thus, Tsix:Xist duplexes are detected primarily from Xi.
Duplexes may form only on Xi, or they may form on both Xs but be stable only on Xi. The latter is especially intriguing, considering the inverse kinetic relationship between the appearance of long dsRNA versus xiRNA. Could dsRNA be processed to xiRNA on Xa? Several observations favored this idea. First, dsRNA was selectively lost from Xa. Second, xiRNA production depended on Tsix, a gene expressed from Xa on d4-d10. Finally, despite lacking Xi, XY cells produced xiRNAs.
Because dsRNAs are substrates for Dcr, we tested Dcr's role by deleting Dcr's RNAseIII domain in female ES cells (15) (Fig. S2). Because Dcr-/- cells are known to grow poorly (15), we introduced a Dcr transgene expressed at ≪5% of wildtype levels (Fig. 3A,B) and improved growth of Dcr-deficient clones (henceforth dubbed Dcr-/-). Northern analysis revealed diminished xiRNA levels (Fig. 3C,D), suggesting that xiRNA production depends on Dcr. All tested Dcr-deficient clones behaved similar. Significantly, Xist expression prematurely increased 5- to 10-fold in pre-XCI cells (Fig. 3B), implying increased Xist transcription or greater RNA stability. Male Dcr-/- clones likewise showed significant Xist de-repression on d4 (Fig. S3). Thus, Dcr regulates xiRNA levels and antagonizes Xist upregulation in ES cells.
Figure 3. Dcr deficiency impairs xiRNA production and XCI.
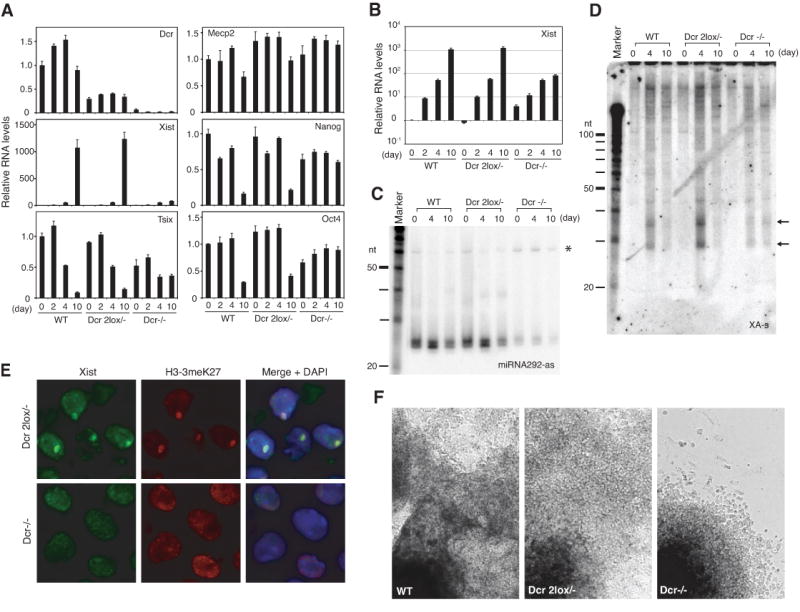
(A) Quantitative realtime RT-PCR of indicated transcripts normalized to β-actin.
(B) Xist quantitation plotted on a log scale.
(C,D) Northern analyses of miRNA292-as control (C) and xiRNAs (D) in mutant cells. Note accumulation of miRNA292-as precursors (*) in Dcr-/- cells.
(E) Immuno-RNA FISH for Xist and H3-3meK27 on d10. DAPI, blue.
(F) Phase contrast images of d10 EB.
RNA immunoFISH analysis showed that Dcr has additional XCI effects. Despite elevated Xist levels, Xist could not ‘coat’ the X nor induce heterochromatic changes (Fig. 3E). On d10, Xist RNA accumulation occurred in only 0.4% of cells (n = 278) and H3-K27 trimethylation (H3-3meK27) in 0.7% (n = 278). By contrast in Dcr 2lox/- controls, Xist accumulated in 56.8% and H3-K27 trimethylation in 83.1% (n = 148). Moreover, the X-linked Mecp2 gene failed to be dosage compensated in Dcr-/- cells, whereas it appropriately decreased 1.5- to 2-fold in controls (Fig. 3A). Therefore, in addition to local effects on Xist, Dcr also affected Xi globally, as formation of Xist and H3-3meK27 domains were compromised without Dcr.
Because XCI and cell differentiation are linked (16, 17), the XCI defects might be explained by Dcr's pleiotropic effects on differentiation (15, 18) rather than specific effects on XCI. Indeed, Dcr-/- clones differentiated poorly and continued to express Oct4 and Nanog pluripotency factors on d10 (Fig. 3A,F, Fig. S3). To determine if Dcr specifically affects XCI, we truncated Tsix by inserting a polyA cassette in Dcr-/- cells (Fig. S4), reasoning that disabling Tsix – which negatively regulates Xist — might overcome the failure to accumulate Xist RNA. As expected, Dcr-/- Tsix-/+ double mutants (Dcr-TST) and Tsix-/+ controls (TST) showed truncated Tsix expression from Xmus and highly skewed XCI patterns (Fig. 4A). Although Dcr-TST cells continued to differentiate poorly (Fig. S5), total Xist levels were restored to nearly WT levels during differentiation (Fig. 4B). Furthermore, disabling Tsix partially restored Xist localization to Xi (Fig. 4C). Therefore, Dcr's effect on XCI can be genetically separated from its effect on cell differentiation.
Figure 4. Tsix genetically interacts with Dcr.

(A) Allele-specific RT-PCR analysis. All RT- reactions were negative (not shown). Xist d0 samples (asterisks) were 10-fold overloaded to visualize low expression.
(B) Realtime RT-PCR of indicated transcripts, each normalized to β-actin.
(C) Immuno-RNA FISH for Xist and H3-3meK27 domains (arrowheads) on d10.
(D) Frequency of aberrant H3-3meK27 enrichment in the Xist+ subpopulation of indicated cell lines. n=100-150.
(E) Model: intersection of RNAi and XCI.
(F) Methylation-sensitive restriction analysis of the Xist promoter. Genomic DNA was digested with EcoRV or EcoRV+HpaII. % uncut (methylated) DNA at HpaII is plotted.
Additionally, to the extent that Xist levels and localization were restored in Dcr-TST cells, H3-K27 trimethylation was only partly rescued in Xist+ cells (Fig. 4D). In WT and TST controls, Xist accumulation was almost always accompanied by robust H3-K27 trimethylation. In contrast, 30-40% of Xist+ Dcr-TST cells displayed weak or no H3-3meK27 foci, implying that H3-K27 trimethylation also depends on Dcr. These data showed that Xist accumulation and H3-K27 methylation are genetically separable. We conclude that Dcr intersects XCI in several ways. Locally, Dcr controls xiRNA and Xist expression. Globally, it regulates Xist accumulation and H3-K27 trimethylation on Xi.
In aggregate, our data suggest specific effects of RNAi on XCI (Fig. 4E). Dcr and Tsix/Xist genetically interact, as a second-site mutation in Tsix partially suppresses the Dcr-/- effect on Xist. We propose that Tsix:Xist duplexes initially form on both Xs. During XCI, continued expression of Tsix on Xa would lead to dsRNA processing to xiRNAs, which would locally repress Xist in cis – an idea reminiscent of transcriptional gene silencing (TGS) (6, 19-21). Consistent with ‘allele-specific TGS’ at Xist, RNA-directed DNA methylation by Tsix has been proposed (10). Indeed, here we found that abrogating Dcr and/or Tsix resulted in decreased methylation at the 5′ end of Xist (Fig. 4F). By our model, extremely low Tsix and Xist expression might be sufficient to maintain TGS on Xa in post-XCI cells (19). On Xi, chromosome-wide accumulation of Xist RNA and H3-K27me3 depends on Dcr. These ideas support the emerging concept of nuclear RNAi processes in mammals (20, 21). Because Dcr is not known to cleave RNAs to 25-42 nt, the observed effects on XCI may be partially indirect. Alternatively, Dcr may have novel properties yet to be discovered in mammals. XCI now provides a new system in which to investigate RNAi processes.
Supplementary Material
References and Notes
- 1.Lyon MF. Nature. 1961;190:372. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- 2.Brown CJ, et al. Cell. 1992;71:527. doi: 10.1016/0092-8674(92)90520-m. [DOI] [PubMed] [Google Scholar]
- 3.Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N. Nature. 1996;379:131. doi: 10.1038/379131a0. [DOI] [PubMed] [Google Scholar]
- 4.Lee JT, Lu N. Cell. 1999;99:47. doi: 10.1016/s0092-8674(00)80061-6. [DOI] [PubMed] [Google Scholar]
- 5.Fire A, et al. Nature. 1998;391:806. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 6.Grewal SI, Elgin SC. Nature. 2007;447:399. doi: 10.1038/nature05914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cobb BS, et al. J Exp Med. 2005;201:1367. doi: 10.1084/jem.20050572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wutz A, Rasmussen TP, Jaenisch R. Nat Genet. 2002;30:167. doi: 10.1038/ng820. [DOI] [PubMed] [Google Scholar]
- 9.Ogawa Y, Lee JT. Mol Cell. 2003;11:731. doi: 10.1016/s1097-2765(03)00063-7. [DOI] [PubMed] [Google Scholar]
- 10.Sun BK, Deaton AM, Lee JT. Mol Cell. 2006;21:617. doi: 10.1016/j.molcel.2006.01.028. [DOI] [PubMed] [Google Scholar]
- 11.Buzin CH, Mann JR, Singer-Sam J. Development. 1994;120:3529. doi: 10.1242/dev.120.12.3529. [DOI] [PubMed] [Google Scholar]
- 12.Shibata S, Lee JT. Hum Mol Genet. 2003;12:125. doi: 10.1093/hmg/ddg010. [DOI] [PubMed] [Google Scholar]
- 13.Avner P, et al. Genet Res. 1998;72:217. doi: 10.1017/s0016672398003516. [DOI] [PubMed] [Google Scholar]
- 14.Shibata S, Lee JT. Curr Biol. 2004;14:1747. doi: 10.1016/j.cub.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 15.Murchison EP, Partridge JF, Tam OH, Cheloufi S, Hannon GJ. Proc Natl Acad Sci U S A. 2005;102:12135. doi: 10.1073/pnas.0505479102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monk M, Harper MI. Nature. 1979;281:311. doi: 10.1038/281311a0. [DOI] [PubMed] [Google Scholar]
- 17.Lee JT. Science. 2005;309:768. doi: 10.1126/science.1113673. [DOI] [PubMed] [Google Scholar]
- 18.Kanellopoulou C, et al. Genes Dev. 2005 Feb 15;19:489. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Volpe TA, et al. Science. 2002;297:1833. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- 20.Kim DH, Villeneuve LM, Morris KV, Rossi JJ. Nat Struct Mol Biol. 2006;13:793. doi: 10.1038/nsmb1142. [DOI] [PubMed] [Google Scholar]
- 21.Morris KV, Chan SW, Jacobsen SE, Looney DJ. Science. 2004;305:1289. doi: 10.1126/science.1101372. [DOI] [PubMed] [Google Scholar]
- 22.We thank G. Hannon for the Dcr targeting construct and Dcr2lox/- male ES cells; N. Lau for technical advice; and M. Anguera, J. Erwin, S. Namekawa, B. Payer, and J. Zhao for careful critique of the manuscript. Y.O. is especially indebted to A. Ogawa for her support. This work is funded by the Medical Scientist Training Program (B.K.S.) and the NIH and HHMI (J.T.L.).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.