

Abstract
Peptide amphiphiles (PAs) previously designed in our laboratory are known to self-assemble into nanofibers that exhibit bioactivity both in vitro and in vivo. Self-assembly can be triggered by charge neutralization or salt-mediated screening of charged residues in their peptide sequences, and the resulting nanofibers can form macroscopic gels at concentrations as low as 0.5% by weight. Controlling the kinetics of gelation while retaining the bioactivity of nanofibers could be critical in tailoring these materials for specific clinical applications. We report here on a series of PAs with different rates of gelation resulting from changes in their peptide sequence without changing the bioactive segment. The pre-existence of hydrogen-bonded aggregates in the solution state of more hydrophobic PAs appears to accelerate gelation kinetics. Mutation of the peptide sequence to include more hydrophilic and bulky amino acids suppresses formation of these nuclei and effectively slows down gelation through self-assembly of the nanofiber network. The ability to modify gelation kinetics in self-assembling systems without disrupting bioactivity could be important for injectable therapies in regenerative medicine.
Keywords: Self-assembly, Peptide amphiphile, Gelation kinetics, Bioactive nanofibers
1. Introduction
Self-assembly is an attractive strategy to introduce biomaterials into living tissues due to the versatility it offers for delivery methods and its potential for structural control at the nanoscale. Self-assembling systems could be used in minimally invasive therapies as biomaterials that can be injected as fluids which solidify once inside the body by physiological triggers such as heat or salinity [1-5]. Depending on the application, the desired time to gelation could vary from a time scale of seconds up to several minutes. While shorter gelation times allow for more targeted delivery of the therapeutic, longer gelation times can be desirable in the case where a gelating material must be delivered to an area of the body with little void space and high tortuosity, such as the spinal cord. Too rapid gelation in such applications can lead to clogging of the delivery needle and failure of the material to diffuse throughout the affected area. Such issues of delivery motivated the current work, which attempts to describe the factors that influence gelation speed of nanofiber-forming peptide amphiphiles (PAs).
We previously reported on a class of self-assembling PAs consisting of a lipophilic segment attached to the N- or C-terminus of a short peptide with one or more charged residues, that self-assemble to form nanofibers of 5–8 nm in diameter and up to microns long [5-11]. The lipophilic segment of the PA forms the core of the nanofiber and the peptide portion is nearest to the fiber surface. Electrostatic charges in the structure of PAs and the consequent intermolecular repulsion among molecules prevent long nanofibers from forming spontaneously in solution. If this charge is neutralized or shielded, for instance by the addition of electrolytes or by adjustment of pH, fiber formation is energetically favored by hydrophobic sequestration of the alkyl tails away from water and the potential for hydrogen bonding between the peptide segments. PAs in this class characteristically form assemblies rich in β-sheet-type hydrogen bonding [8,12-14]. These β-sheets are believed to play a role in determining the cylindrical nanofiber morphology, which is robust to major perturbations of the peptide sequence. According to classical models for surfactant self-assembly, as the hydrophilic headgroup becomes bulkier relative to the tail the cylindrical morphology should give way to spherical micelles [15]. This transition does not seem to happen easily in these PAs, presumably due to the large energetic reward for hydrogen bonding in β-sheets [8,16]. The nanofibers formed by our PA designs contain β-sheets that tend to align parallel to the long axis of the nanofibers to an extent which is sequence dependent and have as well some degree of order in their hydrophobic alkyl cores [17]. This robustness enables the incorporation of large varieties of bioactive peptide sequences into the PAs without disrupting fiber formation. Possible bioactive peptides include the extensively studied cell adhesion sequence RGD from fibronectin [18] and the laminin-1 sequence IKVAV [19,20] known to promote neurite outgrowth. PAs containing RGD [4,6,9,11,21,22] and IKVAV [5,8,11,13,23] have been studied and found to be promising biomaterials. In particular an IKVAV-bearing PA has been shown to promote neurite outgrowth and selective and rapid differentiation of mouse neural progenitor cells into neurons [5], a result that has important implications in the use of these biomaterials in spinal cord injury repair.
It has been established that the final mechanical properties of PA nanofiber gels can be tuned by modulating interactions among the self-assembling monomers [9,12,24]. Previous work in our laboratory demonstrated that PA mechanical properties can be tuned by adjusting the strength of intermolecular ionic bonding, which enables different mechanical regimes to be accessed without modifying the molecule [9,25]. Paramonov et al. have also shown that it is possible to tune PA mechanical properties by systematic mutation of N-methylation of glycines [12] or by the inclusion of phospholipids in the assembled structure [24], both of which serve to disrupt the hydrogen bonding that is critical for fiber formation and lower the mechanical strength.
In order to maximally exploit the advantages of self-assembly, it is desirable to control not only matrix structure and function but also the kinetics of assembly, which to our knowledge has not been systematically studied for this type of molecule. The kinetics of gelation would be particularly important for biomaterials that are designed for minimally invasive therapies to be injected as liquids and form gels in situ. In this paper we report on three series of epitope-bearing PAs in which the rates of gelation under identical conditions have been varied through modifications in peptide sequence. We also investigate in one of the systems whether these changes in kinetics affect bioactivity. These results could have significant impact on the design of future generations of PAs.
2.Materials and methods
2.1. Materials
Resin for solid phase synthesis and some amino acids were purchased from EMD Biosciences (La Jolla, CA). The remaining amino acids and synthesis reagents were purchased from Sigma–Aldrich (St. Louis, MO) and Anaspec, Incorporated (San Jose, CA). Solvents were purchased from VWR International (West Chester, PA) and Sigma–Aldrich.
2.2. PA synthesis
The PAs were prepared as described previously [9,12] using standard fluorenylmethoxycarbonyl (Fmoc) chemistry on an Applied Biosystems 433A automated peptide synthesizer. All peptides prepared have a C-terminal carboxylic acid and were made by using prederivatized Wang resins. After the peptide portion of the molecule was synthesized, the resin was removed from the automated synthesizer and the N-terminus was capped with palmitic acid. Cleavage and deprotection of the PAs were done with a mixture of triflouroacetic acid, water, and triisopropylsilane in a ratio of 95:2.5:2.5 for 2–3 h at room temperature depending on the sequence. The cleavage mixture and two subsequent dichloromethane washings were filtered into a round-bottom flask. The solution was concentrated in vacuo to a thick viscous solution by rotary evaporation, then triturated with cold diethyl ether. The white precipitate was collected by filtration, washed with copious cold ether, and dried overnight. All PAs were purified on an XBridge® C18 reverse phase column (Waters Corporation, Milford, MA) using high pressure liquid chromatography (HPLC) in a water/acetonitrile gradient containing 0.1 vol% NH4OH to aid solubility and separation. The purified PAs were again rotary evaporated to remove the acetonitrile, then dialyzed against water from a MilliQ water purification system operating at 18.2 MΩ. Finally the PAs were lyophilized and stored in cold, dry conditions until use.
2.3. Rheology
Oscillating rheology was used to quantify the time to gelation and final mechanical properties of the molecules. Each PA was dissolved in aqueous solution at 1% w/v and pH 7.5–8. This solution was mixed 1:1 with Hank’s Buffered Saline Solution (HBSS Sigma), or 20 mm calcium chloride on the stage of a Paar Physica Modular Compact Rheometer (MCR) 300 (Graz, Austria). A 25-mm parallel plate configuration was used and the temperature was held constant at 21 °C. Storage (G′) and loss (G″) moduli and complex viscosity (V*) were measured at 3% strain and 4 Hz for the IKVAV PAs, at 0.1% strain and 4 Hz for the RGD PAs, and 0.5% strain and 100 Hz for the EEE PAs, in order to carry out measurements in the linear viscoelastic region for each set of molecules. For time tests, measurements began 50 s after mixing due to the requirements of instrument setup. Final storage and loss moduli were measured at 4000 s after the start of measurement.
2.4. Circular dichroism
Circular dichroism measurements were carried out to correlate changes in gelation kinetics with the development of secondary structure. Wavelength scans were taken of the IKVAV-bearing PAs in water and 1:1 water:HBSS at 0.1 nm intervals between 250 nm and 185 nm at a concentration of 0.5 mg/mL with a 0.1 cm path length quartz cuvette. For the RGD PAs, measurements were taken in water and in 50 mm CaCl2. For the EEE PAs, measurements were taken in water and in 20 mm CaCl2. All samples were allowed to equilibrate for 2–3 h prior to measurement. The percentages of secondary structures were estimated by averaging the results of matrix-based data decomposition with CONTIN, SELCON, and CDSSTR algorithms using two distinct sets of basis vectors. CDPro software [26] was used for these calculations. Measurements were taken on a Jasco J-715 circular dichroism instrument at room temperature using a 1 mm cuvette. Data was taken at a resolution of at least 0.5 nm and averaged over at least 10 acquisition cycles.
2.5. Substrate preparation for cell studies
12-mm round glass coverslips (Carolina Biological) were distributed into 24-well cell culture plates (VWR). For the PA substrates, dilute (0.1 mg/mL) PA solutions were mixed 1:1 with HBSS and pipetted onto the coverslips in 100 μL aliquots. The solutions were allowed to dry overnight in a laminar flow hood under ambient conditions, after which the coverslips were rinsed once by pipetting 500 μL of sterile distilled water into each well, and aspirating the water to remove excess salt. Poly-d-lysine (PDL) and PDL-laminin were used as control substrates. For the PDL substrates, 500 μL of 0.1 mg/mL aqueous PDL (<300 kDa, Sigma) solution was pipetted onto the coverslips. After 2 h, the excess PDL was aspirated and the coverslips were rinsed three times with sterile distilled water to remove soluble polymer. The PDL-laminin substrates were treated identically, then 1 μg/mL laminin in PBS was added after the final water rinse. These substrates were also allowed to dry overnight.
2.6. Cell culture
Neural tissue was harvested from the lateral and medial ganglionic emminences of E13 mouse embryos, dissociated, and plated in Petri dishes. Neural progenitors were separated from other neural cell types by growing them as neurospheres through two passages as previously described [25,26]. Briefly, the lateral ganglionic eminence was dissected, triturated and grown in suspension using non-adherent flasks (Petri flasks, VWR) containing 45 ml of neural stem cell medium (DMEM/F12 (Gibco), 10 ml B27 supplement (Gibco), 5 ml N2 medium (Gibco), 5 ml of 100× Penicillin/Streptomycin/Glutamine solution (Gibco), 0.5 ml 2 mg/ml heparin (Sigma) and 20 ng/ml EGF (BD Bioscience)) at 37 °C and 5% CO2. After 3 days, the spherical cell clumps (neurospheres) were dissociated by centrifugation and incubation with trypsin/EDTA, quenching with trypsin inhibitor, and triturating through a pipette tip. Cells were plated in PA gels at a density of 5 ×105 cells/ml.
After plating, the cells were incubated at 37 °C, 95% humidity, and 5% CO2 for three days, after which cell viability was determined using calcein and ethidium homodimer-1 stains (Molecular Probes Live/Dead kit). The differentiation state of the cells was also assayed at three days by immunolabeling fixed coverslips with antibodies against the neuronal marker β-tubulin and glial marker glial fibrillary acid protein (GFAP) using a modification of the procedure used by Silva et al. [5]. Fluorescent micrographs were acquired at 200× magnification and the NIH-developed software program ImageJ (http://rsb.info.nih.gov/ij/) was used in the counting of the immunopositive cells.
2.7. Turbidity
Turbidity (absorption) was measured over time using a Cary 500 UV–vis spectrophotometer. Solutions of each PA at 0.5% by weight were mixed with an equal volume of HBSS in a 10-mm path length quartz cuvette and monitored for 60 min at 10 s intervals. Measurements were obtained at 600 nm.
2.8. Zeta potential
The zeta potential of dilute PA suspensions (0.05 mg/mL) was calculated at various salt concentrations between 0 and 1 m using the measured mobility, which was automatically converted to zeta potential according to the Smoluchowski equation [27]. The measurements were made using a phase analysis light-scattering instrument (Brookhaven Instruments ZetaPALS, Holtsville NY).
3. Results and discussion
Fig. 1 shows the chemical structures of the seven PAs studied here, containing four distinct regions: a bioactive epitope at the C-terminus, a tripeptide spacer, a β-sheet forming region to facilitate self-assembly at the N-terminus, and an N-terminal palmityl segment. The synthesis of the PAs was confirmed by matrix-assisted laser desorption–ionization time-of-flight (MALDI-TOF) mass spectroscopy and HPLC. All PAs were found by transmission electron microscopy (TEM) to form nanofibers similar to those previously described. These data are provided in Supplementary information. Three of these PAs contain the bioactive pentapeptide IKVAV. PA1 has been previously shown to cause both process outgrowth and selective differentiation of multipotent neural progenitor cells into neurons, and its gelation and self-assembly properties have been previously described [8,28]. PA2 also contains the IKVAV epitope, but has a different β-sheet forming region, an alternating SLSL sequence was substituted for the uniformly hydrophobic AAAA β-sheet segment of PA1. PA3 is structurally identical to PA2 with the exception that AAA, which is considered to be molecularly less flexible than the sequence GGG [29,30], was used as the spacer sequence. PAs 4 and 5 bear the bioactive RGD epitope and are otherwise analogs of PA1 and PA3, respectively. Similarly, PAs 6 and 7 bear an acidic, non-epitope sequence but are otherwise analogous. Although these three series of PAs do not compose a comprehensive library of possible sequences, they are diverse enough to suggest that the observed results have a good degree of generality. This is explored in depth later in this section.
Fig. 1.
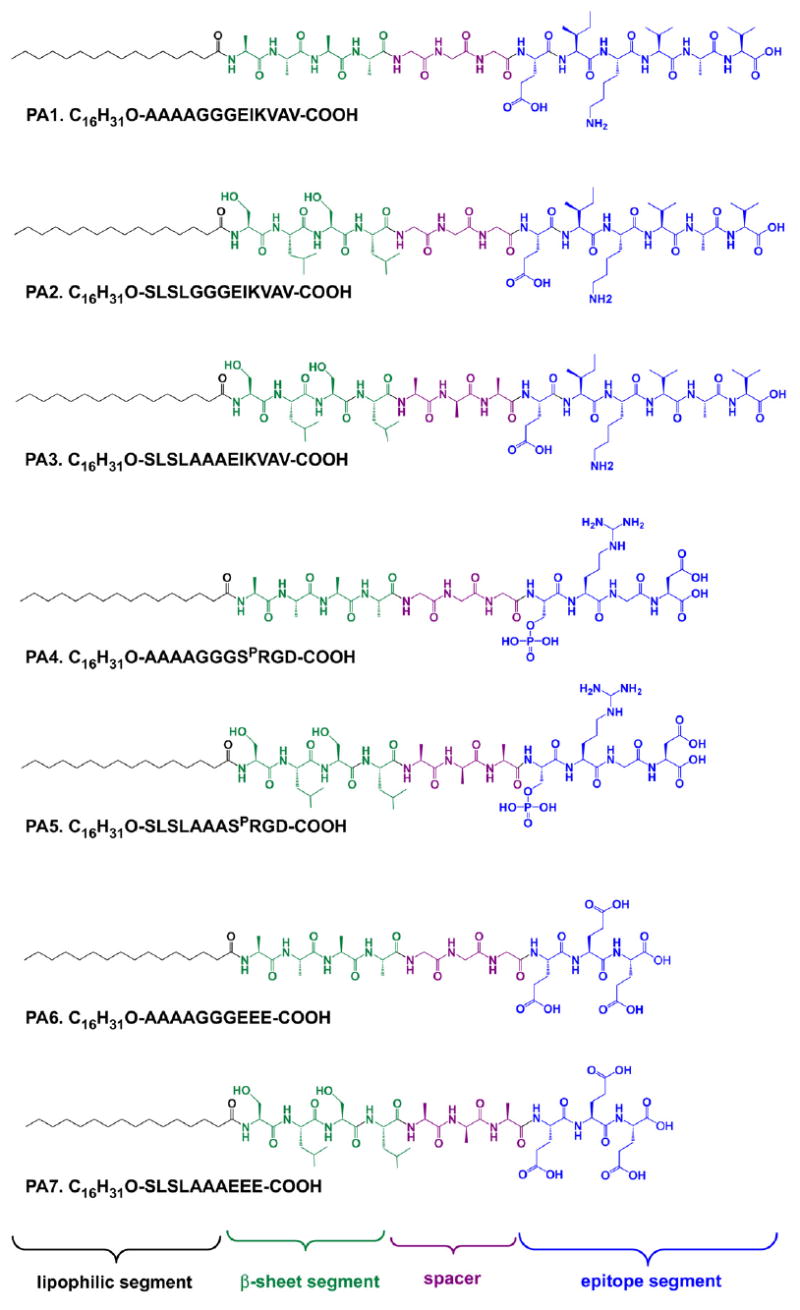
Chemical structures of peptide amphiphiles (PAs) investigated in this work, discussed in this paper.
The changes made in PAs 2 and 3 compared to PA1 were intended to increase polarity and steric hindrance in the self-assembling molecules to decrease the driving force toward self-assembly. The polar side chains in serine increase the molecule’s overall polarity, and leucine residues make these molecules bulkier than the amino acids in a similar position in PA1. In PA3, additional steric bulk and stiffness are provided by the substitution of alanines for a G3 spacer. Since the β-sheet forming region is buried inside the fiber [7,8,11,15,4], these changes were hypothesized not to significantly affect any biological activity mediated by the IKVAV sequence, but they should have an impact on energetic barriers to nanofiber formation. It has been amply demonstrated [31] that hydrogen bonding in the N-terminal region of the peptide is crucial to the energetic favorability of PA fiber formation, and this capability is retained in all of the PA variants described here. What has changed between PAs 1 and 2 is the driving force toward self-assembly, which should be lowered by the addition of two amino acids with hydrophilic side chains. The conformational flexibility relative to PA1 is also decreased in PA3 and, to a lesser extent, in PA2. These changes were designed to slow the kinetics of gelation without changing the geometry of the final nanostructure.
The oscillating rheology measurements shown in Fig. 2A illustrate that gelation occurred more rapidly for PA1 than for PA2 or PA3 with the addition of the physiological saline buffer HBSS. Despite the low viscosity of the initial solutions, in all cases G′ > G″ for all measured time points in the experiment. Thus the commonly used definition of gel point of G″/G′ = 1, which represents the percolation of a fibrous network in which both elastic and viscous moduli scale linearly [32], is already surpassed at the outset of the experiment and is not a useful demarcation between the solution state and the gel state. For this reason, the gel point was instead defined as the point at which the damping factor (G″/G′) dropped to a value below 0.1 [33]. This conservative value of the damping factor, which indicates that the elastic character of the gel is an order of magnitude more prominent than the viscous (liquid-like) character, was chosen for rigor, but any arbitrarily chosen value of G″/G′ shows the same behavior. For PA1, this gel point occurred after less than 5 min on average, as opposed to over 15 min for PA2 and nearly an hour for PA3. These data are consistent with the visual differences observed in gelation kinetics among these PAs. To assess the generality of this trend, the gelation speed of PAs 4–7 were also tested (Fig. 2B, C). As mentioned above, these are the RGD- and EEE-bearing analogs of PAs 1 and 3. These PAs do not form gels with HBSS, presumably due to the larger formal charge on these molecules relative to the solution’s ionic strength. As previously reported [9], however, PA4 forms a gel in 50 mm CaCl2, and the other PAs were also found to form a gel with this solution. Although a macroscopic gel was clearly formed and again G′ > G″ at all time points, G″/G′ never reached 0.1 for either of these PAs. Because the final G″/G′ values of these PAs were very different, gelation was defined for these experiments as the point when G″/G′ reached 95% of its final value. The kinetics of gelation follow similar trends for all three sets of PAs, with the gelation of the PAs with the SLSL-AAA motif occurring at a slower rate than for the PAs containing the sequence AAAA-GGG. This is interesting because it suggests that the effect is potentially general to a variety of PAs. Final complex modulus data on these PAs are provided in the Supplemental data.
Fig. 2.
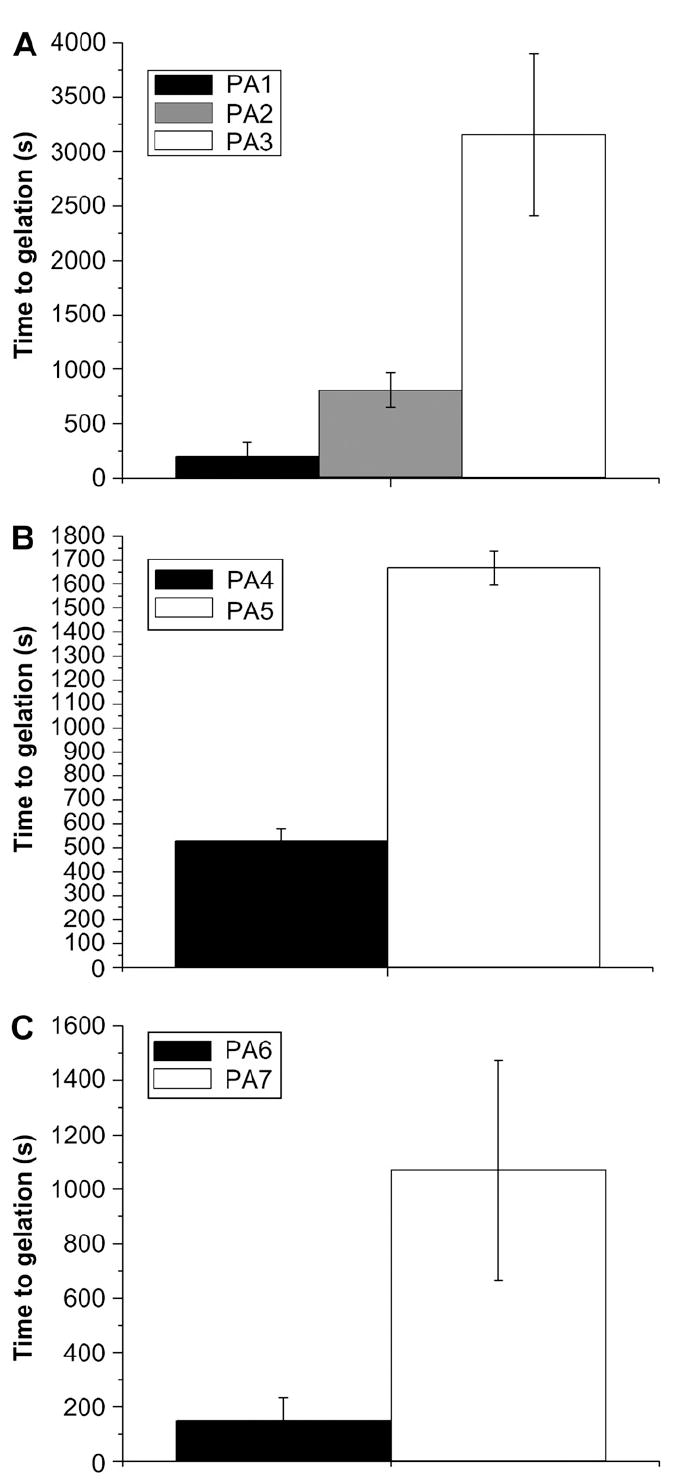
Time to gelation for IKVAV-bearing PAs upon the addition of HBSS (A) and RGD-and EEE-bearing PAs upon the addition of calcium chloride. In all cases, the SLSLAAA-based PAs gelled much more slowly. Error bars represent the standard deviation of three runs.
In Fig. 3, molecular bulk (quantified by molecular weight) and polarity (number of polar groups normalized by molecular weight) are plotted against gelation time, demonstrating that, as hypothesized, gelation time increases with the bulkiness and number of polar groups in the PA molecule. In both cases a positive trend is observed. Since the self-assembly of these systems is known to involve β-sheet formation, we obtained circular dichroism (CD) measurements to understand the origin of the observed changes in kinetics. These measurements, shown in Figs. 4-6, suggest the presence of variable amounts of hydrogen-bonded PA aggregates in solution even before the triggering of self-assembly by the addition of electrolytes. Fig. 4 shows the CD spectra and quantified secondary structure data for the IKVAV-bearing PAs. Even in aqueous, salt-free solution, the secondary structures of the IKVAV-bearing PAs are all dominated by hydrogen-bonded β-sheet structures. The relative contribution of α-helix versus β-sheet secondary structure may be overestimated in the quantification of data because the aggregation of PA molecules is expected to cause a light scattering induced red shift in the negative CD peak [34]. There is little change in the nature of the secondary structures after addition of HBSS to the PA solutions, other than a slight increase in the ratio of β-sheet to α-helical character in all cases and a greater red shift of the negative peak location attributable to the growth of nanofibers. Overall, however, the structural features remain the same. We interpret this as an indication that even before the addition of HBSS, there is significant hydrogen bonding among molecules in all of the IKVAV-PAs. Further formation of these hydrogen bonds presumably occurs during long nanofiber formation after HBSS addition. Although this is consistent with existing models of PA nanofiber formation, the data do not explain the differences in gelation times among the three PAs.
Fig. 3.

Effect of molecular bulk (top) and polarity (bottom) on gelation time. A positive trend is observed in both cases, supporting the stated hypothesis.
Fig. 4.
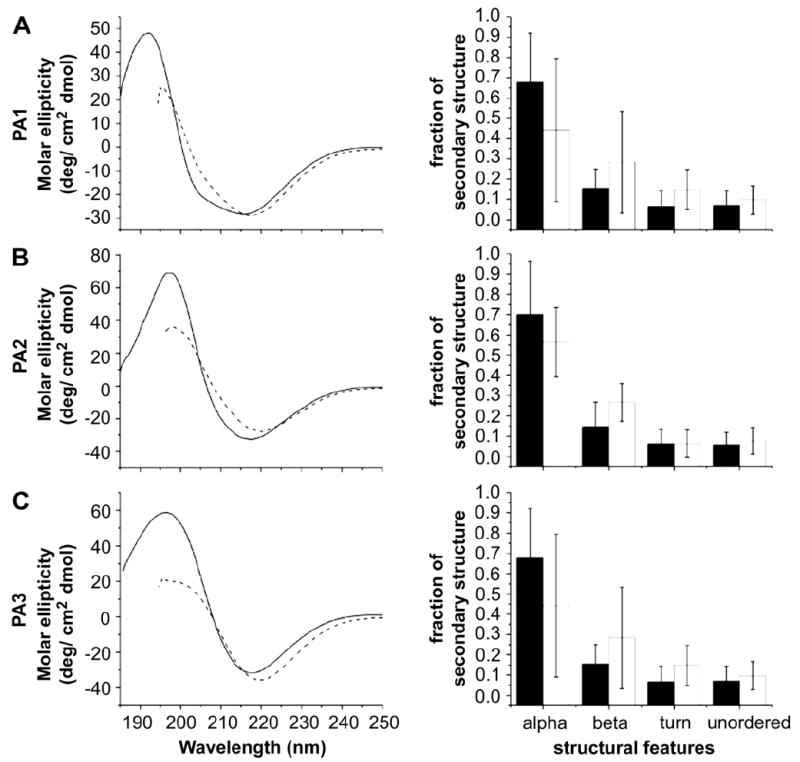
CD spectra and quantified secondary structure for IKVAV-bearing PAs before (solid line/black bars) and after (dotted line/white bars) addition of HBSS. The amount of each secondary structure present in the PAs is statistically similar before and after addition of the gelator.
Fig. 6.

CD spectra and quantified secondary structure for the EEE-bearing PAs. In spite of the different headgroup, the structural changes in the EEE PAs are very similar to those seen in the RGD PAs. PA6 shows similar secondary structure characteristics before and after the addition of calcium chloride (indicated by solid lines/black bars and dotted lines/ white bars, respectively). PA7 shows a significant random coil component prior to calcium chloride addition that disappears afterward. In the bar graph quantifying the percentage of each secondary structure type, a single asterisk (*) denotes that the percentage of that structure type post-salt addition is significantly different from the original percentage to a 95% confidence interval; a double asterisk (**) denotes a difference to the 99% confidence interval. The amount of three out of four components changes significantly.
The CD data for the RGD-bearing PAs, shown in Fig. 5, on the other hand, suggest that a greater number of aggregates are present in PA4 and they may be responsible for its more rapid gelation relative to PA5. The spectra for PA4 before and after salt addition show a similar pattern to those of the IKVAV-bearing PAs. However, the slow-gelling PA5 shows a significant amount of random coil component prior to salt addition that disappears once self-assembly is triggered. This points to a possible role for the pre-existing β-sheet structures in nucleating nanofiber formation, in which case PA5 would be expected to gel more slowly due to the absence of these nuclei. A similar trend is observed for the EEE-bearing PAs 6 and 7 (Fig. 6). Although the data do not account for the difference in gelation times for PAs 1–3, the IKVAV epitope itself is, as was previously noted, more hydrophobic than RGD, and previous studies have shown that it has a propensity for β-sheet formation [35,36]. To probe the possibility of differential nucleation tendencies among the IKVAV-bearing PAs without this confounding factor, we obtained turbidity measurements for PAs 1–3. Turbidity measurements are often used in aggregating systems to probe kinetics or deduce the presence of nucleating bodies [37,38]. This technique is based on the principle that aggregates scatter light and so can be detected by absorption measurements. The turbidity measurements performed on PAs 1–3 and the relationship between initial turbidity and time to gelation for each PA are shown in Fig. 7. All three PAs have non-zero turbidity prior to addition of HBSS, indicating the presence of aggregates. Most significantly, the initial amount of turbidity indicating the presence of aggregates correlates with faster gelation rates for the PAs investigated. PA1, with the fastest gelation rate, revealed the greatest turbidity and the slowest gelling PA3 the least. This trend, together with the CD data for the RGD PAs presented above, is strongly indicative of a nucleation-based mechanism for self-assembly in which pre-existing, hydrogen-bonded PA aggregates form the nucleating bodies. We suggest that these aggregates are energetically favored due to the energy lost in hydrogen bond formation and hydrophobic collapse of the alkyl segments, but are prevented from growing into high aspect ratio nanofibers by electrostatic barriers. The extension of the nanofibers by self-assembly can be likened to the phenomenon of flocculation in colloids, in which the aggregation of smaller particles into large masses is controlled by changes in electrostatic charges [39,40]. The more aggregates that are present in the solution, the faster the network will reach percolation and thus gelation as a result of a greater number of nanofibers growing simultaneously.
Fig. 5.
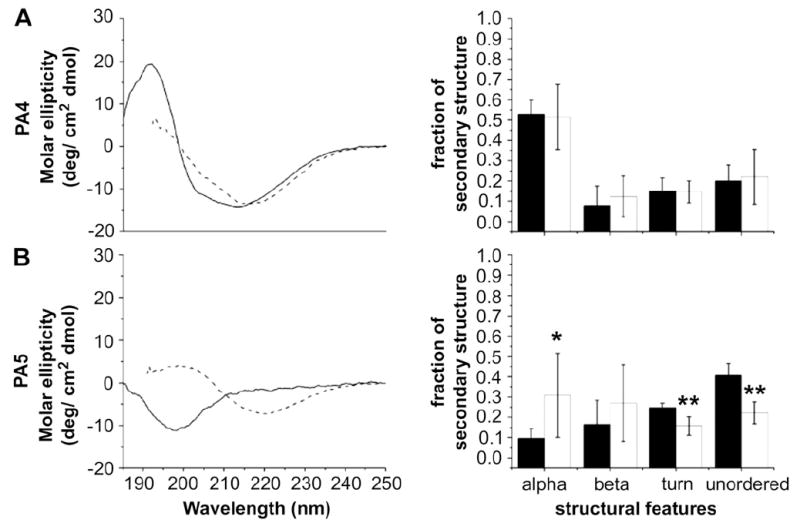
CD spectra and quantified secondary structure for RGD-bearing PAs. PA4 shows similar secondary structure characteristics before and after the addition of calcium chloride (indicated by solid lines/black bars and dotted lines/white bars, respectively). PA5 shows a significant random coil component prior to calcium chloride addition that disappears afterward. In the bar graph quantifying the percentage of each secondary structure type, error bars denote standard deviation; a single asterisk (*) denotes that the percentage of that structure type post-salt addition is significantly different from the original percentage to a 95% confidence interval; a double asterisk (**) denotes a difference to the 99% confidence interval. The amount of three out of four components changes significantly.
Fig. 7.
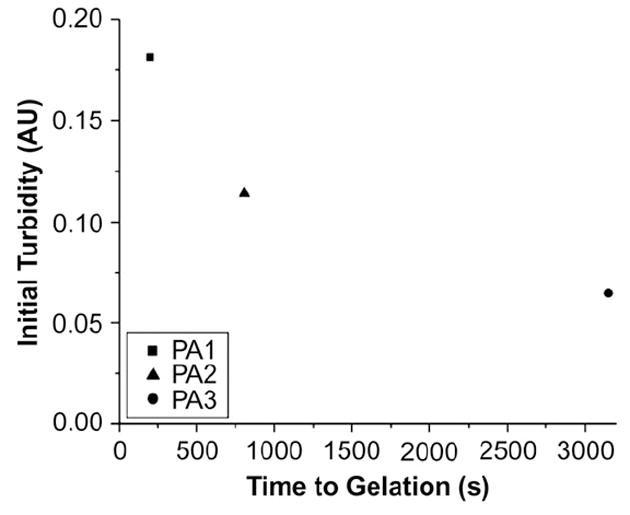
Time to gelation plotted against initial turbidity for PAs 1–3. The pre-existing turbidity is predictive of the speed of gelation, which strongly suggests a nucleation mechanism for self-assembly.
Nucleation-based mechanisms for self-assembly have been previously reported in the context of various chiral molecules, including amyloid-forming peptides [41,42], helical strand-forming tartaric acid-related components [43], and, most recently, π-conjugated organic molecules [44]. The latter work has suggested a nucleation-based self-assembly mechanism for at least one such system, showing the distinct stages of hierarchical self-assembly through sensitive spectroscopy measurements and identifying a role for solvent-mediated interactions.
Since the rate of growth of certain aggregate-forming materials, such as colloids, is controlled by electrostatic charge on the aggregate, ζ-potential measurements were taken to verify the change of sequence was causing a different effective charge to be present on the monomer. As shown in Fig. 8, the ζ-potentials of PAs 3 and 1 are essentially identical, showing that the lessened aggregation of PA3 relative to PA1 is not due to electrostatic contributions. These data, along with the CD spectra and turbidity measurements, support the idea that pre-assembly aggregation of PA molecules is discouraged by greater hydrophilicity of the peptide sequences. The bulkiness of the amino acid sequence may also play a role, as demonstrated by the difference in gelation time between PAs 2 and 3. In this case PA3 is actually more hydrophobic, but the absence in this molecule of the very flexible G3 sequence may suppress the formation of nuclei by decreasing the ability of the PAs to pack into nanofibers. Similar effects may be occurring in the bulkier sequences of PA5, explaining the predominance of coil-like conformations in this PA prior to the process of nanofiber formation that accompanies gelation.
Fig. 8.
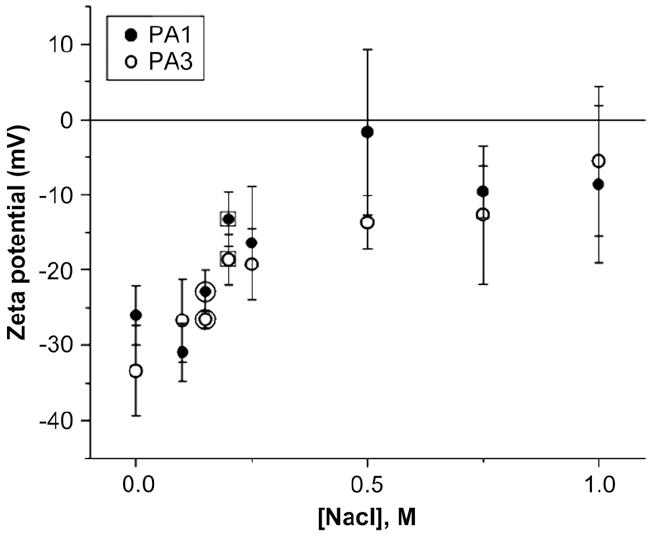
ζ-Potential series. The ζ-potentials of PAs 1 and 3 are both negative and similar, showing that the changes in gelation time are not attributable to electrostatics. The circled data points were taken with HBSS and the squared data points with DMEM.
Because the change in amino acid sequence could result in bioactivity changes as well as mechanical properties, the survival and differentiation of mouse neural progenitor cells were assayed as well. As shown in Fig. 9, survival of mouse neural progenitor cells on PA1 and PA3 substrates was comparable to that observed on standard cell culture surfaces consisting of poly-d-lysine (PDL) or PDL-laminin. It was previously reported [5] that PA1 supports the selective differentiation of these cells into neurons rather than astrocytes, and as also shown in Fig. 9 this property is retained in PA3 in spite of the structural changes to the PA. Most importantly, this shows that the bioactivity of the PAs is, at least in this case, not affected by changes to the C-terminal sequence of the peptide. A great attribute of the PA designs discussed here is the possibility of independently changing parts of the peptide sequence in PAs that control bioactivity vs. other properties such as stiffness and gelation kinetics.
Fig. 9.
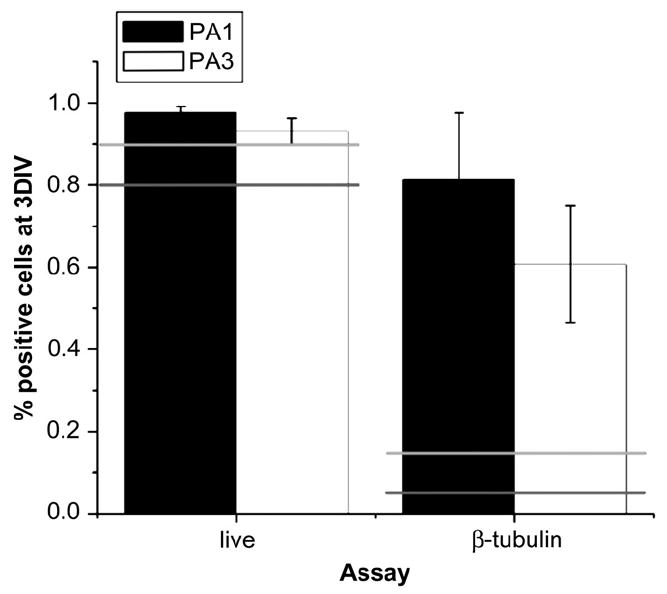
Viability (‘live’) and neuronal differentiation (‘β-tubulin’) of neural progenitor cells in PAs 1 and 3. Error bars represent standard deviation. Light gray and dark gray horizontal lines represent the viability and differentiation values on PDL and PDL-laminin controls, respectively. Viability in both gels is similar to that in the controls, but differentiation into neurons is enhanced in both cases.
The results described in this paper suggest a mechanism for the self-assembly of peptide amphiphile nanofiber scaffolds in which fiber formation is nucleated by pre-existing, hydrogen-bonded aggregates in solution. The number of these nuclei, and thus the rate of gelation, can be controlled by changes in the hydrophobicity and bulkiness of amino acid residues in the PA monomers. In the case of the IKVAV PAs investigated here, the structural changes in the peptide sequence that affect nucleus formation and gelation rate do not appear to affect neurobioactivity. This information could be used in the future to design PAs for central nervous system regeneration and other applications in regenerative medicine. For injection-based therapies such as those envisioned for these PAs, in the spinal cord, brain or elsewhere, the gelation time is a vital parameter that can control the extent of diffusion of these biomaterials within tissues. There are also more general potential applications for PAs with variable gelation kinetics, including the use of mixed systems or the possibility of generating gradients in bioactivity.
4. Conclusions
The self-assembly kinetics of epitope-bearing, fiber-forming peptide amphiphiles can be modified by molecular design of the PA. The pre-existence of hydrogen-bonded aggregates in the solution state of more hydrophobic PAs appears to accelerate gelation through growth of nanofibers. Mutation of the sequence to include more hydrophilic and bulky amino acids in the peptide sequence suppresses formation of these nuclei and effectively slows self-assembly of the nanofiber network. It appears possible, at least in one system investigated here, to decouple bioactivity from molecular design to tune self-assembly kinetics. The ability to modify gelation kinetics in this way without disrupting bioactivity could be important to in vivo applications where injectable therapies are envisioned.
Supplementary Material
Supplementary material can be found, in the online version, at doi:10.1016/j.biomaterials.2008.07.049.
Acknowledgments
This work was mainly supported by NIH grant 5 R01 EB003806 from the National Institute of Biomedical Imaging and Bioengineering. Additional support (for J.A.K.) was obtained from NIH grants NS20013-21 and P50 NS54287. The authors are also grateful to Professor Wesley Burghardt for the use of his laboratory’s rheometer and to Professor Aaron Packman for the use of his laboratory’s ZetaPALS instrument and to the NUANCE facility (Northwestern University) for TEM access.
References
- 1.Klok H-A, Hwang JJ, Hartgerink JD, Stupp SI. Self-assembling biomaterials: L-lysine-dendron-substituted cholesteryl-(l-lactic acid)(n) Macromolecules. 2002;35(16):6101–11. [Google Scholar]
- 2.Hwang J, Iyer S, Li L, Claussen R, Harrington D, Stupp S. Self-assembling biomaterials: liquid crystal phases of cholesteryl oligo(l-lactic acid) and their interactions with cells. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(15):9662–7. doi: 10.1073/pnas.152667399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langer R, Tirrell DA. Designing materials for biology and medicine. Nature. 2004;428(6982):487–92. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 4.Beniash E, Hartgerink JD, Storrie H, Stendahl JC, Stupp SI. Self-assembling peptide amphiphile nanofiber matrices for cell entrapment. Acta Biomaterialia. 2005;1(4):387–97. doi: 10.1016/j.actbio.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, Kessler JA, et al. Selective differentiation of neural progenitor cells by high-epitope density nanofibers. Science. 2004;303(5662):1352–5. doi: 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]
- 6.Bull SR, Guler MO, Bras RE, Meade TJ, Stupp SI. Self-assembled peptide amphiphile nanofibers conjugated to MRI contrast agents. Nano Letters. 2005;5(1):1–4. doi: 10.1021/nl0484898. [DOI] [PubMed] [Google Scholar]
- 7.Claussen RC, Rabatic BM, Stupp SI. Aqueous self-assembly of unsymmetric peptide bolaamphiphiles into nanofibers with hydrophilic cores and surfaces. Journal of the American Chemical Society. 2003;125(42):12680–1. doi: 10.1021/ja035882r. [DOI] [PubMed] [Google Scholar]
- 8.Niece KL, Hartgerink JD, Donners JJJM, Stupp SI. Self-assembly combining two bioactive peptide-amphiphile molecules into nanofibers by electrostatic attraction. Journal of the American Chemical Society. 2003;125(24):7146–7. doi: 10.1021/ja028215r. [DOI] [PubMed] [Google Scholar]
- 9.Stendahl JC, Rao MS, Guler MO, Stupp SI. Intermolecular forces in the self-assembly of peptide amphiphile nanofibers. Advanced Functional Materials. 2006;16:499. [Google Scholar]
- 10.Hartgerink JD, Beniash E, Stupp SI. Self-assembly and mineralization of peptide-amphiphile nanofibers. Science. 2001;294:1684–8. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 11.Hartgerink JD, Beniash E, Stupp SI. Supramolecular chemistry and self-assembly special feature: peptide-amphiphile nanofibers: a versatile scaffold for the preparation of self-assembling materials. Proceedings of the National Acadamy of Sciences of the United States of America. 2002;99:5133–8. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paramonov SE, Jun H-W, Hartgerink JD. Self-assembly of peptide-amphiphile nanofibers: the roles of hydrogen bonding and amphiphilic packing. Journal of the American Chemical Society. 2006;128(22):7291–8. doi: 10.1021/ja060573x. [DOI] [PubMed] [Google Scholar]
- 13.Tovar JD, Claussen RC, Stupp SI. Probing the interior of peptide amphiphile supramolecular aggregates. Journal of the American Chemical Society. 2005;127(20):7337–45. doi: 10.1021/ja043764d. [DOI] [PubMed] [Google Scholar]
- 14.Behanna HA, Donners JJJM, Gordon AC, Stupp SI. Coassembly of amphiphiles with opposite peptide polarities into nanofibers. Journal of the American Chemical Society. 2005;127(4):1193–200. doi: 10.1021/ja044863u. [DOI] [PubMed] [Google Scholar]
- 15.Israelachvili J. Intermolecular and surface forces. London: Academic Press; 1992. [Google Scholar]
- 16.Guler MO, Claussen RC, Stupp SI. Encapsulation of pyrene within self-assembled peptide amphiphile nanofibers. Journal of Materials Chemistry. 2005;15(42):4507–12. [Google Scholar]
- 17.Jiang H, Guler MO, Stupp SI. The internal structure of self-assembled peptide amphiphiles nanofibers. Soft Matter. 2007;1(4):454–62. doi: 10.1039/b614426h. [DOI] [PubMed] [Google Scholar]
- 18.Pierschbacher MD, Ruoslahti E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. 1984;309(5963):30–3. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- 19.Sephel GC, Tashiro K-I, Sasaki M, Greatorex D, Martin GR, Yamada Y, et al. Laminin A chain synthetic peptide which supports neurite outgrowth. Biochemical and Biophysical Research Communications. 1989;16(2):821–9. doi: 10.1016/0006-291x(89)92384-x. [DOI] [PubMed] [Google Scholar]
- 20.Tashiro K-i, Sephel GC, Weeks B, Sasaki M, Martin GR, Kleinman HK, et al. A synthetic peptide containing the IKVAV sequence from the A chain of laminin mediates cell attachment, migration, and neurite outgrowth. Journal of Biological Chemistry. 1989;264(27):16174–82. [PubMed] [Google Scholar]
- 21.Jiang HZ, Stupp SI. Dip-pen patterning and surface assembly of peptide amphiphiles. Langmuir. 2005;21(12):5242–6. doi: 10.1021/la0501785. [DOI] [PubMed] [Google Scholar]
- 22.Storrie H, Guler MO, Abu-Amara SN, Volberg T, Rao M, Geiger B, et al. Supramolecular crafting of cell adhesion. Biomaterials. 2007;28(31):4608–16. doi: 10.1016/j.biomaterials.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 23.Tysseling-Mattiace VM, Sahni V, Niece KL, Birch D, Czeisler C, Fehlings MG, et al. Self-assembling nanofibers inhibit glial scar formation and promote axon elongation after spinal cord injury. Journal of Neuroscience. 2008;28(14):3814–23. doi: 10.1523/JNEUROSCI.0143-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paramonov SE, Jun H-W, Hartgerink JD. Modulation of peptide-amphiphile nanofibers via phospholipid inclusions. Biomacromolecules. 2006;7(1):24–6. doi: 10.1021/bm050798k. [DOI] [PubMed] [Google Scholar]
- 25.Ozbas B, Kretsinger J, Rajagopal K, Schneider JP, Pochan DJ. Salt-triggered peptide folding and consequent self-assembly into hydrogels with tunable modulus. Macromolecules. 2004;37:7331–7. [Google Scholar]
- 26.Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Analytical Biochemistry. 2000;287(2):252–60. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 27.Spasic AM, Hsu J-P, editors. Finely dispersed particles: micro-, nano-, and atto-engineering. Boca Raton, FL: Taylor and Francis Group, LLC; 2006. [Google Scholar]
- 28.Silva GA, Czeisler C, Niece KL, Beniash E, Kessler JA, Stupp SI. Selective differentiation of neural progenitor cells by high-epitope density nanofibers. Science. 2004;303(5662):1352–5. doi: 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]
- 29.Florio T, Paludi D, Villa V, Principe DR, Corsaro A, Millo E, et al. Contribution of two conserved glycine residues to fibrillogenesis of the 106–126 prion protein fragment. Evidence that a soluble variant of the 106–126 peptide is neurotoxic. Journal of Neurochemistry. 2003;85(1):62–72. doi: 10.1046/j.1471-4159.2003.01664.x. [DOI] [PubMed] [Google Scholar]
- 30.D’Aquino JD, Gomez J, Hilser VJ, Lee KH, Amzel LM, Freire E. The magnitude of the backbone conformational entropy change in protein folding. Proteins. 1996;25:143–56. doi: 10.1002/(SICI)1097-0134(199606)25:2<143::AID-PROT1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 31.Guler MO, Soukasene S, Hulvat JF, Stupp SI. Presentation and recognition of biotin on nanofibers formed by branched peptide amphiphiles. Nano Letters. 2005;5(2):249–52. doi: 10.1021/nl048238z. [DOI] [PubMed] [Google Scholar]
- 32.Gennes P-Gd. Scaling concepts in polymer physics. Ithaca, NY: Cornell University Press; 1979. [Google Scholar]
- 33.Kumar G, Bristow JF, Smith PJ, Payne GF. Enzymatic gelation of the natural polymer chitosan. Polymer. 2000;41(6):2157–68. [Google Scholar]
- 34.Steim JM, Fleischer S. Aggregation-induced red shift of the cotton effect of mitochondrial structural protein. Proceedings of the National Acadamy of Sciences of the United States of America. 1967;1967(58):1292–8. doi: 10.1073/pnas.58.4.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamada M, Kadoya Y, Kasai S, Kato K, Mochizuki M, Nishi N, et al. Ile-lys-val-ala-val (IKVAV)-containing laminin alpha1 chain peptides form amyloid-like fibrils. FEBS Letters. 2002;530:48–52. doi: 10.1016/s0014-5793(02)03393-8. [DOI] [PubMed] [Google Scholar]
- 36.Nomizu M, Utani A, Shiraishi N, Kibbey MC, Yamada Y, Roller PP. The all-d-configuration segment containing the Ikvav sequence of laminin a-chain has similar activities to the all-l-peptide in vitro and in vivo. Journal of Biological Chemistry. 1992;267(20):14118–21. [PubMed] [Google Scholar]
- 37.Ferrone F. Analysis of protein aggregation kinetics. In: Wetzel R, editor. Amyloid, prions, and other protein aggregates. Vol. 309. Academic Press; 1999. pp. 256–74. [DOI] [PubMed] [Google Scholar]
- 38.Okubo T, Miyamoto T, Otake A, Suda M, Tsuchida A. Seed polymerization of tetraethyl ortho-silicate in the presence of rod-like colloidal particles of anionic palygorskite and cationic b-FeO(OH) Colloid and Polymer Science. 2004 February 4;282:1341–6. [Google Scholar]
- 39.Nicolai T, Durand D, Gimel J-C. Aggregation and gelation. In: Brown W, editor. Light scattering: principles and development. New York: Oxford University Press; 1996. pp. 211–20. [Google Scholar]
- 40.Jones RAL. Oxford master series in condensed matter physics. Oxford: Oxford University Press; 2002. Soft condensed matter; pp. 49–68. [Google Scholar]
- 41.Jarrett JT, Lansbury PT., Jr Amyloid fibril formation requires a chemically discriminating nucleation event: studies of an amyloidogenic sequence from the bacterial protein OsmB. Biochemistry. 1992;31(49):12345–52. doi: 10.1021/bi00164a008. [DOI] [PubMed] [Google Scholar]
- 42.Sokolowski F, Modler AJ, Masuch R, Zirwer D, Baier M, Lutsch G, et al. Formation of critical oligomers is a key event during conformational transition of recombinant Syrian hamster prion protein. Journal of Biological Chemistry. 2003;278(42):40481–92. doi: 10.1074/jbc.M304391200. [DOI] [PubMed] [Google Scholar]
- 43.Gulik-Krzywicki T, Fouquey C, Lehn J. Electron microscopic study of supramolecular liquid crystalline polymers formed by molecular-recognition-directed self-assembly from complementary chiral components. Proceedings of the National Academy of Science. 1993;90(1):163–7. doi: 10.1073/pnas. 90.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jonkheijm P, van der Schoot P, Schenning A, Meijer EW. Probing the solvent-assisted nucleation pathway in chemical self-assembly. Science. 2006;313(5783):80–3. doi: 10.1126/science.1127884. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material can be found, in the online version, at doi:10.1016/j.biomaterials.2008.07.049.