

Abstract
For several decades the 2,3-dihydroquinazolinone (DHQZ) heterocycle has been known to possess a variety of important biological and medicinal properties. Despite the many interesting facets of these molecules, synthetic access to nonracemic DHQZ analogues has remained elusive. Herein, we disclose a synthetic route that allows access to either enantiomer of a variety of DHQZ derivatives. We illustrate the utility of this chemistry with the asymmetric preparation and biological evaluation of a new chiral fluorescent tubulin binding agent with extremely potent antiproliferative properties against human cancer cells. A computational rationale for the increased potency of the (S)-enantiomer over the (R)-enantiomer is given, based on the crystal structure of α,β-tubulin complexed with colchicine. Taking advantage of the inherent fluorescence of these molecules, confocal images of GMC-5-193 (compound 7) in the cytoplasm of human melanoma cells (MDA-MB-435) cells are presented.
Introduction
The 2,3-dihydroquinazolinone (DHQZa) family of compounds has a rich pharmacology with reported antibiotic,1 antidefibrillatory, 2 antispermatogenic,3 vasodilatory,4 and analgesic5 efficacy. We have been interested in 2-biphenyl substituted DHQZ analogues as potent tubulin inhibitors with impressive antiproliferative activity against a number of human cancer cell lines (Figure 1). DHQZ analogues that are substituted at the 2-position contain a stereocenter at the aminal carbon. Through computational docking experiments and molecular dynamics (Figure 2), we rationalized that the (S)-enantiomer should bind to tubulin better than the (R)-enantiomer. To test this hypothesis, we set out to synthesize enantiopure 2-aryl substituted DHQZ analogues.
Figure 1.

Structure of 2-biphenyl-2,3-dihydroguinazoline.
Figure 2.

Binding model of (S)-(+)-7 (A) and (R)-(−)-7 (B) with the α-tubulin and β-tubulin polypeptide backbones. Residue side chains at the colchicine site (Cys β241, Lys β254, and Asn α101) and 7 are shown in stick with the carbon atoms of colchicine colored green and the carbon atoms of 7 colored white. Yellow dashed lines indicate potential intermolecular hydrogen bonds.
Racemic synthesis of 2-aryl DHQZ analogues involves condensation of an arylaldehyde with anthraquinoline. Despite efforts by Lee,6 Priego,7 and possibly others, the enantiomeric synthesis or chiral separation of racemic DHQZ compounds has yet to be reported. To the best of our knowledge, there are no general synthetic methods for obtaining enantiopure 2-aryl DHQZ analogues.
Our efforts began with the synthesis of similar amino acid based DHQZ diastereomers according to the methods of Priego et al.,8 followed by auxiliary removal from the amide moiety. Initially, we envisioned two routes, as shown in Scheme 1. The first route was semisuccessful in that, after separation of the diastereomers, hydroxyl–iodine exchange of the serine-based auxiliary followed by reaction with magnesium successfully cleaved the auxiliary from the amide. However, when the enantiomeric purity was assessed by polarimetry and chiral HPLC, complete racemization of the desired stereocenter was observed. We then tried a similar strategy that employed milder conditions. The free acid of a phenylalanine-based chiral auxiliary was treated with diphenylphosphorylazide (DPPA) and heated to 60 °C under modified Curtius conditions.8 The isocyanate product was quenched with water in situ, resulting in decarboxylation and collapse of the unstable free aminal. While the auxiliary was successfully cleaved, to our dismay the desired stereocenter had once again racemized.
Scheme 1.

Synthesis of Compound 6 Resulting in Racemization
We rationalized that the aminal stereocenter is sensitive to racemization when a negative charge or even a transient negative charge is placed on the amide nitrogen atom. With this in mind, we hypothesized that the undesired racemization was occurring through a mechanism similar to that illustrated in Figure 3. To circumvent the racemization of our stereocenter, we sought a similar approach in which the chiral auxiliary is not directly bound to the heterocycle. We envisioned that a chiral auxiliary strategically grafted on the adjacent aromatic ring would provide a substrate that is not directly attached to the sensitive heterocycle but retains close proximity to the new stereocenter (Scheme 2).
Figure 3.

Proposed mechanism of racemization for compound 6.
Scheme 2.

Synthesis of Compounds 3 and 4
Removal of the auxiliary without affecting the sensitive aminal stereocenter as shown in Scheme 3 would then give a general method to nonracemic analogues of our target compounds. Furthermore, we would be able to verify that the (S)-enantiomer of our target compound binds more tightly than the (R)-enantiomer to tubulin as predicted by our computational simulations.
Scheme 3.

Synthesis of Nonracemic Compound 6
Results and Conclusions
A series of chiral salicylaldehyde-derived esters were prepared, condensed with anthranilamide under acid-catalyzed conditions, and screened for their ability to give separable diastereomers. We found that the Boc-protected amino acid derived auxiliaries furnished the best results in this regard. The results for the screen including the yields of the major diastereomers formed and the diastereomeric ratios are tabulated in Table 1.
Table 1.
Yield and Diastereomeric Ratios for Selected Chiral Auxiliaries
 | ||
|---|---|---|
| R | %Yield | D/R |
| 26 | 71:29 | |
| 47 | 55:45 | |
| 75 | 83:17 | |
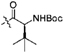 |
85 | 91:9 |
The phenylglycine-derived auxiliary gave moderate selectivity but diminished yield (see Table 1). Increasing the steric bulk of the auxiliary from methyl to isopropyl to tert-butyl significantly enhanced the diastereoselectivity and the yield of the reaction. Fortuitously, the diastereomers were separable both by chromatography and by solubility differences. During most of these cyclizations, the major diastereomer precipitated from solution as a single pure isomer. In all cases examined thus far, the major diastereomer exists as a crystalline solid while the minor diastereomers appear to be an amorphous foam or a viscous oil after purification. These dramatic solubility differences obviate the need for the tedious purifications and/or multiple columns often required for purifying diastereomers with similar Rf values. These conditions may be ideally suited for preparative synthesis on a larger scale.
In order to probe the scope and limitations of this key stereocenter formation, we condensed nonracemic aldehyde 1 in Scheme 2 with a variety of anthranilamide analogues that encompass electron-donating, electron-withdrawing, and bulky functional groups. The results of this screen are tabulated in Table 2. We found, as expected, that the enantiomer of 1 gave the opposite configuration at the aminal stereocenter (entry a). We also noticed that the most deleterious effects on yield and selectivity occurred with an electron-donating substituent (entry d) at R5. A similar phenomenon was also observed by Lee and co-workers.7 Examination of the electron-withdrawing nitro groups (Table 2, entries e and f) show similar diastereoselectivity but with a reduced yield. It should be noted that the nitro containing molecules required reaction conditions 40–50 °C higher than that of the other compounds listed.
Table 2.
Yield and Diastereomeric Ratios for Compounds 3 and 4
| entry | R3 | R4 | R5 | R6 | auxilliary config | % yield | dr | |
|---|---|---|---|---|---|---|---|---|
| a | H | H | H | H | R | 71 | 11 | 89 |
| b | H | H | H | H | S | 85 | 91 | 9 |
| c | H | OMe | H | H | S | 78 | 85 | 15 |
| d | H | H | OMe | H | S | 36 | 75 | 25 |
| e | H | NO2 | H | H | S | 66 | 85 | 15 |
| f | H | H | NO2 | H | S | 58 | 89 | 11 |
| g | Me | H | H | H | S | 62 | 79 | 21 |
| h | H | H | H | Me | S | 34 | 83 | 17 |
| i | H | H | Cl | H | S | 59 | 83 | 17 |
Having successfully isolated several pure diastereomers, we were now well positioned to determine the absolute configuration of the stereocenter. X-ray crystallographic analysis of compound 3 (see Supporting Information) shows that the S configuration of the chiral auxiliary resulted in a heterocycle with an R configuration at the aminal carbon.
To remove the auxiliary and isolate the target DHQZ enantiomers, we sought conditions that would cleave the ester but not racemize the sensitive aminal stereocenter. Treatment of the DHQZ analogues with several hydride sources resulted in a yellow color, presumably from the DHQZ anion formed by deprotonation of the amide nitrogen. These reagents proved to be incompatible with the DHQZ diastereomers, resulting in complete racemization of the aminal stereocenter. To our delight, we found that reaction with hydrazine did not produce a yellow color and resulted in cleavage of the ester at ambient temperature to give the phenol with no detectable racemization.
The resulting phenol derived from ester 4 (see Scheme 3) was treated with N-phenyl-bis-triflimide in the presence of Hunig’s base to selectively give the aryl triflate 5.9 The nonracemic triflate was then reduced with ammonium formate and Pd(dppf)Cl2 to give the desired enantiomerically pure DHQZ 6, as indicated by chiral HPLC. To test whether this compound was sensitive to base-catalyzed racemization as we hypothesized, we treated a 5.4 mM solution of the nonracemic aminal (−)-2,3-dihydro-2-phenyl-4(1H)-quinazolinone in THF with 50 µL of 6 M NaOH solution. Indeed, the measured optical rotation was zero within 10 min, signifying complete racemization of the aminal stereocenter.
Studies in our laboratory showed that racemic compound 7 had remarkably potent antiproliferative activity toward a number of human cancer cell lines (see Table 3). Since the isolation or resolution of the enantiomers of DHQZ 7 or other DHQZ analogues has not been reported, the biological effects of each enantiomer had not been previously assessed. To obtain this information, we applied this newly developed synthetic methodology to prepare both stereocenters of compound 7 as outlined in Scheme 4.
Table 3.
Biological Evaluation of Racemic-7, (R)-(−)-7, and, (S)-(+)-7a
| % inhibition of 3H-colchicine binding |
|||||
|---|---|---|---|---|---|
| compd | MDA-MB-435 GI50, nM | HCT-116 GI50, nM | inhibition of tubulin assembly, IC50, µM | 5 µM compd | 50 µM compd |
| racemic-7 | 100 | 300 | 1.7 ± 0.3 | 17 ± 2 | 51 ± 0.6 |
| (R)-(−)-7 | 1100 | 930 | 16 ± 4 | 2.1 ± 10 | 5.5 ± 0.9 |
| (S)-(+)-7 | 0.1 | 100 | 1.1 ± 0.2 | 28 ± 1 | 66 ± 1 |
| CA-4 | 1.3 ± 0.04 | 99 ± 0.9 | not done | ||
In the colchicine binding experiment, the tubulin concentration was 1 µM and the [3H]colchicine was 5 µM. Paclitaxel GI50 for MDA-MB-435 is 4 nM (see ref 12).
Scheme 4.

Syntheses of Compound (S)-(+)-7
Suzuki-Miyaura coupling10 of o-tolylboronic acid with 3-bromoacetoxybenzene gave phenol 8, which was converted to aldehyde 9 via regioselective formylation using the method of Wang and co-workers.11 Coupling N-Boc-d-tert-leucine or N-Boc-l-tert-leucine to the phenol gave aldehyde 10a or 10b, respectively. Condensation of aldehyde 10 with 5-nitroanthranilamide gave the diastereomeric DHQZ analogues 11. The three-step sequence for removal of the auxiliary was performed as follows: treatment with hydrazine, followed by triflation, and palladium-mediated reduction of the resulting triflate to give enantiomers (−)-7 or (+)-7, depending on the configuration of the N-Boc-leucine employed. The absolute configuration was assigned by X-ray crystallography with the S configuration belonging to the (+)-7 enantiomer.
The evaluation of racemic 7, (S)-(+)-7, and (R)-(−)-7 for binding to the colchicine site on tubulin was performed. The results revealed, as predicted, that the S enantiomer of 7 was more effective than both the R enantiomer and the racemic mixture of 7 as an inhibitor of the binding of colchicine to tubulin (Table 3).
Inhibitory Effects of Enantiomers on Cell Growth and on Tubulin Polymerization and the Binding of [3H]Colchicine to Tubulin
Racemic 7 and each enantiomer, (S)-(+)-7 and (R)-(−)-7, were separately assayed against two different human cancer cell lines for their antiproliferative effects. As Table 3 shows, both cell lines appear to be significantly more sensitive to (S)-(+)-7 when compared to the racemate. Interestingly, the “less active” (R)-(−)-7 still retains significant cytotoxicity, though much less so than either (S)- (+)-7 or the racemate. Particularly noteworthy is the picomolar level growth inhibition induced by (S)-(+)-7 on MDA-MB-435 cells, approximately 10 times more active than paclitaxel.12
Similarly, (S)-(+)-7 was significantly more potent than (R)-(−)-7 as an inhibitor of tubulin assembly and as an inhibitor of colchicine binding to tubulin (Table 3). Although we found no significant effect of (R)-(−)-7 on colchicine binding, this enantiomer did have a reproducible, weak inhibitory effect on tubulin assembly. It should be noted that (S)-(+)-7 was as potent as combretastatin A-4 (CA-4) as an inhibitor of tubulin assembly but much less active as an inhibitor of colchicine binding to tubulin.
The effects of (S)-(+)-7 on the MDA-MB-435 and HCT-116 cell proliferation revealed a 1000-fold difference compared to (R)-(−)-7 (Table 3). This might be explained by a selectivity of a multidrug resistance (MDR) mechanism including p-glycoprotein toward (R)-(−)-7 over (S)-(+)-7 in regard to substrate selectivity. Detailed studies addressing the chiral interactions of (R)-(−)-7 and (S)-(+)-7 with p-glycoprotein will provide more insight into this important resistance mechanism.
Molecular Modeling Studies
Our studies support that the (S)-(+)-7 enantiomer was more effective at inhibiting tubulin polymerization and the binding of [3H]colchicine to tubulin, as well as human cancer cell proliferation than (R)-(−)-7. These findings could be rationalized by molecular docking simulations. Initially the docking procedure was validated by comparing the binding mode obtained by docking the known tubulin depolymerization agents colchicine, podophyllotoxin, and combretastatin A-4. Automatic docking simulations of the R and S configuration of compound 7 with α,β-tubulin were conducted and produced several potential binding conformations. Since inhibition of [3H]colchicine binding, as shown in Table 3, provides strong evidence that compound 7 and its S enantiomer bind to the colchicine site, we selected one conformer for the S, as well as one conformer for the R, that closely resembled the bound conformation of colchicine. Independent use of two programs, Autodock13 and FlexX,14 also provided the same docking pose for the 5-nitroanthranilamide interacting in similar conformational space on the α,β-tubulin interface as our simulated docking results.
Docking inhibitors into a protein binding site derived from a crystal structure of the protein complexed with another ligand (in this case, colchicine) may not have the correct binding mode because the potential ligand may induce conformational changes on the protein side chains. Thus, we further refined the binding modes of compound 7 (R and S) using molecular dynamics (MD) simulations. Initially, the tubulin–compound 7 (R and S) complexes were subjected to energy minimization to relieve any unfavorable interactions on the model structure. The minimized complex structure was then subjected to MD simulation of 300 ps, performed in the NVE ensemble. Since the tubulin system is large, the residues within 15 Å of the ligand were allowed to relax while fixing all other residues during the simulation. Low energy minimum structures were collected every 10 ps giving a total of 30 structures which were energy minimized and further analyzed. To evaluate the readjustment of the active site residues for the calculated tubulin-bound compound 7 (R and S), the root-mean square displacement (rmsd) between the atoms of the initial tubulin structure and the corresponding atoms of each tubulin-compound complex was evaluated. In the case of backbone atoms superimposition, the rmsd was lower than 0.37 Å, while for side chains the maximum rmsd found was about 1.38 Å. These data suggest that ligand-induced conformational changes are more pronounced for side chains compared with the polypeptide back-bone functional groups. Finally, the global energy minimum of the 30 collected structures was selected as the final binding model.
Upon analysis of 30 low energy minima structural models for the (R)-(−)-7 and (S)-(+)-7 conformations, most of the ligand-interacting residues were reoriented. In particular, for both low energy conformations of (R)-(−)-7 and (S)-(+)-7, we observed significant conformational reorientation of the Q-α11, N-α101, and K-β254 side chains and the formation of hydrogen bonds (H-bonds) with the nitro and NH groups. These residues were not within the H-bond distance in the starting docked model. The side chain reorientation of these residues is not surprising because these residues are exposed and not interacting with other active site residues. However, this suggests that ligand-induced conformational changes may be critical in the protein active site, and side chains may undergo rearrangement during ligand binding. In the final model shown in Figure 2A for the S conformation, we noticed a network of seven H-bonds between compound 7 and Q-α11, N-α101, S-α178, T-α179, N-β249, K-β254, Y-β224, and N-β258. We also noticed that the biphenyl rings were buried inside the β-site of the hydrophobic pocket containing β-tubulin residues L255, L248, A250, A316, V318, K352, A354, and I378. This hydrophobic region of compound 7 for both the R and S enantiomers overlaps with the colchicine trimethoxyphenyl group. However, significant differences in the interaction of the 5-nitroanthranilamide moiety with the tubulin active site residues for the R and S enantiomers were observed. The 5-nitroanthranilamide moiety of the S enantiomer also appears to be more planar than the R enantiomer in the final model with increased H-bonds for (S)-(+)-7 with tubulin (seven H-bonds compared to four possible H-bonds for (R)-(−)-7) (Figure 2A and Figure 2B). Specifically, (R)-(−)-7 H-bond interactions with Y-β224, S-α178, and T-α179 appear to be absent. These differential binding interactions provide a rationale for the greater inhibitory activity of (S)-(+)-7 compared with (R)-(−)-7 in inhibiting clochicine binding to tubulin and tubulin assembly.
Intracellular Localization of Compound 7
A notable property of compound 7 is its inherent fluorescence, which allowed us to examine its intracellular localization in human cancer cells. Treatment of the MDA-MB-435 human melanoma cells with a 5 µM solution of compound 7 for 6 h was followed by fixation of the cells, which were then subjected to photo-excitation and visualization by confocal microscopy using a two-photon laser. Propidium iodide staining (in red) was used to visualize the nucleus (Figure 4A), and differential interference contrast (DIC) was used to reveal the morphology of the cells. Cytoplasmic images clearly show that compound 7 (in green) localized in the cytoplasm (Figure 4C and Figure 4D), as would be expected for an antitubulin agent.
Figure 4.

Confocal image of fluorescent racemic 7 in human melanoma cells (MDA-MB- 435): (A) propidium iodine staining of the nucleus; (B) DIC image with 0.5 million MDA-MB-435 cells; (C) cytoplasmic fluorescence of racemic 7; (D) merged DIC, propidium iodide, and fluorescent 7 image. (E) MDA-MB-435 cells were treated for 1 h with 5 M racemic 7. Excitation was at 715 nm, and absorbance was read at bandpath filter of 480–520 nm. Images were taken with a two photon confocal system at 60× magnification.
In this study, we developed methodology to synthetically resolve and determine the stereochemistry of each enantiomer of 2-substitutued DHQZ compounds. Using molecular modeling, we rationalized important interactions of each enantiomer with the colchicine binding site. Evaluation of compound 7 for inhibition of colchicine binding, tubulin assembly, and cancer cell proliferation demonstrated that the (S)-(+)-7 enantiomer is the more active stereoisomer. Finally, we provided evidence using the inherent fluorescent property of compound 7, that it localizes to the cytoplasm of human melanoma cancer cells.
Experimental Section
Chemistry: General Methods
Reagents were purchased from Aldrich or Lancaster and were used as received. The solvents were not dried prior to use. Melting points were determined in open capillary tubes on an Electrothermal melting point apparatus and are uncorrected. 1H (300 or 500 MHz) and 13C NMR (75 or 125 MHz) spectra were measured at 25 °C on compounds in solution in CDCl3 or DMSO-d6 on a Varian 300 or 500 MHz spectrometer. Chemical shifts (δ) are given in ppm downfield from tetramethylsilane, an internal standard. Mass spectra were obtained on a Finnagan LcQ Classic. Elemental analyses were performed by Atlantic Microlabs, and microanalytical data were within ±0.4% of the calculated values. Reactions were monitored by TLC using Merck 60 F254 silica gel aluminum sheets (20 cm × 20 cm).
N-Boc-l-tert-leucine(salicylaldehyde) Ester (1a)
l-tert-Leucine (5.0 g, 38.12 mmol) was added to a solution of 38.1 mL (8.1 mmol) of 1 N NaOH in 50 mL of dioxane. Di-tert-butyldicarbonate (8.32 g, 38.12 mmol) was added, and the mixture was stirred at room temperature for 14 h. The reaction mixture was concentrated and partitioned between 100 mL of EtOAc and 100 mL of water. The aqueous phase was acidified to a pH of ~2 with concentrated HCl. The resulting cloudy, aqueous solution was extracted with three 100 mL portions of EtOAc, and the combined organic extract was dried over MgSO4. Filtration and removal of the solvent under reduced pressure gave 6.7 g (29.0 mmol) of the Boc-protected amino acid as a viscous clear oil, which was immediately taken up in 50 mL of DMF. To this solution was added 6.6 g (31.9 mmol) of DCC. After 30 min, 4.31 g (31.9 mmol) of HOBt was added. After an additional 30 min, 4.0 mL (37.7 mmol) of salicylaldehyde was added and the reaction mixture was stirred for 12 h and then quenched with 100 mL of water. The reaction mixture was extracted with EtOAc (3 × 100 mL), and the combined organic phase was dried over MgSO4. Filtration and removal of the solvent gave the crude product. Purification by flash chromatography eluting with 7:1 hexanes–EtOAc gave the aldehyde product. Further purification by recrystallization from hexanes gave 4.7 g (48% over two steps) of the product as colorless crystals. [α]D −2.1 (c 1.0, CHCl3); mp 104–106 °C; 1H NMR (300 MHz, CDCl3) δ 10.22 (s, 1H), 7.92–7.95 (dd, J ) 7.5 Hz, 1.5 Hz, 1H), 7.60–7.65 (dt, J = 7.5, 1.8 Hz, 1H), 7.36–7.41 (t, J = 7.8 Hz, 1H), 7.19–7.26 (d, J = 8.1 Hz, 1H), 5.12–5.15 (d, J = 0.9 Hz, 1H), 4.34–4.38 (d, J = 9.3 Hz, 1H), 1.47 (s, 9H), 1.14 (s, 9H); 13C (75 MHz, CDCl3) δ 188.7, 171.0, 156.0, 152.4, 135.4, 129.6, 128.5, 126.8, 123.1, 80.6, 62.6, 34.6, 28.5, 26.9; ESI m/z (rel intensity) 335.2 (100).
N-Boc-d-tert-leucine(salicylaldehyde) Ester (1b)
d-tert-Leucine (1.0 g, 7.6 mmol) was added to a solution of 7.6 mL (7.6 mmol) of 1 N NaOH in 20 mL of dioxane. Di-tert-butyldicarbonate (1.8 g, 8.0 mmol) was added, and the reaction mixture was stirred at room temperature for 14 h. The reaction mixture was concentrated and partitioned between 100 mL of EtOAc and 100 mL of water. The aqueous phase was acidified to a pH of ~2 with concentrated HCl. The resulting cloudy, aqueous solution was extracted with three 50 mL portions of EtOAc, and the combined organic extract was dried over MgSO4. Filtration and removal of the solvent under reduced pressure gave 0.75 g (3.5 mmol) of the Boc-protected amino acid as a viscous clear oil, which was immediately taken up in 25 mL of DMF. To this solution was added 0.75 g (3.6 mmol) of DCC. After 30 min, 0.49 g (3.6 mmol) of HOBt was added. After an additional 30 min, 0.48 mL (4.5 mmol) of salicylaldehyde was added and the reaction mixture was stirred for 12 h and then quenched with 100 mL of water. The reaction mixture was extracted with EtOAc (3 × 50 mL), and the combined organic phase was dried over MgSO4. Filtration and removal of the solvent under reduced pressure gave the crude product. Purification by flash chromatography eluting with a 7:1 solution of hexanes–EtOAc gave the aldehyde product. Further purification by recrystallization from hexanes gave 0.58 g (53% over two steps) of the purified product as colorless crystals. [α]d +2.1 (c 1.0, CHCl3); mp 104–106 °C; 1H NMR (300 MHz, CDCl3) δ 10.22 (s, 1H), 7.91–7.95 (dd, J = 10.8 Hz, 1.5 Hz, 1H), 7.60–7.65 (dt, J = 8.1, 1.8 Hz, 1H), 7.36–7.41 (t, J = 7.8 Hz, 1H), 7.19–7.22 (d, J = 8.1 Hz, 1H), 5.13–5.16 (d, J = 0.9 Hz, 1H), 4.34–4.37 (d, J = 9.0 Hz, 1H), 1.47 (s, 9H), 1.14 (s, 9H); 13C (75 MHz, CDCl3) δ 188.8, 171.0, 156.0, 152.4, 135.5, 129.7, 128.5, 126.8, 123.2, 80.6, 62.6, 34.7, 28.6, 27.0; ESI m/z (rel intensity) 335.2 (100).
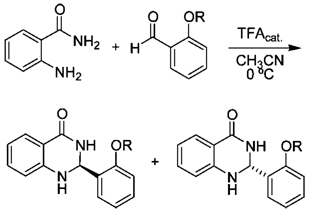
General Procedure A. Synthesis of Dihydroquinazolinone Diastereomers
To a solution of the anthranilamide (1 equiv) and the aldehyde (1 equiv) in 8 mL/mmol of MeCN at 0 °C was added 1–2 drops of trifluoroacetic acid. After 1.5 h, the precipitate was collected by filtration and recrystallized from THF–hexanes.
2S-(2′-N-Boc-d-tert-leucine phenyl ester)-4-quinazolinone (4a)
The title compound was prepared according to general procedure A from anthranilamide (0.13 g, 0.93 mmol) and aldehyde 1a (0.30 g, 0.93 mmol) to give 0.29 g (71%) of the resulting dihydroquinazolinone as colorless crystals. [α]d +120 (c 0.1, THF); mp 236–237 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.00 (s, 1H), 7.49–7.61 (m, 3H), 7.36–7.42 (t, J = 7.7 Hz, 1H), 7.25–7.30 (t, J = 7.2 Hz, 1H), 7.18–7.23 (t, J = 7.8 Hz, 1H), 7.10–7.13 (d, J = 7.8 Hz, 1H), 6.64–6.73 (m, 3H), 5.95 (s, 1H), 4.08–4.11 (d, J = 9.9 Hz, 1H), 1.33 (s, 9H), 1.06, (s, 9H); 13C (75 MHz, DMSO-d6) δ 171.3, 164.3, 157.09, 148.4, 148.3, 134.1, 133.8, 130.3, 128.5, 128.1, 126.8, 123.1, 118.0, 115.5, 115.1, 79.7, 64.0, 61.7, 34.0, 28.8, 27.4; ESI m/z (rel intensity) 453.2 (100). Anal. (C25H31N3O5 · 0.1H2O) C, H, N. C: calcd, 65.95; found, 65.80. H: calcd, 6.91; found, 6.86. N: calcd, 9.23; found, 9.19.
2R-(2′-N-Boc-l-tert-leucine phenyl ester)-4-quinazolinone (3b)
The title compound was prepared according to general procedure A from anthranilamide (0.20 g, 1.49 mmol) and aldehyde 1 (0.50 g, 1.49 mmol) to give 0.57 g (85%) of the resulting dihydroquinazolinone as colorless crystals. [α]d −126 (c 0.1, THF); mp 236–237 °C; 1H NMR (300 MHz, DMSO-d6) δ 7.98 (s, 1H), 7.59–7.62 (dd, J = 7.8, 1.5 Hz, 1H), 7.54–7.57 (dd, J = 4.5, 1.2 Hz, 1H), 7.48–7.50 (d, J = 6.6 Hz, 1H), 7.36–7.42 (t, J = 7.8 Hz, 1H), 7.18–7.30 (m, 2H), 7.11–7.13 (d, J = 7.8 Hz, 1H), 6.64–6.73 (m, 3H), 5.95 (s, 1H), 4.09–4.11 (d, J = 6.9 Hz, 1H), 1.33 (s, 9H), 1.07, (s, 9H); 13C (75 MHz, DMSO-d6) δ 171.2, 164.2, 157.0, 148.4, 148.2, 134.0, 133.7, 130.3, 128.5, 128.1, 126.7, 123.0, 118.0, 115.5, 115.1, 79.6, 64.0, 61.6, 34.0, 28.8, 27.3; ESI m/z (rel intensity) 453.2 (100). Anal. (C25H31N3O5) C, H, N. C: calcd, 66.21; found, 66.12. H: calcd, 6.89; found, 6.92. N: calcd, 9.27; found, 9.14.
2R-(2′-N-Boc-d-tert-leucine phenyl ester)-7-methoxy-4-quinazolinone (3c)
The title compound was prepared according to general procedure A from 4-methoxyanthranilamide (0.20 g, 1.19 mmol) and aldehyde 1 (0.40 g, 1.19 mmol) to give 0.45 g (78%) of the resulting dihydroquinazolinone as colorless crystals. [α]d −105 (c 0.1, THF); mp 228–230 °C; 1H NMR (300 MHz, DMSO-d6) δ 7.79 (s, 1H), 7.50–7.54 (m, 3H), 7.36–7.41 (t, J = 7.5 Hz, 1H), 7.24–7.29 (t, J = 7.5 Hz, 1H), 7.11–7.13 (d, J = 8.1 Hz, 1H), 6.67 (s, 1H), 6.22–6.27 (m, 2H), 5.92 (s, 1H), 4.09–4.11 (d, 1H), 3.67 (s, 3H), 1.35 (s, 9H), 1.07 (s, 9H); 13C (75 MHz, DMSO-d6) δ 171.2, 164.3, 164.2, 157.1, 149.8, 148.3, 134.1, 130.2, 130.0, 128.3, 126.7, 123.0, 108.9, 105.7, 98.5, 79.7, 64.0, 61.7, 55.8, 34.0, 28.8, 27.4; ESI m/z (rel intensity) 483.2 (100). Anal. (C26H33N3O6) C, H, N. C: calcd, 64.58; found, 64.58. H: calcd, 6.88; found, 6.99. N: calcd, 8.69; found, 8.93.
2R-(2′-N-Boc-d-tert-leucine phenyl ester)-6-methoxy-4-quinazolinone (3d)
To a 0 °C solution of the 5-methoxyanthranilamide (0.25 g, 1.51 mmol) and aldehyde 1 (0.51 g, 1.51 mmol) in 8 mL of MeCN was added 1–2 drops of trifluoroacetic acid. After 1.5 h, the solvent was removed under reduced pressure and the resulting residue purified by flash chromatography eluting with 2:1 hexanes–EtOAc to give 0.26 g (36%) of the resulting dihydroquinazolinone as a light-green solid. [α]d −104 (c 0.1, THF); mp 192–194 °C; 1H NMR (300 MHz, DMSO-d6) δ 7.73–7.75 (d, J = 7.5 Hz, 1H), 7.40–7.41 (d, J = 3.3 Hz, 1H), 7.34–7.37 (dd, J = 7.8, 1.5 Hz, 1H), 7.24–7.29 (t, J = 7.5 Hz, 1H), 7.11–7.14 (d, J = 7.8 Hz, 1H), 6.87–6.911 (dd, J = 8.7, 4.8 Hz, 1H), 6.70 (s, 1H), 6.62–6.65 (d, 1H), 6.15 (s, 1H), 5.26–5.29 (d, J = 7.5 Hz, 1H), 5.04 (s, 1H), 4.21–4.23 (d, J = 7.8 Hz, 1H), 3.78 (s, 3H), 1.39 (s, 9H), 1.15 (s, 9H); 13C (75 MHz, DMSO-d6) δ 171.6, 165.5, 156.3, 153.2, 148.0, 141.4, 132.6, 130.2, 128.3, 126.9, 123.0, 122.4, 117.0, 116.1, 110.4, 80.8, 63.0, 62.6, 56.0, 34.1, 28.5, 27.1; ESI m/z (rel intensity) 483.2 (100). Anal. (C26H33N3O6) C, H, N. C: calcd, 64.58; found, 64.53. H: calcd, 6.88; found, 6.71. N: calcd, 8.69; found, 8.75.
2R-(2′-N-Boc-d-tert-leucine phenyl ester)-7-nitro-4-quinazolinone (3e)
To a room temperature solution of 4-nitroanthranilamide (0.22 g, 1.19 mmol) and chiral aldehyde 1 (0.40 g, 1.19 mmol) in 8 mL of MeCN was added 1–2 drops of trifluoroacetic acid. After 24 h, the precipitate was collected and recrystallized from THF–hexanes to give 0.39 g (66%) of the resulting dihydroquinazolinone as a yellow solid. [α]d −167 (c 0.1, THF); mp 240–241 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.47 (s, 1H), 7.84–7.86 (d, J = 8.4 Hz, 1H), 7.58–7.61 (m, 2H), 7.42–7.50 (m, 3H), 7.30–7.35 (t, J = 6.6 Hz, 2H), 7.15–7.17 (d, J = 7.8 Hz, 1H), 6.13 (s, 1H), 4.08–4.10 (d, J = 6.6 Hz, 1H), 1.28 (s, 9H), 1.07 (s, 9H); 13C (75 MHz, DMSO-d6) δ 171.2, 162.7, 157.0, 151.4, 148.9, 148.4, 132.8, 130.7, 129.9, 128.6, 127.0, 123.1, 120.0, 111.9, 109.7, 79.6, 63.9, 61.6, 33.9, 28.7, 27.3; ESI m/z (rel intensity) 498.2 (100). Anal. (C25H30N4O7 · 0.1H2O) C, H, N. C: calcd, 60.02; found, 59.70. H: calcd, 6.08; found, 6.04. N: calcd, 11.24; found, 10.97.
2R-(2′-N-Boc-d-tert-leucine phenyl ester)-6-nitro-4-quinazolinone (3f)
To a 50 °C solution of 5-nitroanthranilamide (0.27 g, 1.49 mmol) and aldehyde 1 (0.50 g, 1.49 mmol) in 8 mL of MeCN was added 1–2 drops of trifluoroacetic acid. After 24 h, the precipitate was collected and recrystallized from THF–hexanes to give 0.43 g (58%) of the resulting dihydroquinazolinone as a yellow solid. [α]d −182 (c 0.1, THF); mp 211–213 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.49 (s, 1H), 8.42 (d, J = 2.7 Hz, 1H), 8.15 (s, 1H), 8.07–8.11 (dd, J = 9.0, 2.7 Hz, 1H), 7.58–7.61 (d, J = 7.5 Hz, 1H), 7.43–7.47 (t, J = 6.3 Hz, 2H), 7.31–7.37 (t, J = 7.2 Hz, 1H), 7.14–7.16 (d, J = 8.1 Hz, 1H), 6.83–6.86 (d, J = 9.3 Hz, 1H), 6.21 (s, 1H), 4.06–4.08 (d, J = 6.9 Hz, 1H), 1.28 (s, 9H), 1.06 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 171.1, 162.0, 156.8, 153.0, 148.2, 137.8, 132.4, 130.7, 129.3, 128.5, 127.0, 124.6, 122.9, 114.9, 113.3, 79.4, 63.6, 61.3, 33.7, 28.5, 27.1; ESI m/z (rel intensity) 498.2 (100). Anal. (C25H30N4O7) C, H, N. C: calcd, 60.23; found, 60.35. H: calcd, 6.07; found, 6.25. N: calcd, 11.24; found, 11.19.
2R-(2′-N-Boc-d-tert-leucine phenyl ester)-8-methyl-4-quinazolinone (3g)
To a 0 °C solution of 3-methylanthranilamide (0.05 g, 0.30 mmol) and aldehyde 1 (0.10 g, 0.30 mmol) in 8 mL of MeCN was added 1–2 drops of trifluoroacetic acid. After 2 h the solvent was removed under reduced pressure, and the resulting residue was purified by flash chromatography eluting with 2:1 hexanes–EtOAc, followed by recrystallization from THF–hexanes to give 0.090 g (62%) of the resulting dihydroquinazolinone as a colorless solid. [α]d −109 (c 0.1, THF); mp 177–179 °C; 1H NMR (300 MHz, DMSO-d6) δ 7.76–7.82 (m, 2H), 7.37–7.43 (dt, J = 7.8, 1.5 Hz, 1H), 7.29–7.34 (t, J = 7.5 Hz, 1H), 7.13–7.16 (d, J = 7.2 Hz, 1H), 7.05–7.07 (d, J = 7.2 Hz, 1H), 6.71–6.76 (t, J = 7.5 Hz, 1H), 6.14–6.17 (d, J = 8.4 Hz, 2H), 5.10–5.12 (d, J = 8.7 Hz, 1H), 4.63 (s, 1H), 4.22–4.25 (d, J = 8.1 Hz, 1H), 2.08 (s, 3H), 1.30 (s, 9H), 1.12 (s, 9H); 13C (75 MHz, DMSO-d6) δ 172.3, 165.5, 156.0, 148.2, 145.5, 135.1, 132.9, 130.8, 128.6, 127.5, 126.6, 122.5, 122.3, 118.8, 115.1, 81.1, 62.7, 62.4, 34.3, 28.4, 27.0, 16.8; ESI m/z (rel intensity) 467.2 (100). Anal. (C26H33N3O5) C, H, N. C: calcd, 66.79; found, 66.42. H: calcd, 7.11; found, 7.23. N: calcd, 8.99; found, 8.62.
2R-(2′-N-Boc-d-tert-leucine phenyl ester)-5-methyl-4-quinazolinone (3h)
To a 0 °C solution of 6-methylanthranilamide (0.18 g, 1.19 mmol) and aldehyde 1 (0.40 g, 1.19 mmol) in 8 mL of MeCN was added 1–2 drops of trifluoroacetic acid. After 2 h the solvent was removed under reduced pressure, and the resulting residue was purified by flash chromatography eluting with 2:1 hexanes–EtOAc, 2:1, followed by recrystallization from THF–hexanes to give 0.19 g (34%) of the resulting dihydroquinazolinone as a colorless solid. [α]d −143 (c 0.1, THF); mp 148–150 °C; 1H NMR (300 MHz, DMSO-d6) δ 7.73–7.74 (d, J = 4.5 Hz, 1H), 7.34–7.37 (t, J = 4.5 Hz, 1H), 7.25–7.27 (t, J = 3.3 Hz, 1H), 7.08–7.09 (d, J = 4.8 Hz, 1H), 7.03–7.07 (t, J = 4.5 Hz, 1H), 6.56–6.57 (d, J = 4.5 Hz, 1H), 6.49–6.50 (d, J = 4.8 Hz, 1H), 6.34 (s, 1H), 6.05 (s, 1H), 5.16–5.18 (d, J = 4.5 Hz, 1H), 5.07 (s, 1H), 4.18–4.20 (d, J = 4.8 Hz, 1H), 2.63 (s, 3H), 1.35 (s, 9H), 1.11 (s, 9H); 13C (75 MHz, DMSO-d6) δ 171.4, 165.6, 156.0, 148.3, 147.8, 142.1, 132.7, 132.2, 130.0, 128.2, 126.7, 122.7, 122.0, 113.6, 113.2, 80.5, 62.6, 61.6, 33.8, 28.2, 26.8, 22.3; ESI m/z (rel intensity) 467.2 (100). Anal. (C26H33N3O5) C, H, N. C: calcd, 66.79; found, 66.85. H: calcd, 7.11; found, 7.22. N: calcd, 8.99; found, 8.86.
2R-(2′-N-Boc-d-tert-leucine phenyl ester)-6-chloro-4-quinazolinone (3i)
The title compound was prepared according to general procedure A from 5-chloroanthranilamide (0.05 g, 0.30 mmol) and aldehyde 1 (0.10 g, 0.30 mmol) to give 0.11 g (77%) of the resulting dihydroquinazolinone as colorless crystals. [α]d −123 (c 0.1, THF); mp 207–209 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.21 (s, 1H), 7.59–7.60 (d, J = 4.8 Hz, 1H), 7.54 (d, J = 1.2 Hz, 1H), 7.48–7.49 (d, J = 3.9 Hz, 1H), 7.41–7.44 (t, J = 4.5 Hz, 1H), 7.30–7.33 (t, J = 4.5 Hz, 1H), 7.26–7.28 (d, J = 5.1 Hz, 1H), 7.12–7.14 (d, J = 4.8 Hz, 1H), 6.93 (s, 1H), 6.76–6.78 (d, J = 4.8 Hz, 1H), 5.99 (s, 1H), 4.08–4.10 (d, J = 4.2 Hz, 1H), 1.31 (s, 9H), 1.07 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 171.3, 163.2, 157.0, 148.5, 147.3, 133.7, 133.1, 130.6, 128.7, 127.1, 126.9, 123.0, 121.7, 117.2, 116.8, 79.6, 63.9, 61.7, 34.0, 28.8, 27.4; ESI m/z (rel intensity) 487.2 (100). Anal. (C25H30ClN3O5) C, H, N. C: calcd, 61.53; found, 61.43. H: calcd, 6.20; found, 6.13. N: calcd, 8.61; found, 8.52.
General Procedure B. Ester Cleavage and Phenol Triflation
To a solution of the ester (1 equiv) in 30 mL/mmol of THF was added hydrazine monohydrate (3 equiv). The mixture was stirred at room temperature until ester cleavage was complete as visualized by TLC (3–12 h). The solvent was removed under reduced pressure, and the residue was taken up in 15 mL of chloroform. The solution was brought to 0 °C for 2 h, and the resulting precipitate was collected by filtration. The mother liquor was concentrated and taken up in 5–10 mL of chloroform and brought to 0 °C for 2 h and then filtered.
The combined precipitates were confirmed by 1H NMR to be the corresponding phenol. The phenol (1 equiv) was then taken up in 10 mL of THF and reacted with N-phenyl-bis(trifluorosulfonimide) (2 equiv) in the presence of diisopropylethylamine (10 equiv). After triflation was complete, as judged by TLC visualization (3–12 h), the reaction mixture was concentrated and partitioned between 50 mL of EtOAc and 50 mL of water. The aqueous phase was extracted with EtOAc, and the combined phase was dried over MgSO4. Filtration, removal of the solvent under reduced pressure, and purification of the resulting residue by flash chromatography eluting with 1:1 hexanes–EtOAc gave the product as a viscous oil.
2R-(2′-Trifluoromethane sulfonyl phenyl ester)-4-quinazolinone (5a)
The title compound was synthesized according to general procedure B from 4a (0.40 g, 0.88 mmol) to give 0.20 g (96%) of the phenol. [α]d −128 (c 0.1, THF); 1H NMR (300 MHz, DMSO-d6) δ 9.84 (s, 1H), 7.92 (s, 1H), 7.58–7.61 (d, J = 7.8 Hz, 1H), 7.30–7.33 (d, J = 7.5 Hz, 1H), 7.17–7.22 (dt, J = 7.2, 1.2 Hz, 1H), 7.10–7.15 (t, J = 6.6 Hz, 1H), 6.82–6.85 (d, J = 8.1 Hz, 1H), 6.73–6.80 (m, 3H), 6.61–6.66 (t, J = 7.5 Hz, 1H), 5.98 (s, 1H). Subsequently, 0.22 g (94%) of the triflate was isolated as a clear viscous oil. [α]d −123 (c 0.1, THF); 1H NMR (300 MHz, CDCl3) δ 7.93 (dd, J = 7.5, 2.1 Hz, 1H), 7.83–7.86 (dd, J = 7.8, 1.5 Hz, 1H), 7.42–7.50 (m, 2H), 7.26–7.34 (m, 2H), 6.84–6.89 (t, J = 7.8 Hz, 1H), 6.66–6.68 (d, J = 8.1 Hz, 1H), 6.63 (s, 1H), 6.24 (s, 1H), 4.78 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 165.1, 146.8, 146.5, 134.6, 132.9, 131.7, 130.1, 129.6, 128.7, 122.0, 120.0, 115.3, 115.0, 62.4 (CF3 signal not detected); ESI m/z (rel intensity) 372.0 (100).
2S-(2′-Trifluoromethane sulfonyl phenyl ester)-4-quinazolinone (5b)
The title compound was synthesized according to general procedure B from 3b (0.15 g, 0.34 mmol) to give 0.076 g (94%) of the phenol. [α]d +122 (c 0.1, THF); 1H NMR (300 MHz, DMSO-d6) δ 9.92 (s, 1H), 8.01 (s, 1H), 7.67–7.69 (d, J = 7.8 Hz, 1H), 7.39–7.41 (d, J = 7.5 Hz, 1H), 7.18–7.30 (m, 2H), 6.91–6.93 (d, J = 8.1 Hz, 1H), 6.81–6.88 (m, 3H), 6.70–6.75 (t, J = 7.5 Hz, 1H), 6.06 (s, 1H).
Subsequently, 0.10 g (84%) of the triflate was isolated as a clear viscous oil. [α]d +120 (c 0.1, THF); 1H NMR (300 MHz, CDCl3) δ 7.96–7.98 (d, J = 7.5 Hz, 1H), 7.89–7.92 (d, J = 7.2 Hz, 1H), 7.50–7.54 (m, 2H), 7.35–7.40 (m, 2H), 6.90–6.95 (t, J = 7.5 Hz, 1H), 6.70–6.73 (d, J = 8.1 Hz, 1H), 6.56 (s, 1H), 6.29 (s, 1H), 4.75 (bs, 1H); 13C NMR (75 MHz, CDCl3) δ 165.11, 146.7, 146.5, 134.6, 132.9, 131.7, 130.1, 129.6, 128.7, 122.1, 120.1, 115.3, 115.1, 62.5 (CF3 signal not detected); ESI m/z (rel intensity) 372.0 (100).
2S-(2′-Trifluoromethane sulfonyl phenyl ester)-7-methoxy-4-quinazolinone (5c)
The title compound was synthesized according to general procedure B from 3c (0.30 g, 0.62 mmol) to give 0.15 g (89%) of the phenol. [α]d −93 (c 0.1, THF); 1H NMR (300 MHz, DMSO-d6) δ 9.85 (bs, 1H), 8.30 (s, 1H), 7.71 (s, 1H), 7.50–7.53 (d, J = 8.7 Hz, 1H), 7.27–7.30 (d, J = 7.8 Hz, 1H), 7.10–7.25 (t, J = 6.6 Hz, 1H), 6.72–6.85 (m, 2H), 6.20–6.30 (m, 2H), 5.94 (s, 1H), 3.68 (s, 3H). Subsequently, 0.14 g (77%) of the triflate was isolated as a clear viscous oil. [α]d −92 (c 0.1, THF); 1H NMR (300 MHz, CDCl3) δ 7.93–7.94 (dd, J = 7.2, 2.4 Hz, 1H), 7.77–7.80 (d, J = 8.7 Hz, 1H), 7.43–7.53 (m, 2H), 7.30–7.33 (dd, J = 7.5, 1.8 Hz, 1H), 6.52 (s, 1H), 6.41–6.45 (dd, J = 8.7, 2.1 Hz, 1H), 6.22 (s, 1H), 6.16–6.17 (d, J = 2.1 Hz, 1H), 4.87 (bs, 1H), 3.79 (s, 3H); 13C (75 MHz, CDCl3) δ 165.0, 164.9, 148.5, 146.4, 133.2, 131.6, 130.6, 130.0, 129.6, 122.0, 108.7, 107.2, 98.9, 62.5, 55.6 (CF3 signal not detected); ESI m/z (rel intensity) 402.1 (100).
2S-(2′-Trifluoromethane sulfonyl phenyl ester)-6-methoxy-4-quinazolinone (5d)
The title compound was synthesized according to general procedure B from 3d (0.16 g, 0.32 mmol) to give 0.080 g (88%) of the phenol. [α]d −165 (c 0.1, THF); 1H NMR (300 MHz, DMSO-d6) δ 9.86 (s, 1H), 7.99 (s, 1H), 7.37–7.39 (d, 7.8 Hz, 1H), 7.15–7.20 (m, 2H), 6.77–6.96 (m, 4H), 6.41 (s, 1H), 5.97 (s, 1H), 3.72 (s, 3H). Subsequently, 0.10 g (100%) of the triflate was isolated as a clear viscous oil. [α]d −78 (c 0.1, THF); 1H NMR (300 MHz, CDCl3) δ 7.94–7.97 (dd, J = 6.9, 2.4 Hz, 1H), 7.50–7.55 (dt, J = 6.6, 1.5 Hz, 2H), 7.45–7.46 (d, 2.7 Hz, 1H), 7.38–7.40 (m, 2H), 7.01–7.04 (dd, J = 8.7, 2.7 Hz, 1H), 6.70–6.73 (d, J = 9 Hz, 1H), 6.59 (bs, 1H), 6.24 (s, 1H), 3.82 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 165.6, 154.0, 146.6, 140.9, 132.5, 131.8, 129.9, 129.6, 127.1, 123.5, 122.2, 117.2, 110.8, 62.8, 56.0 (CF3 signal not detected); ESI m/z (rel intensity) 402.1 (100).
2S-(2′-Trifluoromethane sulfonyl phenyl ester)-7-nitro-4-quinazolinone (5e)
The title compound was synthesized according to general procedure B from 3e (0.17 g, 0.34 mmol) to give 0.090 g (91%) of the phenol. [α]d −233 (c 0.1, THF); 1H NMR (300 MHz, DMSO-d6) δ 9.96 (s, 1H), 8.41 (s, 1H), 7.81–7.84 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 1.8 Hz, 1H), 7.46 (s, 1H), 7.37–7.40 (dd, J = 8.7, 2.4 Hz, 1H), 7.25–7.28 (d, J = 8.4 Hz, 1H), 7.13–7.18 (t, J = 8.1 Hz, 1H), 6.76–6.87 (m, 2H), 6.08 (s, 1H). Subsequently, 0.12 g (100%) of the triflate was isolated as a clear viscous oil. [α]d −132 (c 0.1, THF); 1H NMR (300 MHz, CDCl3) δ 8.84 (s, 1H), 8.00–8.03 (d, J = 8.4 Hz, 1H), 7.86–7.89 (d, J = 7.5 Hz, 1H), 7.62–7.64 (d, J = 8.4 Hz, 1H), 7.48–7.58 (m, 3H), 6.76 (s, 1H), 6.33 (s, 1H), 5.17 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 158.7, 147.2, 142.3, 141.6, 127.5, 127.2, 125.7, 125.0, 122.5, 118.6, 117.7, 109.4, 105.3, 57.8 (CF3 signal not detected); ESI m/z (rel intensity) 417.0 (100).
2S-(2′-Trifluoromethane sulfonyl phenyl ester)-6-nitro-4-quinazolinone (5f)
The title compound was synthesized according to general procedure B from 3f (0.46 g, 0.95 mmol) to give 0.23 g (85%) of the phenol. [α]d −292 (c 0.1, THF); 1H NMR (300 MHz, DMSO-d6) δ 9.98 (s, 1H), 8.43–8.45 (m, 2H), 8.36 (s, 1H), 8.04–8.08 (dd, J = 9.0, 2.7 Hz, 1H), 7.24–7.27 (d, J = 7.5 Hz, 1H), 7.15–7.21 (t, J = 7.5, 1.5 Hz, 1H), 6.80–6.88 (m, 3H), 6.18 (s, 1H). Subsequently, 0.34 g (100%) of the triflate was isolated as a yellow viscous oil. [α]d −194 (c 0.1, THF); 1H NMR (300 MHz, CDCl3) δ 9.77 (s, 1H), 8.77 (s, 1H), 8.20–8.23 (d, J = 9.0 Hz, 1H), 7.88–7.90 (d, J = 6.6 Hz, 1H), 7.47–7.60 (m, 2H), 7.03 (s, 1H), 6.76–6.80 (dd, J = 9.0, 2.7 Hz, 1H), 6.43 (s, 1H), 6.08 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 162.7, 151.0, 146.0, 139.8, 134.9, 131.9, 129.7, 129.4, 126.6, 125.5, 122.8, 122.1, 114.6, 62.2 (CF3 signal not detected); ESI m/z (rel intensity) 417.0 (100).
2S-(2′-Trifluoromethane sulfonyl phenyl ester)-8-methyl-4-quinazolinone (5g)
The title compound was synthesized according to general procedure B from 3g (0.50 g, 1.09 mmol) to give 0.20 g (73%) of the phenol. [α]d −97 (c 0.1, THF); 1H NMR (300 MHz, DMSO-d6) δ 9.97 (s, 1H), 7.98 (s, 1H), 7.48–7.50 (d, J = 7.5 Hz, 1H), 7.16–7.19 (dd, J = 7.2, 1.2 Hz, 1H), 7.08–7.13 (t, J = 7.5 Hz, 2H), 6.82–6.84 (d, J = 8.1 Hz, 1H), 6.71–6.76 (t, J = 7.5 Hz, 1H), 6.57–6.62 (t, 7.5 Hz, 1H), 6.07 (s, 1H), 5.93 (s, 1H), 2.07 (s, 3H). Subsequently, 0.18 g (100%) of the triflate was isolated as a clear viscous oil. [α]d −88 (c 0.1, THF); 1H NMR (300 MHz, CDCl3) δ 7.95–7.98 (dd, J = 7.5, 2.1 Hz, 1H), 7.74–7.76 (d, J = 7.8 Hz, 1H), 7.50–7.55 (m, 2H), 7.22–7.39 (m, 3H), 7.17 (s, 1H), 6.79–6.84 (t, J = 8.1 Hz, 1H), 6.29 (s, 1H), 2.15 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 165.8, 146.5, 145.1, 135.4, 133.2, 131.6, 130.1, 129.6, 126.5, 122.8, 122.0, 119.4, 115.2, 62.3, 16.7 (CF3 signal not detected); ESI m/z (rel intensity) 386.1 (100).
2S-(2′-Trifluoromethane sulfonyl phenyl ester)-5-methyl-4-quinazolinone (5h)
The title compound was synthesized according to general procedure B from 3h (0.22 g, 0.47 mmol) to give 0.10 g (81%) of the phenol. [α]d −114 (c 0.1, THF); 1H NMR (300 MHz, DMSO-d6) δ 9.83 (s, 1H), 7.81 (s, 1H), 7.38–7.41 (dd, J = 7.5, 1.2 Hz, 1H), 7.15–7.21 (dt, J = 8.1, 1.5 Hz, 1H), 7.06–7.11 (t, J = 7.8 Hz, 1H), 6.81–6.90 (m, 2H), 6.64–6.69 (m, 2H), 6.48–6.51 (d, J = 7.5 Hz, 1H), 5.91 (s, 1H), 2.57 (s, 3H). Subsequently, 0.10 g (100%) of the triflate was isolated as a clear viscous oil. [α]d −116 (c 0.1, THF); 1H NMR (300 MHz, CDCl3) δ 7.96–7.99 (dd, J = 6.9, 2.7 Hz, 1H), 7.47–7.52 (m, 2H), 7.31–7.35 (m, 2H), 7.16–7.21 (t, J = 7.5 Hz, 1H), 6.67–6.71 (m, 2H), 6.56–6.58 (d, J = 8.1 Hz, 1H), 6.16 (s, 1H), 2.64 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 165.7, 148.4, 146.7, 142.7, 133.4, 132.7, 131.6, 130.3, 129.5, 126.5, 123.8, 121.9, 113.4, 61.9, 22.5 (CF3 signal not detected); ESI m/z (rel intensity) 386.1 (100).
2S-(2′-Trifluoromethane sulfonyl phenyl ester)-6-chloro-4-quinazolinone (5i)
The title compound was synthesized according to general procedure B from 3i (0.24 g, 0.51 mmol) to give 0.11 g (78%) of the phenol. [α]d −172 (c 0.1, THF); 1H NMR (300 MHz, DMSO-d6) δ 9.82 (s, 1H), 8.07 (s, 1H), 7.45 (s, 1H), 7.20–7.22 (d, J = 6.6 Hz, 1H), 7.14–7.17 (d, J = 7.2 Hz, 1H), 7.04–7.08 (t, J = 7.2 Hz, 1H), 6.92 (s, 1H), 6.68–6.78 (m, 3H), 5.92 (s, 1H). Subsequently, 0.11 g (100%) of the triflate was isolated as a clear viscous oil. [α]d −97 (c 0.1, THF); 1H NMR (300 MHz, CDCl3) δ 9.37 (s, 1H), 7.86–7.89 (dd, J = 7.2, 3.2 Hz, 1H), 7.83 (d, J = 2.4 Hz, 1H), 7.46–7.55 (m, 2H), 7.24–7.24 (m, 1H), 6.63–6.69 (m, 2H), 6.24 (s, 1H), 4.79 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 164.5, 146.4, 145.2, 134.8, 132.0, 129.8, 129.7, 128.3, 127.1, 125.3, 123.2, 122.3, 116.7, 62.5 (CF3 signal not detected); ESI m/z (rel intensity) 406.0 (100).
General Procedure C. Reduction of Aryl Triflate
Triethylamine (15 equiv) and Pd(dppf)Cl2 (0.02 equiv) were added to a solution of formic acid (10 equiv) in DMF (75 mL/mmol). After the resulting red solution was thoroughly degassed, the corresponding aryl triflate (1 equiv) was added (the formic acid must be completely neutralized before adding the triflate, otherwise racemization may occur), and the reaction mixture was heated at 90–100 °C until the starting material was completely consumed as judged by TLC (1.5 h). The dark solution was added to 100 mL of H2O and extracted with EtOAc. The combined organic extracts were dried over MgSO4 and filtered, and the solvent was removed under reduced pressure. The resulting residue was purified by flash chromatography eluting with 1:1 hexanes–EtOAc and further purified by recrystallization from THF–hexanes.
General Procedure D. Reduction of Aryl Triflate for Nitro-Containing Compounds
Triethylamine (15 equiv) and Pd(dppf)Cl2 (0.02 equiv) were added to a solution of formic acid (1.0 equiv) in 75 mL/mmol of DMF (excess formic acid results in the reduction of the nitro functional group). After the resulting red solution was thoroughly degassed, the corresponding aryl triflate (1 equiv) was added (the formic acid must be completely neutralized before adding the triflate, otherwise racemization may occur), and the reaction mixture was heated at 90–100 °C until the starting material was completely consumed as judged by TLC (1.5 h). The dark solution was added to 100 mL of H2O and extracted with EtOAc. The combined organic extracts were dried over MgSO4 and filtered, and the solvent was removed under reduced pressure. The resulting residue was purified by flash chromatography eluting with 1:1 hexanes–EtOAc and further purified by recrystallization from THF–hexanes.
2R-(Phenyl)-4-quinazolinone (6a)
The title compound was synthesized according to general procedure C from triflate 5a (0.075 g, 0.20 mmol) to give 0.044 g (98%) of the resulting dihydroquinazolinone as a colorless solid. [α]d −158 (c 0.1, THF); mp 211–213 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.33 (s, - 1H), 7.63–7.64 (d, J = 3.0 Hz, 1H), 7.50–7.52 (d, J = 7.5 Hz, 2H), 7.33–7.40 (m, 3H), 7.23–7.26 (t, J = 7.0 Hz, 1H), 7.14 (s, 1H), 6.76–6.78 (d, J = 8.0 Hz, 1H), 6.66–6.69 (t, J = 8.0 Hz, 1H), 5.77 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 164.1, 148.4, 142.1, 133.8, 128.9, 128.8, 127.8, 127.3, 117.6, 115.4, 114.9, 67.1; ESI m/z (rel intensity) 224.1 (100). Anal. (C14H12N2O) C, H, N. C: calcd, 74.98; found, 74.86. H: calcd, 5.39; found, 5.25. N: calcd, 12.49; found, 12.50. The enantiopurity was 100% as confirmed by chiral HPLC (ChiralPak AS column with 100% MeCN; flow of 0.2 mL/min; retention time (6b), 32.2 min; retention times of the components of the racemate, 29.9 and 32.3 min).
2S-(Phenyl)-4-quinazolinone (6b)
The title compound was synthesized according to general procedure C from triflate 5b (0.070 g, 0.19 mmol) to give 0.038 g (90%) of the resulting dihydroquinazolinone as a colorless solid. [α]d +160 (c 0.1, THF); mp 211–213 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.32 (s, 1H), 7.60, 7.63 (d, J = 7.8 Hz, 1H), 7.47–7.50 (d, J = 6.6 Hz, 2H), 7.31–7.40 (m, 3H), 7.20–7.25 (t, J = 7.2 Hz, 1H), 7.12 (s, 1H), 6.73–6.76 (d, J = 8.1 Hz, 1H), 6.64–6.68 (t, J = 7.2 Hz, 1H), 5.75 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 164.4, 148.5, 142.3, 134.1, 129.2, 129.0, 128.1, 127.5, 117.8, 115.6, 115.1, 67.2; ESI m/z (rel intensity) 224.1 (100). Anal. (C14H12N2O) C, H, N. C: calcd, 74.98; found, 74.84. H: calcd, 5.39; found, 5.35. N: calcd, 12.49; found, 12.46. The enantiopurity was 100% as confirmed by chiral HPLC (ChiralPak AS column with MeCN 100%; flow of 0.2 mL/min; retention time (6a), 29.5 min; retention times of the components of the racemate, 29.9 and 32.3 min).
2R-(Phenyl)-7-methoxy-4-quinazolinone (6c)
The title compound was synthesized according to general procedure C from triflate 5c (0.12 g, 0.30 mmol) to give 0.049 g (79%) of the resulting dihydroquinazolinone as a colorless solid. [α]d −135 (c 0.1, THF); mp 180–181 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.10 (s, 1H), 7.52–7.53 (d, J = 5.7 Hz, 1H), 7.46–7.48 (d, J = 4.5 Hz, 2H), 7.32–7.39 (m, 3H), 7.11 (s, 1H), 6.25–6.26 (m, 2H), 5.7 (s, 1H), 3.69 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 164.0, 149.9, 142.3, 129.6, 128.8, 128.7, 127.4, 109.0, 105.5, 98.4, 67.2, 55.7; ESI m/z (rel intensity) 254.1 (100). Anal. (C15H14N2O2) C, H, N. C: calcd, 70.85; found, 70.49. H: calcd, 5.55; found, 5.49. N: calcd, 11.02; found, 10.87. The racematic mixture of 6c could not be resolved into its separate diastereomeric components.
2R-(Phenyl)-6-methoxy-4-quinazolinone (6d)
The title compound was synthesized according to general procedure C from triflate 5d (0.090 g, 0.22 mmol) to give 0.042 g (91%) of the resulting dihydroquinazolinone as a colorless solid. [α]d −200 (c 0.1, THF); mp 206–208 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.35 (s, 1H), 7.51–7.53 (d, J = 7.5 Hz, 2H), 7.36–7.43 (m, 3H), 7.17 (s, 1H), 6.93–6.97 (dd, J = 8.7, 3.0 Hz, 1H), 6.74–6.78 (m, 2H), 5.71 (s, 1H), 3.70 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 164.4, 152.1, 143.0, 142.2, 129.1, 129.0, 127.7, 122.2, 116.9, 116.4, 110.5, 67.6, 56.0; ESI m/z (rel intensity) 254.1 (100). Anal. (C15H14N2O2) C, H, N. C: calcd, 70.85; found, 70.85. H: calcd, 5.55; found, 5.49. N: calcd, 11.02; found, 10.91. The components of the racemate of 6d were not separable under any conditions we examined.
2R-(Phenyl)-7-nitro-4-quinazolinone (6e)
The title compound was synthesized according to general procedure D from triflate 5e (0.03 g, 0.07 mmol) to give 0.015 g (78%) of the resulting dihydroquinazolinone as a yellow solid. [α]d −279 (c 0.1, THF); mp 219–221 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.73 (s, 1H), 7.82–7.84 (d, J = 5.1 Hz, 1H), 7.76 (s, 1H), 7.57 (d, J = 1.5 Hz, 1H), 7.48–7.49 (d, J = 4.2 Hz, 2H), 7.36–7.43 (m, 4H), 5.91 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ 162.3, 151.2, 148.6, 141.5, 129.6, 129.2, 129.0, 127.2, 119.7, 111.4, 109.3, 66.8; ESI m/z (rel intensity) 269.1 (100). Anal. (C14H11N3O3 · 0.2H2O) C, H, N. C: calcd, 61.63; found, 61.59. H: calcd, 4.21; found, 4.00. N: calcd, 15.40; found, 15.29. The enantiopurity was 100% as confirmed by chiral HPLC (ChiralPak AS column with MeCN 100%; flow of 0.4 mL/min; retention time (6e), 12.0 min; retention times of the components of the racemate, 12.0 and 16.0 min).
2R-(Phenyl)-6-nitro-4-quinazolinone (6f)
The title compound was synthesized according to general procedure D from triflate 5f (0.20 g, 0.48 mmol) to give 0.044 g (58%) of the resulting dihydroquinazolinone as a yellow solid. [α]d −240 (c 0.1, THF); mp 238–240 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.75 (s, 1H), 8.57 (s, 1H), 8.42 (d, J = 1.5 Hz, 1H), 8.08–8.10 (d, J = 5.4 Hz, 1H), 7.36–7.47 (m, 5H), 6.81–6.83 (d, J = 5.7 Hz, 1H), 6.01 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ 161.8, 152.6, 141.5, 137.5, 129.5, 129.4, 129.1, 127.0, 124.6, 114.7, 113.1, 66.7; ESI m/z (rel intensity) 269.1 (100). Anal. (C14H11N3O3 · 0.2H2O) C, H, N. C: calcd, 61.63; found, 61.57. H: calcd, 4.21; found, 4.02. N: calcd, 15.61; found, 15.24. The enantiopurity was 100% as confirmed by chiral HPLC (ChiralPak AS column with MeCN 100%; flow of 0.4 mL/min; retention time (6f), 12.6 min; retention times of the components of the racemate, 12.5 and 15.0 min).
2R-(Phenyl)-8-methyl-4-quinazolinone (6g)
The title compound was synthesized according to general procedure C from triflate 5g (0.18 g, 0.47 mmol) to give 0.069 g (77%) of the resulting dihydroquinazolinone as a colorless solid. [α]d −150 (c 0.1, THF); mp 176–178 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.40 (s, 1H), 7.48–7.50 (d, J = 4.5 Hz, 1H), 7.44–7.46 (d, J = 4.5 Hz, 2H), 7.32–7.35 (t, J = 4.5 Hz, 2H), 7.27–7.29 (m, 1H), 7.12–7.13 (d, J = 4.2 Hz, 1H), 6.56–6.61 (t, J = 4.5 Hz, 2H), 5.72 (s, 1H), 2.12 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 164.1, 145.9, 143.0, 134.5, 128.7, 128.5, 126.9, 125.7, 122.8, 117.2, 115.6, 65.9, 17.4; ESI m/z (rel intensity) 238.1 (100). Anal. (C15H14N2O) C, H, N. C: calcd, 75.61; found, 75.55. H: calcd, 5.92; found, 5.87. N: calcd, 11.76; found, 11.68. The components of the racemate of 6g were not separable under any conditions we examined.
2R-(Phenyl)-5-methyl-4-quinazolinone (6h)
The title compound was synthesized according to general procedure C from triflate 5h (0.09 g, 0.23 mmol) to give 0.035 g (78%) of the resulting dihydroquinazolinone as a colorless solid. [α]d −172 (c 0.1, THF); mp 134–135 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.22 (s, 1H), 7.53–7.56 (d, J = 6.6 Hz, 2H), 7.40–7.42 (d, J = 7.2 Hz, 3H), 7.09–7.13 (t, J = 7.5 Hz, 1H), 7.05 (s, 1H), 6.68–6.71 (d, J = 7.5 Hz, 1H), 6.50–6.52 (d, J = 6.9 Hz, 1H), 5.67 (s, 1H), 3.70 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 165.2, 150.1, 142.1, 141.2, 132.9, 129.1, 129.0, 127.7, 121.7, 114.3, 113.7, 67.1, 22.8; ESI m/z (rel intensity) 238.1 (100). Anal. (C15H14N2O) C, H, N. C: calcd, 75.61; found, 75.63. H: calcd, 5.92; found, 5.94. N: calcd, 11.76; found, 11.74. The components of the racemate of 6h were not separable under any conditions we examined.
2R-(Phenyl)-6-chloro-4-quinazolinone (6i)
The title compound was synthesized according to general procedure C from triflate 5i (0.11 g, 0.27 mmol) to give 0.045 g (59%) of the resulting dihydroquinazolinone as a colorless solid. [α]d −111 (c 0.1, THF); mp 243–245 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.53 (s, 1H), 7.59 (d, J = 2.1 Hz, 1H), 7.51–7.54 (d, J = 6.6 Hz, 2H), 7.38–7.45 (m, 4H), 7.30–7.33 (dd, J = 8.7, 2.4 Hz, 1H), 6.81–6.84 (d, J = 8.7 Hz, 1H), 5.83 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ 163.2, 147.3, 142.0, 133.8, 129.3, 129.1, 127.6, 127.2, 121.5, 117.1, 116.8, 67.2; ESI m/z (rel intensity) 258.1 (100). Anal. (C14H11ClN2O) C, H, N. C: calcd, 65.00; found, 64.91. H: calcd, 4.29; found, 4.19. N: calcd, 10.83; found, 10.81. The components of the racemate of 6i were not separable under any conditions we examined.
5-(2′-Tolyl)-2-formylphenol (9)
A solution of 3.95 g (18.4 mmol) of 3-bromophenylacetate, 2.50 g (18.4 mmol) of 2-tolylboronic acid, 8.97 g (27.6 mmol) of Cs2CO3, and 0.42 g (0.37 mmol) of Pd(PPh3)4 in 50 mL of dioxane was thoroughly degassed and brought to reflux until the starting materials were consumed (12 h) as judged by TLC. EtOH (10 mL) was added, and the reaction mixture was stirred for 1 h. The reaction mixture was concentrated under reduced pressure and partitioned between 100 mL of saturated NH4Cl and 100 mL of EtOAc. The aqueous phase was extracted with EtOAc. The combined organic extracts were dried over MgSO4 and filtered, and the solvent was removed under reduced pressure. Purification by flash chromatography eluting with 7:1 hexanes–EtOAc gave 3.17 g (94%) of the phenol 8 as a brown oil.
To a solution of 3.17 g (17.2 mmol) of phenol 8 in 25 mL of THF at 0 °C was added 5.7 mL (3 M solution in Et2O, 17.2 mmol) of MeMgBr. After 30 min, a precipitate formed, and all the THF was removed under reduced pressure. Benzene (25 mL) was added and removed under reduced pressure. Then another 25 mL portion of benzene was added. Then 6.61 g (73.4 mmol) of paraformaldehyde and 6.1 mL (44.1 mmol) of Et3N were added, and the mixture was brought to reflux for 24 h. The reaction was quenched by addition of 150 mL of saturated NH4Cl and extracted with three 75 mL portions of EtOAc. The combined organic extracts were dried over MgSO4 and filtered, and the solvent was removed under reduced pressure. Purification by distillation (220 °C, 1 mmHg) gave 2.4 g (66%) of the aldehyde as a viscous pale-yellow oil. 1H NMR (300 MHz, CDCl3) δ 11.22 (s, 1H), 9.97 (s, 1H), 7.62–7.65 (d, J = 7.8 Hz, 1H), 7.27–7.37 (m, 4H), 7.03–7.07 (m, 2H), 2.36 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 196.5, 161.7, 151.5, 140.6, 135.3, 133.7, 130.9, 129.5, 128.5, 126.3, 121.6, 119.6, 118.5, 20.7; ESI m/z (rel intensity) 212.1 (100).
N-Boc-d-tert-leucine[5-(2′-tolyl)-2-formyl]phenyl Ester (10)
To a room temperature solution of 1.07 g (5.0 mmol) of Boc-(l)-tert-leucine was added 1.02 g (5.0 mmol) of DCC. After 45 min, 0.67 g (5.0 mmol) of HOBt was added. After an additional 45 min, 1.00 g (4.7 mmol) of aldehyde 9 was added and the mixture was stirred overnight. Water (150 mL) was added, and the mixture was extracted with three 75 mL portions of EtOAc. The combined organic extracts were dried over MgSO4 and filtered, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography eluting with 6:1 hexanes–EtOAc, and 1.20 g (59%) of the product was obtained as a viscous oil. 1H NMR (300 MHz,CDCl3) δ 10.29 (s, 1H), 8.00–8.03 (d, J = 8.1 Hz, 1H), 7.28–7.40 (m, 5H), 7.21 (s, 1H), 5.19–5.22 (d, J = 8.7 Hz, 1H), 4.40–4.43 (d, J = 8.4 Hz, 1H), 2.32 (s, 3H), 1.51 (s, 9H), 1.18 (s, 9H); ESI m/z (rel intensity) 425.2 (100).
2S-[4′-(2″-Tolyl)-2′-N-Boc-d-tert-leucine phenyl ester)-6-nitro-4-quinazolinone (11)
The title compound was prepared according to general procedure A from 5-nitroanthranilamide (0.27 g, 1.50 mmol) and aldehyde 10 (0.64 g, 1.50 mmol) to give 0.33 g (37%) of the resulting product as a yellow foam. [α]d −210 (c 0.1, THF); 1H NMR (300 MHz, DMSO-d6) δ 8.79–8.80 (d, J = 2.7 Hz, 1H), 8.06–8.10 (dd, J = 9.0, 2.7 Hz, 1H), 7.62–7.64 (m, 2H), 7.16–7.28 (m, 6H), 6.83 (s, 1H), 6.74–6.77 (d, J = 9.0 Hz, 1H), 6.48 (s, 1H), 5.18–5.20 (d, J = 6.0 Hz, 1H), 4.11–4.14 (m, 1H), 2.90 (s, 3H), 1.44 (s, 9H), 1.17 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 171.0, 164.2, 156.8, 151.3, 147.0, 144.2, 140.0, 139.1, 135.5, 131.0, 130.8, 129.9, 129.5, 128.2, 127.5, 127.0, 126.2, 125.7, 123.4, 115.0, 113.0, 81.2, 63.4, 61.8, 33.7, 28.6, 27.1, 20.6; ESI m/z (rel intensity) 588.3 (100).
2S-[4′-(2″-tolyl)Phenyl]-6-nitro-4-quinazolinone ((S)-(+)-7)
The title compound was prepared by making the triflate according to general procedure C, followed by reduction of the triflate according to general procedure D. Reaction of ester 11 (0.33 g, 0.56 mmol) with hydrazine (0.82 mL, 1.68 mmol) gave 0.17 g (80%) of the corresponding phenol as a yellow solid. [α]d −288 (c 0.1, THF); 1H NMR (300 MHz, DMSO-d6) δ 10.17 (s, 1H), 8.49–8.57 (m, 2H), 8.12–8.17 (dd, J = 9.0, 2.7 Hz, 1H), 7.28–7.40 (m, 5H), 7.18–7.20 (m, 1H), 6.85–6.92 (m, 3H), 6.30 (s, 1H), 2.27 (s, 3H).
To 0.080 g (0.22 mmol) of the phenol was added 0.16 g (0.44 mmol) of N-phenyl-bis(trifluoromethanesulfonimide) and 0.38 mL (2.2 mmol) of diisopropylethylamine to give 0.11 g (100%) of the triflate as a viscous yellow oil. [α]d −156 (c 0.1, THF); 1H NMR (300 MHz, CDCl3) δ 8.87 (s, 1H), 8.29–8.31 (d, J = 6.6 Hz, 1H), 7.98–8.01 (d, J = 7.8 Hz, 1H), 7.56–7.59 (d, J = 8.1 Hz, 1H), 7.36–7.51 (m, 4H), 7.23–7.28 (m, 2H), 6.85–6.88 (d, J = 9.0 Hz, 1H), 6.56 (s, 1H), 5.79 (s, 1H), 2.38 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 163.1, 151.0, 146.8, 145.8, 140.5, 138.9, 135.5, 131.2, 130.8, 130.5, 130.2, 129.9, 129.4, 129.0, 127.3, 126.6, 125.9, 123.4, 123.2, 115.0, 62.5, 20.6 (CF3 signal not detected).
The triflate was reduced according to general procedure D using formic acid (0.013 mL, 0.35 mmol), triethylamine (0.17 mL, 1.20 mmol), and Pd(dppf)Cl2 (0.004 g, 0.005 mmol) to give 0.058 g (67%) of dihydroquinazolinone 7 as yellow crystals. [α]d −213 (c 0.1, THF); mp 258–260 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.86 (s, 1H), 8.69 (s, 1H), 8.51 (s, 1H), 8.15–8.19 (dd, J = 9.0, 2.4 Hz, 1H), 7.58–7.61 (d, J = 8.4 Hz, 2H), 7.44–7.47 (d, J = 8.1 Hz, 2H), 7.29–7.32 (m, 3H), 7.21–7.22 (m, 1H), 6.90–6.93 (d, J = 9.0 Hz, 1H), 6.14 (s, 1H), 2.27 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 162.0, 152.9, 142.7, 141.4, 140.5, 137.9, 135.4, 131.1, 130.2, 130.1, 129.8, 128.2, 127.3, 126.7, 125.0, 115.0, 113.3, 67.0, 20.9; ESI m/z (rel intensity) 359.1 (100). Anal. (C21H17N3O3) C, H, N. C: calcd, 70.18; found, 70.05. H: calcd, 4.77; found, 4.65. N: calcd, 11.69; found, 11.71. The enantiopurity was 100% as confirmed by chiral HPLC (ChiralPak AS column with MeCN 100%; flow of 0.2 mL/min; retention time ((−)-7), 24.1 min; retention times of the components of the racemate, 24.3 and 27.0 min).
N-Boc-l-tert-leucine [5-(2′-tolyl)-2-formyl]phenyl Ester (13)
To a room temperature solution of 1.50 g (6.9 mmol) of Boc-d-tert-leucine (1.50 g, 6.91 mmol) was added 1.57 g (7.6 mmol) of DCC. After 45 min, 1.03 g (7.6 mmol) of HOBt was added. After an additional 45 min, 1.10 g (5.2 mmol) of aldehyde 9 was added, and the reaction mixture was stirred overnight. Water (150 mL) was added, and the mixture was extracted with EtOAc. The combined organic extracts were dried over MgSO4 and filtered, and the solvent was removed under reduced pressure. Purification by flash chromatography eluting with 6:1 hexanes–EtOAc gave 0.80 g (38%) of the product as a viscous oil. 1H NMR (300 MHz,CDCl3) δ 10.25 (s, 1H), 7.96–7.99 (d, J = 8.1 Hz, 1H), 7.17–7.36 (m, 5H), 7.16 (s, 1H), 5.14–5.17 (d, J = 8.1 Hz, 1H), 4.35–4.38 (d, J = 8.4 Hz, 1H), 2.29 (s, 3H), 1.47 (s, 9H), 1.13 (s, 9H); ESI m/z (rel intensity) 425.2 (100).
2R-[4′-(2″-tolyl)-2′-N-Boc-l-tert-leucine phenyl ester)-6-nitro-4-quinazolinone (14)
The title compound was prepared according to general procedure A from 5–nitroanthranilamide (0.28 g, 1.55 mmol) and aldehyde 10 (0.70 g, 1.55 mmol) to give 0.44 g (50%) of the product as a yellow foam. [α]d +213 (c 0.1, THF); 1H NMR (300 MHz, DMSO-d6) δ 8.78–8.79 (d, J = 2.4 Hz, 1H), 8.05–8.09 (dd, J = 9.0, 2.4 Hz, 1H), 7.61–7.63 (m, 2H), 7.16–7.27 (m, 6H), 6.84 (s, 1H), 6.74 –6.77 (d, J = 9.3 Hz, 1H), 6.48 (s, 1H), 5.18–5.20 (d, J = 5.7 Hz, 1H), 4.10–4.13 (dd, J = 7.2, 2.4 Hz, 1H), 2.25 (s, 3H), 1.44 (s, 9H), 1.11 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 171.0, 163.9, 156.8, 151.1, 145.9, 144.3, 140.0, 139.2, 135.6, 130.9, 130.8, 129.9, 129.5, 128.2, 127.6, 127.0, 126.2, 125.8, 123.4, 115.0, 113.0, 81.3, 63.3, 61.8, 34.1, 28.6, 27.1, 20.6; ESI m/z (rel intensity) 588.3 (100).
2R-[4′-(2″-Tolyl)phenyl]-6-nitro-4-quinazolinone ((R)-(−)-7)
The title compound was prepared by making the triflate according to general procedure C, followed by reduction of the triflate according to general procedure D. Reaction of 0.22 g (0.38 mmol) of ester 11 with 0.056 mL (1.15 mmol) of hydrazine gave 0.14 g (95%) of the corresponding phenol as a yellow solid. [α]d +293 (c 0.1, THF); 1H NMR (300 MHz, DMSO-d6) δ 10.13 (s, 1H), 8.45–8.52 (m, 3H), 8.08–8.12 (dd, J = 9.0, 2.7 Hz, 1H), 7.22–7.36 (m, 5H), 7.13–7.16 (m, 1H), 6.81–6.87 (m, 3H), 6.26 (s, 1H), 2.23 (s, 3H).
To 0.11 g (0.28 mmol) of the phenol was added 0.20 g (0.56 mmol) of N-phenyl-bis(trifluoromethanesulfonimide) and 0.49 mL (2.8 mmol) of diisopropylethylamine to give 0.14 g (100%) of the triflate as a viscous yellow oil. [α]d +155 (c 0.1, THF); 1H NMR (300 MHz, CDCl3) δ 8.77–9.78 (d, J = 2.7 Hz, 1H), 8.17–8.21 (dd, J = 9.0, 2.7 Hz, 1H), 7.87–7.89 (d, J = 8.1 Hz, 1H), 7.45–7.48 (dd, J = 8.1, 1.5 Hz, 1H), 7.25–7.38 (m, 4H), 7.16–7.19 (d, J = 7.2 Hz, 1H), 7.07 (s, 1H), 6.73–6.6.76 (d, J = 9.0 Hz, 1H), 6.44 (s, 1H), 5.65 (s, 1H), 2.26 (s, 3H); 13C NMR (75 MHz,CDCl3) δ 163.0, 150.9, 146.7, 145.7, 140.4, 138.8, 135.4, 131.1, 130.7, 130.3, 130.0, 129.8, 129.3, 128.9, 127.3, 126.5, 125.8, 123.3, 123.1, 114.9, 62.3, 20.5 (CF3 signal not detected).
The triflate was reduced according to general procedure D with formic acid (0.010 mL, 0.27 mmol), triethylamine (0.36 mL, 2.56 mmol), and Pd(dppf)Cl2 (0.004 g, 0.005 mmol) to give 0.050 g (54%) of 7 as yellow crystals. [α]d +209 (c 0.1, THF); mp 258–260 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.66 (s, 1H), 8.49 (s, 1H), 8.13–8.17 (dd, J = 9.3, 2.7 Hz, 1H), 7.55–7.58 (d, J = 7.8 Hz, 2H), 7.42–7.45 (d, J = 8.1 Hz, 2H), 7.27–7.31 (m, 3H), 7.17–7.20 (m, 1H), 6.87–6.90 (d, J = 9.0 Hz, 1H), 6.11 (s, 1H), 2.25 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 162.0, 152.8, 142.6, 141.3, 140.4, 137.8, 135.3, 131.1, 130.2, 130.1, 130.0, 128.2, 127.4, 126.7, 124.9, 115.0, 113.3, 66.9, 20.9; ESI m/z (rel intensity) 359.1 (100). Anal. (C21H17N3O3 · 0.1H2O) C, H, N. C: calcd, 69.83; found, 69.75. H: calcd, 4.80; found, 4.65. N: calcd, 11.63; found, 11.67. The enantiopurity was 100% as confirmed by chiral HPLC (ChiralPak AS column with MeCN 100%; flow of 0.2 mL/min; retention time (15), 27.2 min; retention times of the components of the racemate, 24.3 and 27.0 min).
X-ray Crystallography
A crystal of (−)-3 suitable for X-ray diffraction was obtained by slow evaporation of a solution containing 3 and a 1:1 mixture of isopropanol and 1-butanol. The crystal (dimensions 0.37 mm × 0.09 mm × 0.07 mm) was mounted on the Rigaku RAPID diffractometer, and data collection was controlled by the HKL-2000 package.15 The diffraction experiment was carried out at 103 K using Cu Kα X-ray radiation. During data collection ω scans with χ offsets were used. The diffraction images were recorded with Δω = 5°. Indexing, integration, and scaling were performed with HKL2000. The reflections were scaled in space group P43212 with Friedel pairs separated. The collected reflection composed a complete data set up to a resolution 0.81 Å. The data collection and processing statistics are summarized in Table 3. The structure solution and determination was performed with HKL-3000SM.16 The absolute configuration of the aminal carbon of (−)-3 was assigned as the R configuration.
Molecular Modeling
The X-ray crystal structure of α,β-tubulin complexed with colchicine (PDB code 1SA0) was used for the automated molecular docking of compounds (S)-7 and (R)-7. Inconsistencies between the PDB format and the tubulin residues library translation to atomic potential types were corrected manually. The tubulin was minimized using the DISCOVER (Accelrys, San Diego, CA) program’s cff91 force field17 with distance-dependent dielectrics.
Initially, the DHQZ ligand was constructed using Sybyl7.3 (Tripos Inc.) and optimized with the MP2/6-31G** basis set using the Gaussian quantum mechanical program.18 Docking experiments were performed with the AutoDock 3.0.512 and FlexX programs.19 The standard procedures were used for docking simulations using AutoDock and FlexX. In the case of AutoDock, for the protein and ligand torsion angles were identified for 10 independent runs per ligand. A grid of 60 × 60 × 60 points in the x, y, and z directions was built, centered on the center of the mass of the N atom in compound 7 that is in the tubulin active site. A grid spacing of 0.4 Å and a distance-dependent function of the dielectric constant were used for the calculation of the energetic map. The default settings were used for all other parameters. At the end of the docking simulation, ligands with the most favorable free energy of binding were selected. In the case of FlexX, all the parameters were used with the default settings, except the number of solution conformations was set to 90.
The best docked geometry was minimized using the DISCOVER module of Insight II (Accelrys Inc.) with the procedure described below. A consistent valence force field (cff91) was used. The cutoff for nonbonded interaction energies was set to ∞ (i.e., no cutoff) with the dielectric constant set at 4 to account for the dielectric shielding found in proteins. Each minimization was carried out in two steps, first using steepest descent minimization for 200 cycles and then using conjugate gradient minimization, until the average gradient fell below 0.01 kcal/mol. All atoms within 6.0 Å of the inhibitor were allowed to relax during the minimization, whereas those atoms beyond 6.0 Å were held rigid.
The minimized complex was subjected to molecular dynamics simulations using the DISCOVER module of Insight II (Accelrys Inc.). Molecular dynamics simulations performed in the NVE ensemble consisted of an initial equilibration of 25 ps (ps), followed by a production run of 300 ps dynamics at 300 K. The final complex structure at the end of the molecular dynamics simulations was subjected to 2000 steps of steepest descent energy minimization, followed by conjugate gradient energy minimization. A distance-dependent dielectric constant and nonbonded distance cutoff of 12 Å were used. Molecular dynamics simulations were performed using the AMBER8 package,20 with the general Amber force field (GAFF)21 and RESP charge models.22
[3H]Colchicine Binding Assays
The binding of [3H]colchicine to tubulin was measured by the DEAE-cellulose filter method, as described in detail previously.23 The tubulin and [3H]colchicine concentrations were 1.0 µM (0.1 mg/mL) and 5.0 µM, respectively.
Tubulin Polymerization
Tubulin polymerization was followed turbidimetrically at 350 nm in Beckman model DU-7400 and Du7500 spectrophotometers equipped with electronic temperature controllers, as described in detail previously.24 The tubulin concentration was 10 µM (1.0 mg/mL).
Cell Proliferation Assay
HCT-116 colon cancer cells (ATCC) were maintained in McCoy’s 5a medium with 1.5 mM l-glutamine (Invitrogen) supplemented with 5% fetal bovine serum (Atlanta Biologicals). MDA-MB-435 cancer cells (ATCC) were maintained in D-MEM with 1.5 mM l-glutamine supplemented with 5% fetal bovine serum (Optima). Cells were grown at 37 °C in an incubator with a humidified, 5% CO2 atmosphere.
Cells grown to confluency were trypinized, counted, and diluted to 6400 cells per 100 µL, and 3200 cells/well were added to a clear 96-well tissue culture plate (Falcon). Cells were allowed to attach, and 50 µL of media with vehicle or concentrations of drug at 10 nM, 30 nM, 100 nM, 300 nM, 1 µM, or 3 µM was added to each well (n = 8). The cells were incubated for 72 h. The CellTiter 96 AQueous nonradioactive cell proliferation assay (Promega) was used to measure the number of living cells in each well. The absorbance was read at 490 nm on an ELISA plate reader, and the data were evaluated for a dose response curve. The EC50 for each cell line (the amount of compound needed for 50% inhibition of cell proliferation) was calculated using GraphPad Prism 4 software.
Confocal Microscopy
Human melanoma (MDA-MB-435) samples were plated on sterilized microscope slides and incubated for 1 h with 5 µM racemic 7. Excitation at 715 nm and absorbance were read with a band path filter of 480–520 nm. Images were taken with a two-photon confocal system at 60× magnification. The nuclei were stained with propidium iodide, and the differential interference contrast (DIC) image was taken with 0.5 million MDA-MB-435 cells.
Acknowledgment
The authors gratefully acknowledge Tamara Stoops and the MAPS Core facility at the University of Virginia for their assistance with the cytological assays and partial financial support from the Georgetown University Drug Discovery Program. The authors also thank Professor Erick Carreira for suggesting the racemization studies.
Footnotes
Abbreviations: CDCl3, deuterated chloroform; DCC, dicyclohexylcarbodiimide; DPPA, diphenylphosphorylazide; DHQZ, 2,3-dihydroquinazolinone; DIC, differential interference contrast; DMSO, dimethyl sulfoxide; ESI, electrospray ionization; EtOAc, ethyl acetate; Et3N, triethylamine; GAFF, general Amber force field; HCT-116, human colon cancer cells; MDA-MB-435, human melanoma cells; MeCN, acetonitrile; MeMgBr, methylmagnesium bromide; MgSO4, magnesium sulfate; MHz, megahertz; NaOH, sodium hydroxide; NH4Cl, ammonium chloride; NMR, nuclear magnetic resonance; RESP, restrained electrostatic potential; rmsd, root-mean square displacement; THF, tetrahydrofuran.
Supporting Information Available: Results from elemental analysis of key compounds, X-ray crystal structure of 3b, and 1H and 13C NMR spectra of 1a, 1b, 4a, 3c, 3d, 3e, 3f, 3g, 3h, 3i, 6a, 6b, 6c, 6d, 6e, 6f, 6g, 6h, 6i, (R)-11, (S)-(+)-7, and (R)-(−)-7. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Russel HE, Alaimo RJ. Antibacterial 2,3-dihydro-2-(5-nitro-2-thienyl)quinazolin-4-(1H)-ones. J. Med. Chem. 1972;15:335–336. doi: 10.1021/jm00273a034. [DOI] [PubMed] [Google Scholar]
- 2.Bonola G, Da Re P, Magistretti MJ, Massarani E, Setnikar I. 1-Aminoacyl-2,3-dihydro-4(1H)-quinazolinone derivatives with choleretic and antifibrillatory activity. J. Med. Chem. 1968;11:1136–1139. doi: 10.1021/jm00312a007. [DOI] [PubMed] [Google Scholar]
- 3.Neil GL, Li LH, Buskirk HH, Moxley TE. Antitumor effects of the antispermatogenic agent, 2,3-dihydro-2-(1-naphthyl)-4(1H)-quinazolinone. Cancer Chemother. 1972;56:163–173. [PubMed] [Google Scholar]
- 4.Levin JI, Chan PS, Bailey T, Katocs AS, Venkatesan AM. The synthesis of 2,3-dihydro-4(1H)-quinazolinone angiotensin II receptor antagonists. Bioorg. Med. Chem. Lett. 1994;4:1141–1146. [Google Scholar]
- 5.Okumura K, Oine T, Yamada Y, Hayashi G, Nakama M. 4-Oxo-1,2,3,4-tetrahydroquinazolines. I. Syntheses and pharmacological properties of 2-methyl-3-aryl-4-oxo-1,2,3,4-tetrahydroquinazolines and their 1-acyl derivatives. J. Med. Chem. 1968;11:348–352. doi: 10.1021/jm00308a036. [DOI] [PubMed] [Google Scholar]
- 6.Hour MJ, Huang LJ, Kuo SC, Xia Y, Bastow K, Nakanishi Y, Hamel E, Lee KH. 6-Alkylamino- and 2,3-dihydro-3′-methoxy-2-phenyl-4-quinazolinones and related compounds: their synthesis, cytotoxicity, and inhibition of tubulin polymerization. J. Med. Chem. 2000;43:4479–4487. doi: 10.1021/jm000151c. [DOI] [PubMed] [Google Scholar]
- 7.Escalante J, Flores P, Priego JM. Synthesis of 2,3-dihydro-4(1H)-quinazolinones. Heterocycles. 2004;63:2019–2032. [Google Scholar]
- 8.Shioiri T, Ninomiya K, Yamada S. Diphenylphosphoryl azide. New convenient reagent for a modified Curtius reaction and for peptide synthesis. J. Am. Chem. Soc. 1972;94:6203–6205. doi: 10.1021/ja00772a052. [DOI] [PubMed] [Google Scholar]
- 9.Hendrickson JB, Bergeron R. Triflamides: new acylating and triflating reagents. Tetrahedron Lett. 1973;14:4607–4610. [Google Scholar]
- 10.Miyaura N, Yamada K, Suzuki A. A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979;36:3437–3440. [Google Scholar]
- 11.Wang RX, You XZ, Meng QJ, Mintz EA, Bu XR. A modified synthesis of O-hydroxyaryl aldehydes. Synth. Commun. 1994;24:1757–1760. [Google Scholar]
- 12.National Cancer Institute. [accessed Apr 2008];Developmental Therapeutics Program. http://dtp.nci.nih.gov/dtpstandard/servlet/MeanGraph?searchtype=NSC&searchlist=125973&outputformat=HTML&outputmedium=page&chemnameboolean=AND&debugswitch=false&assaytype=&testshortname=NCI+Cancer+Screen+Current+Data&dataarraylength=86&endpt=GI50&button=Mean+Graph&highconc=-6.0.
- 13.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 14.Sybyl7.0. St. Louis, MO: Tripos Inc.; 2003. http://www.tripos.com. [Google Scholar]
- 15.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 16.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution. From diffraction images to an initial model in minutes. Acta Crystallogr., Sect. D. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 17.Maple JR, Hwang M-J, Stockfisch TP, Dinur U, Waldman M, Ewig CS, Hagler AT. Derivation of class II force fields. 1. Methodology and quantum force field for the alkyl functional group and alkane molecules. J. Comput. Chem. 1994;15:162–182. [Google Scholar]
- 18.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Jr, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Gonzalez C, Challacombe M, Gill PMW, Johnson BG, Chen W, Wong MW, Andres JL, Head-Gordon M, Replogle ES, Pople JA. Gaussian 98. Pittsburgh, PA: Gaussian, Inc.; 1998. [Google Scholar]
- 19.Rarey M, Kramer B, Lengauer T, Klebe G. Predicting receptor–ligand interactions by an incremental construction algorithm. J. Mol. Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 20.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. AMBER 8. San Francisco, CA: University of California; 2004. [Google Scholar]
- 21.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 22.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- 23.Verdier-Pinard P, Lai JY, Yoo HD, Yu JR, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Structure–activity analysis of the interaction of curacin A, the potent colchicine site anti-mitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol. Pharmacol. 1998;53:62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 24.Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem. Biophys. 2003;38:1–21. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]