

Abstract
Cytochrome bd catalyzes the two-electron oxidation of either ubiquinol or menaquinol and the four-electron reduction of O2 to H2O. In the current work, the rates of reduction of the fully oxidized and oxoferryl forms of the enzyme by the 2-electron donor ubiquinol-1 and single electron donor TMPD have been examined by stopped-flow techniques. Reduction of the all-ferric form of the enzyme is 1000-fold slower than required for a step in the catalytic cycle, whereas the observed rates of reduction of the oxoferryl and singly-reduced forms of the cytochrome are consistent with the catalytic turnover. The data support models of the catalytic cycle which do not include the fully oxidized form of the enzyme as an intermediate.
Keywords: cytochrome bd, ubiquinol, respiration
1. Introduction
The cytochorme bd quinol oxidase is one of the two terminal oxidases in the respiratory chain of E. coli, and catalyzes the 2-electron oxidation of either menaquniol or ubiquinol and the 4-electron reduction of oxygen to water [1–5]. This enzyme is a member of a large family of respiratory oxidases that are found in many prokaryotes, both bacteria and archaea. There is no eukaryotic homologue, and the presence of bd-type oxidases in a number of human pathogens makes this enzyme a possible drug target. Cytochrome bd is a transmembrane heterodimer, with one copy each of two proteins coded by the genes CydA (subunit I; 58 kD) and CydB (subunit II; 43 kD)[6]. There is no sequence homology between cytochrome bd and the large superfamily of the heme-copper respiratory oxidases [7], and the bd-type oxidases do not contain copper[6]. During turnover, cytochrome bd does not pump protons but it generates a proton motive force due to the fact that the protons used in the reaction to make H2O come from the bacterial cytoplasm, whereas the protons from the oxidation of QH2, which is oxidized near the opposite side of the membrane, are released to the periplasm [8,9].
Cytochrome bd contains three heme prosthetic groups to transfer electrons from quinol to oxygen: heme b558, heme b595 and heme d. The low spin heme b558 is believed to be the entry site of electrons from quinol and is located near the putative quinol binding site. Heme d is the site where oxygen binds and is reduced to water. The role of the high spin heme b595 is still not clear, but heme b595 is located about 10 Å from the high spin heme d [10] and minimally its role is to provide an electron to facilitate the reduction of oxygen bound to heme d. The membrane potential appears to be generated essentially by proton uptake accompanying the electron transfer from heme b558 to the oxygenated heme b595/heme d center, which generates a product with a high affinity for protons [8].
The isolated cytochrome bd appears to have a single binding site for quinols. Unlike the heme-copper oxidases that utilize ubiquinol as a substrate, such as cytochrome bo3 from E. coli, there is no evidence for a tightly bound quinone which does not readily exchange with the quinone pool in the membrane. Hence, the isolated cytochrome bd has no bound quinone and the fully reduced form of the enzyme contains three electrons for redox chemistry, corresponding to the ferrous forms of each of the three hemes.
Four different states of heme d have been characterized spectroscopically in cytochrome bd. These are 1) ferric (Fe3+); 2) ferrous (Fe2+); 3) ferrous-oxy complex (Fe2+-O2) and 4) oxoferryl (Fe4+=O2−). A putative peroxy complex (supposedly, Fe3+-OOH) has been observed recently as a transient intermediate during oxidation of the fully reduced enzyme by O2 [11], but its structure requires further confirmation. Each of the other four forms of heme d can be generated as stable or metastable forms that can be examined and characterized. These forms suggest a sequence of events at heme d as O2 is converted to water.
| (1) |
The analysis of enzyme turnover, however, must take into account the redox status of the other two heme prosthetic groups, heme b558 and heme b595, which can exist in either the Fe2+ or Fe3+ states, and also the fact the ubiquinol is a 2-electron donor.
The purpose of the current work is to directly test the proposal based on the steady-state kinetics studies [12,13] that the fully oxidized ( ) form of cytochrome bd may not be part of the catalytic cycle (Figure 1).
Figure 1. Catalytic cycle of ubiquinol oxidase activity of cytochrome bd.

The scheme is based on the work of Jünemann et al [12] as modified by Matsumoto et al [13].
The model shown in Figure 1 implies that the rate of reduction of the all-ferric form of the enzyme should be slow. Contrary to this proposal, fast reduction of heme d in the all-ferric enzyme was observed in pulse radiolysis studies [14]. The generation of a strong 1-electron reductant in this experiment resulted in very rapid reduction of heme b558, equilibration within 10 μs between heme b558 and heme b595, followed by electron transfer to heme d with the rate constants of about 3400 s−1 and 590 s−1, for the ferric and the oxoferryl forms, respectively. The 1-electron transfer to heme d in the ferrous-oxy form (Fe2+-O2) was about 100-fold slower than observed with the ferric and oxoferryl forms. Taken at face value, these data suggest that the all-ferric form should be reconsidered as a possible intermediate during steady state analysis, and raises questions about the catalytic competence of the ferrous-oxy complex as an intermediate. An important difference between the steady-state turnover and pulse radiolysis experiments is that the former normally utilizes a 2-electron donor whereas the latter utilizes a 1-electron reductant. This was pointed out by Kobayashi et al [14], in reference to the slow single-electron transfer to the ferrous-oxy complex.
In the current work, a rapid mixing/diode array spectrophotometer was used to monitor the rate of reaction of UQH2-1 and of ascorbate-reduced TMPD with the all-ferric form of cytochrome bd. The kinetics of these reactions were compared with the reactions with the oxoferryl form of the enzyme generated by transient oxidation of the fully-reduced cytochrome bd by a stoichiometric amount of oxygen. The results reveal very slow (seconds) reduction of the all-ferric enzyme by ubiquinol-1, whereas reduction of the oxoferryl form of cytochrome bd is at least 1000-fold more rapid and compatible with the turnover number of the enzyme. With a single-electron donor TMPD, the oxoferryl form of cytochrome bd is also a much better electron acceptor than the all-ferric form, however, the difference is much less pronounced than in the case of 2-electron reductant UQH2-1. The data show directly that the all-ferric form of the enzyme is not likely to be part of the normal catalytic cycle.
2. Materials and Methods
Strains and Plasmids
E. coli strain GO105 (cyd AB::kan, cyo, recA), which lacks both the cytochrome bo3 and cytochrome bd quinol oxidases [15], was used as the host strain for overexpressing the wild type cytochrome bd from plasmid pTK1 [15] which was introduced into the strain.
Cell Growth and Enzyme Purification
Large scale cell growth was carried out in 24 2-liter flasks shaking at 220 rpm 37 °C using an Innova 4330 incubator shaker (New Brunswick Scientific) [16]. Cytochrome bd was purified as described previously [17]. The pooled fractions were concentrated using an Amicon concentrator with a 50 kDa molecular weight cut-off filter and then dialyzed three times against 50 mM sodium phosphate buffer, pH 7.8, containing 5 mM EDTA, 0.05% N-lauroylsarcosine.
Heme Analysis
The heme b content of purified cytochrome bd was measured by the pyridine hemochromogen assay, using an extinction coefficient for the wavelength pair 556.5–540 nm, Δε556.5–540=24 mM−1cm−1 [18]. The heme d content and enzyme concentration were determined from the absolute spectrum of the fully reduced enzyme, using the extinction coefficient Δε628–670 = 25 mM−1cm−1 [8].
Ubiquinol-1 and TMPD Oxidase Activity Assays
The steady state oxidase activity assays were performed as described previously by Zhang et al. [17]. The purified enzyme had a turnover numbers of about 1100 s−1 with 100 μM UQH2-1 + 3 mM DTT and about 160 s−1 with 10 mM ascorbate + 0.5 mM TMPD.
UV-visible Spectroscopic Measurements
Static absorbtion spectra were obtained with a UV-2101PC spectrophotometer (Shimadzu).
Stopped-flow Experiments. were performed at 20°C using an Applied Photophysics stopped flow with a photodiode array detector. The flow system was flushed thoroughly with Argon-saturated buffer (50 mM sodium phosphate, 5 mM EDTA and 0.05% N-lauroylsarcosine, pH 7.8) before the experiments. The whole system was kept anaerobic by flowing Argon during the experiments. The mixing ratio of the two solutions was 1:1. After mixing, 1600 spectra were collected in the 300–1200 nm range over a period ranging from 2s to 1 min. At least three runs were performed for each time scale. The concentrations reported are the initial concentrations before mixing, if not stated otherwise.
Generation of the oxoferryl form of the enzyme
5 μM enzyme was fully reduced by 200 μM ubiquinone-1 + 6 mM dithiothreitol or by 1 mM TMPD + 20 mM ascorbate in an argon-saturated buffer (50 mM sodium phosphate, 5 mM EDTA and 0.05% N-lauroylsarcosine, pH 7.8). The fully reduced enzyme ( ) was then rapidly mixed in the stopped-flow apparatus with an equal volume of a solution containing approximately 5 μM O2 in the same buffer. The reduced cytochrome bd was rapidly oxidized by O2 yielding essentially the oxoferryl form of the enzyme ( ), which was subsequently reduced by the excess reductant present in the solution. The oxidation reaction is too fast to resolve, but the tail end of the reduction kinetics could be observed spectroscopically. Spectroscopic analysis of the product of the reaction of O2 with the reduced enzyme showed that it contained approximately 80% oxoferryl and 20% ferrous-oxy complex (not shown).
Experiments with the All-ferric Form of Cytochrome bd
5 μM of the “as isolated” cytochrome bd, mainly present as ferrous-oxy complex , was incubated for 20 minutes with 16 μM tetrachlorobenzoquinone, a lipophilic strong oxidant [19], in argon-saturated buffer (50 mM sodium phosphate, 5 mM EDTA and 0.05% N-lauroylsarcosine, pH 7.8). The incubation resulted in conversion of the enzyme to the fully oxidized, all ferric form of the enzyme ( ). This form of the enzyme was rapidly mixed in the stopped flow with an equal volume of 200 μM ubiquinone-1 + 6 mM DTT or, alternatively, 1 mM TMPD + 20 mM ascorbate, in order to monitor the rate of reduction of the all-ferric form of the enzyme. A strongly lipophilic tetrachlorobenzoquinone is expected to be an antagonist of UQH2-1 ubiquinol. Indeed, turnover of cytochrome bd with 100 μM UQH2-1 as electron donor in the presence of 16 μM tetrachlorobenzoquinone was decreased by 30–40% (to about 600–800 electrons s−1). There was no effect of tetrachlorobenzoquinone on the ascorbate+TMPD-oxidase activity of the enzyme. Data Analysis. The data were first analyzed with the use of Pro-Kineticist software package “ProK for PC” (Applied Photophysics) and selected data were imported into Origin 7 (Microcal) for further analysis and preparation of the figures.
3. Results
Figure 2A compares the kinetics of reduction of heme d in the oxoferryl and all-ferric forms of cytochrome bd. From the spectra/time surface collected, the kinetics at a wavelength pair 628 nm and 719 nm (ΔA628–719) was extracted to monitor the reduction of heme d. The kinetics of the reduction of the all-ferric form of the enzyme by UQH2-1 is extremely slow and non-homogeneous, reaching only about 25% of the expected maximum in 2 s, the maximal observation period in the experiment shown. This is in contrast to the extremely rapid reduction of heme d in the all-ferric enzyme (k~ 3,400 s−1), reported in the pulsed radiolysis experiments of Kobayashi et al [14]. Reduction of hemes b558 and b595 in the same experiment was even slower that reduction of heme d, with either DTT+ubiquinol or ascorbate+TMPD as electron donors (data not included) and took about about 60 s for completion, whereas heme d reduction was complete in 20 to 30 s. Hence, it is likely that it is electron entry into the enzyme (reduction of b558), rather than intramolecular electron transfer, that limits the rate of reduction of ferric heme d in the all-ferric enzyme.
Figure 2. Kinetics of reduction of heme d in the oxoferryl and the all-ferric forms of cytochrome bd. Panel A. Reduction by UQH2-1.

Heme d in the oxoferryl form is reduced within about 10 ms, compatible with the turnover of the enzyme. No significant reduction of heme d in the all-ferric enzyme is observed in the same time range. Panel B. Reduction by TMPD: The reduction of the all-ferric form is significantly slower than the reduction of heme d in the oxoferryl form of the enzyme. Heme d in the oxoferryl form is reduced by TMPD much more slowly than with UQH2-1 See Materials and Methods for details.
In order to examine the reactivity of the oxoferryl state of cytochrome bd, this form of the enzyme was generated in situ in the stopped flow mixing chamber by oxidation of the fully reduced cytochrome bd with stoichiometric amount of O2.
| (2) |
| (3) |
Formation of the oxycomplex (reaction 2) and subsequent oxidation of the reduced cytochrome bd by bound oxygen (reaction 3) are complete within the mixing time, in agreement with the the known rate constants. At 2.5 μM oxygen (final concentration after mixing), the expected rate for the reaction with O2 is about 2·109 M−1cm−1 2.5 μM O2 = 5 × 103 s−1, τ~200 μs (see [20]), and the oxidation yields largely the oxoferryl state of the enzyme (at least 80%). This was most clearly seen in the spectra from the experiments in which reduced TMPD was used as the reductant (data not shown), since the subsequent reduction of the enzyme in this case is much slower than with UQH2-1.
The oxoferryl form of the enzyme is reduced quite rapidly by UQH2-1, and the reaction is completed in about 10 ms (Figure 2A). During this time, up to 12 time-resolved spectra could be collected. The major part of the transiently oxidized cytochrome bd is actually reduced within the dead time of the mixing apparatus and only the tail end of this rapid reduction could be monitored with the current instrumentation. Global analysis of the spectra/time surface for the resolved part of the reduction process gave a rate constant of ~200–300 s−1, which is close to enzyme turnover rate (~1000 electrons s−1) when multiplied by 4 electrons required to convert enzyme from the oxoferryl to the all ferrous state.
The difference spectrum of the rapid phase of the reduction, as resolved by global analysis, is shown in Figure 3. The spectrum shows clearly simultaneous disappearance of the oxoferryl state (trough around 670 nm – 680 nm) and appearance of reduced heme d (peak at ~630 nm), as well as reduced heme b558 and heme b595 (the peaks at 595 nm, 560 nm and 530 nm in the visible, and an asymmetric Soret band with a maximum at 431 nm and a shoulder around 438 nm). These data imply that the reaction monitored involves reduction of the enzyme by two equivalents of UQH2-1, since four electrons are required to generate the fully reduced form of the enzyme ( ) from the oxoferryl state ( ). The first equivalent of UQH2-1 produces a partially reduced form of the enzyme ( ), so these results imply that the rate of reduction by the second equivalent of UQH2-1 of the partially reduced enzyme ( ) is about as fast as the reaction of the oxoferryl species ( ) with the first equivalent of UQH2-1.
Figure 3. Difference spectrum of the rapidly reduced component of cytochrome bd in the reaction of the oxoferryl form reduced by UQH2-1 found by global analysis of the spectra/time surface.
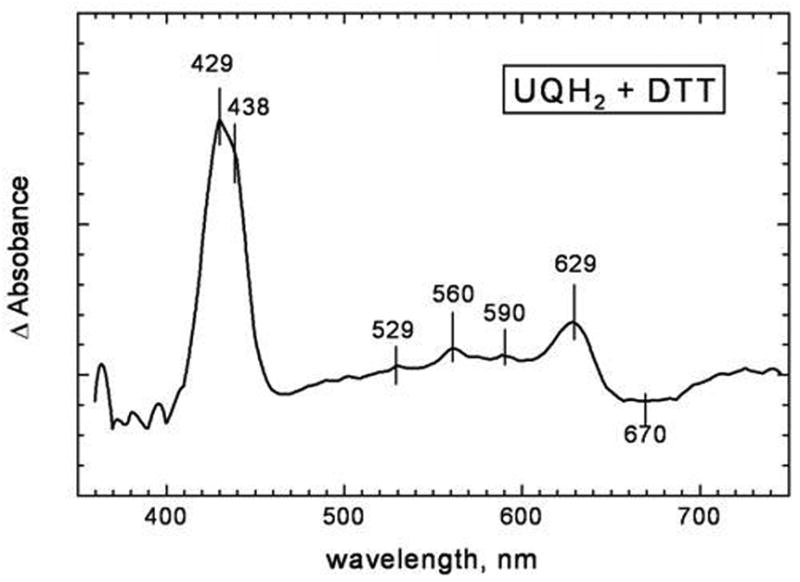
The spectrum is that of major product of the reaction formed in the first 50 ms minus the initial spectrum, which is primarily the oxoferryl form of the enzyme. The conditions correspond to the trace shown in Figure 2A.
The reduction kinetics of the enzyme with a single electron donor TMPD reduced by excess ascorbate was also examined ( Figure 2B). Reduction of the all-ferric enzyme in this case is also very slow. As in the case of ubiquinol, the reaction of reduced TMPD with the oxoferryl form is more rapid than with the all-ferric form of the enzyme, although it is still much slower than observed with UQH2-1. The reduction of the all-ferric cytochrome bd by reduced TMPD occurs on a time scale of 0.1–0.5 s, not in milliseconds. It cannot be excluded that the reaction of cytochrome bd with reduced TMPD is rate-limited by the oxidation of TMPD, i.e., TMPD is a much poorer electron donor than UQH2-1.
4. Discussion
The data clearly show that the reduction of heme d in the oxidized (all-ferric) form of cytochrome bd by its natural 2-electron substrate, ubiquinol, is much too slow to be part of the catalytic cycle. In contast, the reaction of the oxoferryl form of cytochrome bd with ubiquinol is rapid and results in formation of the fully reduced enzyme. Presumably, the reaction proceeds in 2 sequential two-electron steps which are not time-resolved under the conditions of our experiments at a UQH2-1 concentration of 100 μM (the final concentration after mixing).
| (4) |
| (5) |
The rate of reaction 5 must be at least as fast as reaction 4 under the conditions of these experiments, since the appearance of reduced heme d and the reduced hemes b occur in the same time window. However, in the presence of excess of O2, the very rapid rate of binding of O2 with the 1-electron reduced enzyme (microseconds) is expected to result in formation of the 1-electron ferrous-oxy complex.
| (6) |
Hence, when the concentration of O2 is high, reaction 6 would prevail, followed by reaction 7.
| (7) |
The product of reaction 7 has sufficient electrons to split the O-O bond (reaction 3), provided protons are available.
Under microaerophilic growth conditions, in which cytochrome bd is normally expressed in E. coli[21], the reactions of the one-electron reduced enzyme ( ) with O2 (reaction 6) and with UQH2-1 (reaction 5) might occur in the reverse order, which, however, makes no difference to the final outcome of the reaction cycle. It is interesting that the heme d ferrous-oxy complex ( ) is very stable, being the major form of the enzyme as it is isolated [22]. The dissociation of oxygen in the form of superoxide ( ) which would be deleterious to the cell, is negligible. On the other hand, 2-electron reduction of the all-ferric cytochrome bd by UQH2 and its subsequent oxidation by oxygen could give rise to an unstable peroxide adduct of the ferric enzyme that could dissociate releasing hydrogen peroxide, or produce hydroxyl radicals in case of homolytic scission of the O-O bond. Hence, the intermediate states of cytochrome bd with an odd number of electrons relative to the all-ferric form appear to be preferred in the catalytic cycle of the enzyme. These catalytic intermediate forms contain +3, +1 and −1 electrons relative to the all-ferric enzyme: a) +3 electrons = all-ferrous ( ) ; b) +1 electron = oxycomplex ( ); c) − 1 = oxoferryl ( ).
The expectation from the previous work of Jünemann et al [12] and of Matsumoto et al [13], based on steady state kinetics, that the all-ferric form of cytochrome bd is not part of the catalytic cycle of the enzyme is directly verified by pre-steady state kinetics. It remains to be seen why the one-electron reductant formed during the pulsed radiolysis experiments by Kobayashi et al [14] reacts with the all-ferric enzyme rapidly. The explanation might be simply that the N-methylnicotinamide radical and solvated electron generated under the conditions of pulse radiolysis are much stronger reductants than the one-electron donor used in the current work, TMPD ( ) well as than the ubiquinol/ubisemiquinone couple that serves as an initial reductant for cytochrome bd during the reaction of the enzyme with ubiquinol. One might also note, that the all-ferric forms of cytochrome bd were obtained in this work and in ref. [14] by different methods, but it is highly unlikely that a variance in the preparations can account for the ~1000-fold difference in reactivity of heme d.
The observation that UQH2-1 is a poor reductant for the all-ferric form of the enzyme, but a good reductant for the oxoferryl (b5583+b5953+ d4+ - O2−) and one-electron reduced enzyme ( ) suggests that the redox state of heme d plays a role in determining the rate of reduction of heme b558 (and, perhaps, of heme b595 as well). Further work is needed to clarify the mechanism of this effect. Recent work has shown that the redox state of heme b558 and heme b595 regulate the accessibility of heme d by O2 [23], thus, reciprocal regulation of the reactivity of the two hemes b by the redox and/or ligand-binding state of heme d may be a reasonable speculation. Evidently, the interactions between the three hemes play a critical role in determining the sequence of reactions in the catalytic cycle.
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL16101 to RBG), the Wenner-Gren Foundations (to RBG), the Civilian Research & Developement Fund (CRDF grant RUB1-2836-MO-06 to RBG and AAK), the Howard Hughes Medical Institute (International Scholar Award 55005615 to AAK) and the Russian Foundation for Basic Research grant 08-04-00093 (to V.B.B.)
abbreviations
- TMPD
N,N,N′,N′-tetramethyl-p-phenylendiamine
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- UQH2-1
ubiquinol-1
- NMA
N-methylnicotinamide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anraku Y, Gennis RB. The Aerobic Respiratory Chain of Escherichia coli. TIBS. 1987;12:262–266. [Google Scholar]
- 2.Jünemann S. Cytochrome bd Terminal Oxidase. Biochim Biophys Acta. 1997;1321:107–127. doi: 10.1016/s0005-2728(97)00046-7. [DOI] [PubMed] [Google Scholar]
- 3.Mogi T, Tsubaki M, Hori H, Miyoshi H, Nakamura H, Anraku Y. Two Terminal Quinol Oxidase Families in Escherichia coli: Variations on Molecular Machinery for Dioxygen Reduction. J Biochem Mol Biol & Biophys. 1998;2:79–110. [Google Scholar]
- 4.Borisov VB. Cytochrome bd: Structure and Properties. A Review. Biochemistry(Moscow) 1996;61:786–799. [PubMed] [Google Scholar]
- 5.Poole RK, Cook GM. Redundancy of aerobic respiratory chains in bacteria? Routes, reasons and regulation. Adv Microb Physiol. 2000;43:165–224. doi: 10.1016/s0065-2911(00)43005-5. [DOI] [PubMed] [Google Scholar]
- 6.Miller MJ, Gennis RB. The Purification and Characterization of the Cytochrome d Terminal Oxidase Complex of the Escherichia coli Aerobic Respiratory Chain. J Biol Chem. 1983;258:9159–9165. [PubMed] [Google Scholar]
- 7.Green NG, Fang H, Lin R-J, Newton G, Mather M, Georgiou C, Gennis RB. The Nucleotide Sequence of the cyd Locus Encoding the Two Subunits of the Cytochrome d Terminal Oxidase Complex of Escherichia coli. J Biol Chem. 1988;263:13138–13143. [PubMed] [Google Scholar]
- 8.Belevich I, Borisov VB, Zhang J, Yang K, Konstantinov AA, Gennis RB, Verkhovsky MI. Time-resolved Electrometric and Optical Studies on Cytochrome bd Suggest a Mechanism of Electron-proton Coupling in the Di-heme Active Site. Proc Natl Acad Sci USA. 2005;102:3657–3662. doi: 10.1073/pnas.0405683102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jasaitis A, Borisov VB, Belevich NP, Morgan JE, Konstantinov AA, Verkhovsky MI. Electrogenic Reactions of Cytochrome bd. Biochemistry. 2000;39:13800–13809. doi: 10.1021/bi001165n. [DOI] [PubMed] [Google Scholar]
- 10.Arutyunyan AM, Borisov VB, Novoderezhkin VI, Ghaim J, Zhang J, Gennis RB, Konstantinov AA. Strong excitonic interactions in the oxygen-reducing site of bd-type oxidase: the Fe-to-Fe distance between hemes d and b595 is 10 A. Biochemistry. 2008;47:1752–9. doi: 10.1021/bi701884g. [DOI] [PubMed] [Google Scholar]
- 11.Belevich I, Borisov VB, Verkhovsky MI. Discovery of the true peroxy intermediate in the catalytic cycle of terminal oxidases by real-time measurement. J Biol Chem. 2007;282:28514–9. doi: 10.1074/jbc.M705562200. [DOI] [PubMed] [Google Scholar]
- 12.Jünemann S, Butterworth PJ, Wrigglesworth JM. A Suggested Mechanism for the Catalytic Cycle of Cytochrome bd Terminal Oxidase Based on Kinetic Analysis. Biochemistry. 1995;34:14861–14867. doi: 10.1021/bi00045a029. [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto Y, Muneyuki E, Fujita D, Sakamoto K, Miyoshi H, Yoshida M, Mogi T. Kinetic mechanism of quinol oxidation by cytochrome bd studied with ubiquinone-2 analogs. J Biochem (Tokyo) 2006;139:779–88. doi: 10.1093/jb/mvj087. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi K, Tagawa S, Mogi T. Electron Transfer Process in Cytochrome bd-type Ubiquinol Oxidase from Escherichia coli Revealed by Pulse Radiolysis. Biochemistry. 1999;38:5913–5917. doi: 10.1021/bi982088n. [DOI] [PubMed] [Google Scholar]
- 15.Kaysser TM, Ghaim JB, Georgiou C, Gennis RB. Methionine-393 Is an Axial Ligand of the Heme b558 Component of the Cytochrome bd Ubiquinol Oxidase from Escherichia coli. Biochemistry. 1995;34:13491–13501. doi: 10.1021/bi00041a029. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Hellwig P, Osborne JP, Gennis RB. Arginine 391 in Subunit I of the Cytochrome bd Quinol Oxidase from Escherichia coli Stabilizes the Reduced Form of the Hemes and Is Essential for Quinol Oxidase Activity. J Biol Chem. 2004;279:53980–53987. doi: 10.1074/jbc.M408626200. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Hellwig P, Osborne JP, Huang HW, Moënne-Loccoz P, Konstantinov AA, Gennis RB. Site-directed Mutation of the Highly Conserved Region near the Q-loop of the Cytochrome bd Quinol Oxidase from Escherichia coli Specifically Perturbs Heme b595. Biochemistry. 2001;40:8548–8556. doi: 10.1021/bi010469m. [DOI] [PubMed] [Google Scholar]
- 18.Berry EA, Trumpower BL. Simultaneous Determination of Hemes a, b, and c from Pyridine Hemochrome Spectra. Anal Bioch. 1987;161:1–15. doi: 10.1016/0003-2697(87)90643-9. [DOI] [PubMed] [Google Scholar]
- 19.Borisov VB, Smirnova IA, Krasnosel’skaya IA, Konstantinov AA. Oxygenated Cytochrome bd from Escherichia coli Can Be Converted into the Oxidized Form by Lipophilic Electron Acceptors. Biochemistry. 1994;59:437–443. [PubMed] [Google Scholar]
- 20.Hill BC, Hill JJ, Gennis RB. The Room Temperature Reaction of Carbon Monoxide and Oxygen with the Cytochrome bd Quinol Oxidase from Escherichia coli. Biochemistry. 1994;33:15110–15115. doi: 10.1021/bi00254a021. [DOI] [PubMed] [Google Scholar]
- 21.Govantes F, Orjalo AV, Gunsalus RP. Interplay Between Three Global Regulatory Proteins Mediates Oxygen Regulation of the Escherichia coli Cytochrome d Oxidase (cydAB) Operon. Molecular Microbiology. 2000;38:1061–1073. doi: 10.1046/j.1365-2958.2000.02215.x. [DOI] [PubMed] [Google Scholar]
- 22.Lorence RM, Koland JG, Gennis RB. Coulometric and Spectroscopic Analysis of the Purified Cytochrome d Complex of Escherichia coli : Evidence for the Identification of “Cytochrome a1” as Cytochrome b595. Biochemistry. 1986;25:2314–2321. doi: 10.1021/bi00357a003. [DOI] [PubMed] [Google Scholar]
- 23.Belevich I, Borisov VB, Bloch DA, Konstantinov AA, Verkhovsky MI. Cytochrome bd from Azotobacter vinelandii: evidence for high-affinity oxygen binding. Biochemistry. 2007;46:11177–84. doi: 10.1021/bi700862u. [DOI] [PubMed] [Google Scholar]