

Abstract
Purpose
Usher syndrome type II (USH2) is the most common form of Usher syndrome, an autosomal recessive disorder characterized by moderate to severe hearing loss, postpuberal onset of retinitis pigmentosa (RP), and normal vestibular function. Mutations in the USH2A gene have been shown to be responsible for most cases of USH2. To further elucidate the role of USH2A in USH2, mutation screening was undertaken in three Chinese families with USH2.
Methods
Three unrelated Chinese families, consisting of six patients and 10 unaffected relatives, were examined clinically, and 100 normal Chinese individuals served as controls. Genomic DNA was extracted from the venous blood of all participants. The coding region (exons 2–72), including the intron-exon boundary of USH2A, was amplified by polymerase chain reaction (PCR). The PCR products amplified from the three probands were analyzed using direct sequencing to screen sequence variants. Whenever substitutions were identified in a patient, restriction fragment length polymorphism analysis, or single strand conformation polymorphism analysis was performed on all available family members and the control group.
Results
Fundus examination revealed typical fundus features of RP, including narrowing of the vessels, bone-speckle pigmentation, and waxy optic discs. The ERG wave amplitudes of three probands were undetectable. Audiometric tests indicated moderate to severe sensorineural hearing impairment. Vestibular function was normal. Five novel mutations (one small insertion, one small deletion, one nonsense, one missense, and one splice site) were detected in three families after sequence analysis of USH2A. Of the five mutations, four were located in exons 22–72, specific to the long isoform of USH2A.
Conclusions
The mutations found in our study broaden the spectrum of USH2A mutations. Our results further indicate that the long isoform of USH2A may harbor even more mutations of the USH2A gene.
Introduction
Usher syndrome (USH) is an autosomal recessive disorder characterized by retinitis pigmentosa (RP) and sensorineural hearing impairment, with or without vestibular dysfunction. It is a clinically and genetically heterogeneous condition. Clinically, USH is subdivided into three types: USH type I (USH1), USH type II (USH2), and USH type III (USH3). USH1 is the most severe form of this disease characterized by congenital profound hearing loss, prepuberal onset of RP, and vestibular dysfunction. Patients with USH2 experience congenital moderate to severe hearing impairment and postpuberal onset of RP with intact vestibular function. Patients with USH3 show progressive postlingual hearing loss, later onset of RP, and variable vestibular dysfunctions. Of the three clinical types, USH2 is the most common form of USH, and it may account for more than half of the patients with USH [1,2]. To date at least 11 chromosome loci have been mapped for the three clinical USH types. Three of them are assigned to USH2: USH2A (USH2A), USH2C (VLGRI), and USH2D (DFNB31) [1,3-5].
The USH2A gene located on chromosome 1q41 has two alternatively spliced isoforms: a short USH2A isoform a and a much longer USH2A isoform b. In 1998, USH2A isoform a was first identified and found to contain 21 exons encoding a protein of 1,546 amino acids [6]. However, in 2004, van Wijk et al. [7] identified 51 additional exons at the 3′ end of USH2A. The longer USH2A alternative splicing isoform b, comprised of 72 exons, encodes a putative protein of 5,202 amino acids. USH2A isoform a is thought to be an extracellular matrix protein involved in protein–protein or protein-matrix interactions, while USH2A isoform b is a transmembrane protein [3,8]. In the inner ears of mice and rats, USH2A isoform b is a component of the ankle links that connect the differentiating stereocilia of the hair cells [9]. In the photoreceptor, it is an essential partner of a protein network that is likely to be involved in cargo delivery from the inner segment to the outer segment [10]. USH2A is thought to be involved in 74 to 90 percent of cases of USH2 [11]. Moreover, mutations of USH2A could also be responsible for atypical USH and nonsyndromic RP [11-13].
In this study, we performed a mutation screening of USH2A corresponding to USH2A isoform b. The screening was done in three Chinese families who were affected with USH2, and we identified five novel mutations.
Methods
Clinical data and sample collection
This study was granted approval by the Beijing Tongren Hospital Joint Committee on Clinical Investigation and conformed to the tenets of the Declaration of Helsinki. Three Chinese families with USH2 were referred to Beijing Tongren Hospital. After informed consent was obtained from each family member, all participants underwent careful clinical examinations, including best-corrected visual acuity, slit-lamp, and ophthalmoscope. Three probands of the families were given electroretinograms and they also received audiometric and vestibular tests. Full field electroretinogram were performed according to International Society for Clinical Electrophysiology of Vision (ISCEV) protocols. USH2 was diagnosed based on the clinical history, typical RP fundus appearance, sensorineural hearing impairment, and intact vestibular function.
Mutation detection
Blood samples of all participants were obtained by venipuncture and genomic DNA was extracted by standard phenol protocols. Mutation screening was performed in the three families using direct DNA sequence analysis. The coding region (exons 2–72) of USH2A was amplified by polymerase chain reaction (PCR) in the probands of the three families. The pairs of primers for exons 2–21 were used as designed by Weston et al. [11], except those for exons 6 and 7. Those for exons 22–72 were used as described by van Wijk et al. [7], except those for exons 41, 53, 63, 64, and 71 (Appendix 1). For direct sequencing, PCR products were purified (Shenneng Bocai PCR purification kit; Shenneng, Shanghai, China). The purified PCR products were sequenced using an automatic fluorescence DNA sequencer (ABI, Prism 373A; Perkin Elmer, Foster City, CA), according to the manufacturer’s instructions. Nucleotide sequences were compared with the published cDNA sequence of USH2A (GenBank NM_206933.1). For USH2A gene cDNA numbering +1 corresponds to A in the ATG translation initiation codon in RefSeq (AY481573.1).
Restriction fragment length polymorphism (RFLP) analysis
Variations found in the sequencing were confirmed with the restriction endonucleases TaqaI, MslI, and EcoRV (New England Biolabs, Ipswich MA), which were used with all available family members and 100 normal controls. The reaction was performed in a 10 µl volume containing 9.4 µl PCR product, 0.1 μl BSA (100 μg/ml), and 0.5 μl enzyme (4 U/μl). The reaction was incubated overnight at 65 °C for TaqaI and 37 °C for MslI and EcoRV. Afterwards the whole digest was run on a 1% agarose gel and visualized under ultraviolet light.
Single strand conformation polymorphism
For the sequence variants that did not change (create or erase) a restriction site, single strand conformation polymorphism (SSCP) was used with all available family members and the control group. As the PCR fragments used in SSCP analysis were between 150 to 300 bp, two pairs of specific primers were designed for detecting the mutation in exons 43 and 61 (Appendix 1). Amplified DNA was mixed with an equal volume of formamide buffer: 95% formamide, 10 mM EDTA, 0.1% bromophenol blue, and 0.1% xylene cyanol. Denatured samples were electrophoresed on a 16% nondenaturing polyacrylamide gel, with an acrylamide:bisacrylamide ratio of 49:1, for 12–16 h at 300 v and 4 °C, before gels were silver-stained and analyzed.
Prediction of splice scores
A Splice View program was used to assess the possibility that intron sequence variants could create or exclude splice sites. The program is accessible at splice.
Results
We have identified three unrelated Chinese families consisting of six patients diagnosed with USH2 and 10 unaffected relatives. The inheritance pattern in these three families was autosomal recessive (Figure 1). All of the patients experienced night blindness, vision acuity impairment, and moderate to severe hearing loss. Ophthalmoscopic examination demonstrated attenuation of the retinal vessels, bone-speckle pigmentation in the midperiphery of the fundus, and waxy pallor of the optic nerve head (Figure 2). The ERG wave amplitudes of three probands were indistinguishable from baseline (data not shown). Audiometric tests indicated moderate to severe sensorineural hearing impairment. Vestibular function was normal. Detailed clinical information on affected family members is summarized in Table 1.
Figure 1.
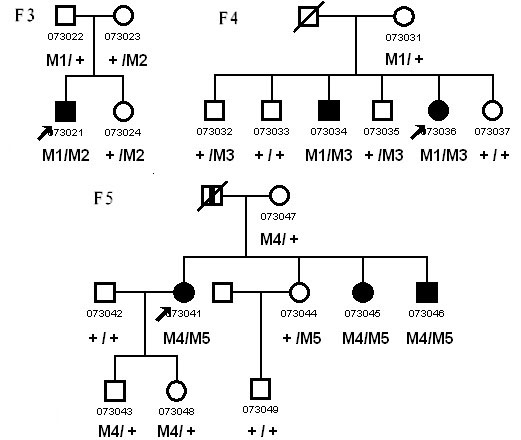
Pedigree of the three Chinese families with Usher syndrome type II who had mutations in USH2A. Males and females are represented by squares and circles, respectively. The symbols for affected family member are filled. The symbols for deceased family members have a slash. The symbols for proband have an arrow. The genotype of each evaluated individual is shown below the individuals’ symbol and identification number. Abbreviations: Wild type (+); c.8559-2A>G (M1); p.T3936P (M2); p.R34fs (M3); p.S2828fs (M4); p.W3150X (M5).
Figure 2.
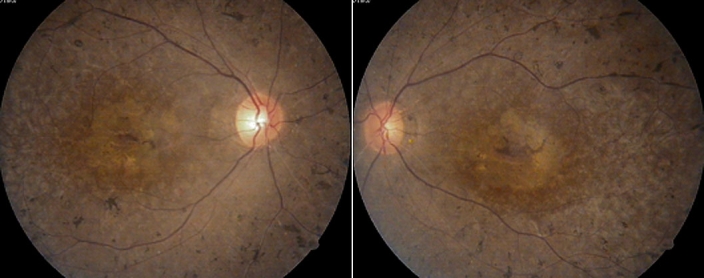
Fundus appearance of a patient with Usher syndrome type II. Fundal photographs from a proband (family F5) show typical retinal degeneration with attenuation of the retinal vessels, irregular pigment clumps in the retina, and waxy pallor of the optic nerve head.
Table 1. Clinical features of the patients from the three Chinese families with Usher syndrome type II.
| Family number | Patient | Best corrected visual acuity (R/L) | Onset age of night blindness (year) | Fundus appearance | Onset age of hearing loss (year) | Hearing impairment | ERG | Vestibular function |
|---|---|---|---|---|---|---|---|---|
| F3 |
73021 |
FC/PL |
14 |
RP |
10 |
Moderate (SP) |
Wave undetectable |
Normal |
| F4 |
73034 |
0.3/0.2 |
15 |
RP |
congenital |
Moderate (SP) |
N/A |
Normal |
| 73036 |
FC/ PL |
14 |
RP |
congenital |
Severe (SP) |
Wave undetectable |
Normal |
|
| F5 |
73041 |
HM/HM |
14 |
RP |
congenital |
Severe (SP) |
Wave undetectable |
Normal |
| 73045 |
HM/HM |
10 |
RP |
congenital |
Severe (SP) |
N/A |
Normal |
|
| 73046 | 0.1/0.1 | 14 | RP | congenital | Moderate (SP) | N/A | Normal |
Abbreviations: R, right eye; L, left eye; FC, finger counting; PL, perception of light; HM, hand motion; SP, slight progressive; N/A, data not available.
Sequencing of USH2A revealed 17 sequence variants in this study, five of which were predicted to be pathogenic mutations (Table 2). All five disease-causing mutations were heterozygous and were first detected in our study (Figure 3). Mutation c.8559–2A>G was identified in two families, and the other four mutations (p.T3936P, p.R34fs, p.S2828fs, and p.W3150X) were only detected in one patient or in in one family. Using RFLP or SSCP analysis, we confirmed the five mutations cosegregated with the USH2 phenotype (Figure 1, Figure 4, and Figure 5), but we did not detect them in 100 normal controls.
Table 2. Novel mutations in USH2A identified in this study.
| DNA change | Exon | Protein change | Type of nucleotide change | Family number | Frequency |
|---|---|---|---|---|---|
| c.99_100insT |
2 |
p.R34fs |
Heterozygous |
F4 |
0/200 |
| c.8483delC |
42 |
p.S2828fs |
Heterozygous |
F5 |
0/200 |
| c.8559–2A>G |
43 |
Exon43DEL |
Heterozygous |
F3,F4 |
0/200 |
| c.9450G>A |
48 |
p.W3150X |
Heterozygous |
F5 |
0/200 |
| c.11806A>C | 61 | p.T3936P | Heterozygous | F3 | 0/200 |
The "Frequency" column, shows the number of chromosomes.
Figure 3.
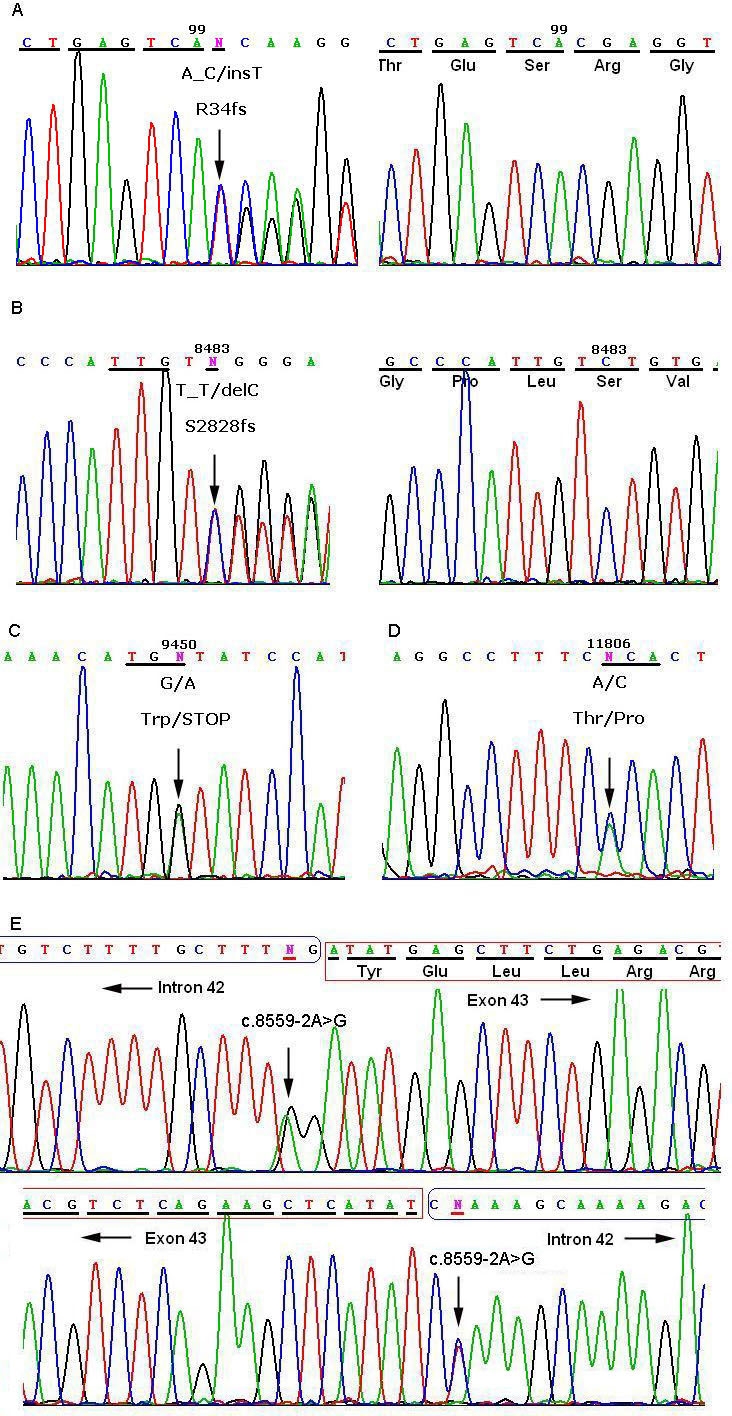
Direct sequencing analysis of the coding region of the USH2A gene. A: Left, heterozygous one base insertion mutation c.99_100insT (p.R34fs) detected in patient 073036; right, the corresponding wild-type sequence. B: left, Sequence shows the heterozygous one base deletion mutation c.8483delC (p.S2828fs) identified in patient 073041 and right, the corresponding wild type sequence. C: Sequence presents the heterozygous nonsense mutation c.9450G>A (p.W3150X) detected in patient 073041. D: Sequence shows the heterozygous missense mutation c.11806A>C (p.T3936P) identified in patient 073021. E: upper, Sequence presents the heterozygous splicing site mutation c.8559-2A>G identified in patient 073021, lower, the corresponding complimentary strand sequence.
Figure 4.
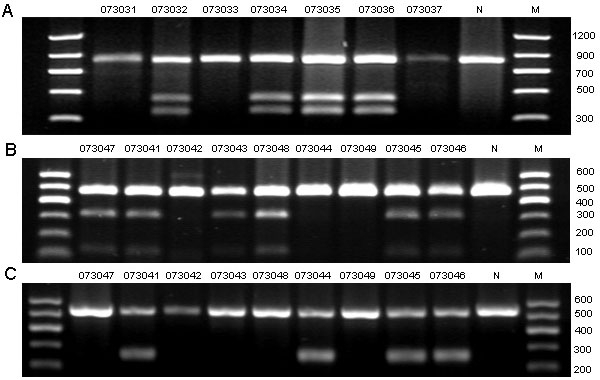
Restriction fragment length analysis on the three mutations detected in this study. A: c.99_100insT created a TaqaI restriction site that cosegregated with the affected individuals and the carriers (370 bp, 470 bp, and 840 bp), but not with unaffected individuals and normal controls (840bp). B: c.8483delC created an MslI restriction site that cosegregated with the affected individuals and the carriers (150 bp, 340 bp, and 490 bp), but not with unaffected individuals and normal control (490 bp). C: c.9450G>A created an EcoRV restriction site that cosegregated with the affected individuals and the carriers (265 bp, and 530 bp), but not with unaffected individuals and normal control (530 bp). Participant identification number is given above each lane. N represents the normal control, and M refers to the DNA ladder in bp.
Figure 5.

Single strand conformation polymorphism analysis for the two mutations identified in the current study. A: Single strand conformation polymorphism (SSCP) analysis for the heterozygous mutation c. 8559–2A>G revealed that the mutant pattern (two bands) cosegregated with the affected individuals and carriers, but not with the unaffected individuals and normal control (one band). B: SSCP analysis for c.11806A>C (p.T3936P) showed that the mutant pattern (three bands) cosegregated with the affected individuals and carriers, but not with the unaffected individuals and normal control (two bands). Participant identification number is listed above each lane. N represents the normal control.
Three different combinations of heterozygous mutations were observed in the three families. In families F3 and F4, one allele harbored the same splice site mutation c.8559–2A>G; the other allele carried a missense mutation c.11806A>C (p.T3936P) and a small insert mutation c.99_100insT (p.R34fs), respectively. Splice site prediction software online (splice) was used, and c.8559–2A>G was predicted to abolish the splice accept site of exon 43, which would lead to the skipping of exon 43. In family F5, one allele harbored a deletion mutation c.8483delC (p. S2828fs), while the other allele carried a nonsense mutation c.9450G>A (p.W3150X).
In addition to the five pathogenic mutations detected in this study, 12 nonpathogenic sequence variants were identified. Four of these were novel, including two silent variants (c.879T>G/p.=and c.1935A>T/p.=), one intron variant (c.10388–27T>C), and one missense variant (c.9340C>T/p.P3114S). Analysis by the online splice prediction software indicated that variant c.10388–27T>C did not disrupt any splice donor site or accept site, and it didn’t create a new splice site. Although p.P3114S was detected in family F5 and cosegregated in family members with the disease in the family, it occurred both in the nonfunction domain and nonconserved region of USH2A isoform b. Furthermore, it was also identified in two individuals among the 100 normal controls. It was classified as a nonpathogenic variant. Table 3 summarizes these variants on the basis of their nature and frequency.
Table 3. Presumed nonpathogenic variants found in USH2A.
| Exon | Nucleotide change | Codon change | Family number | Allele frequency | Source |
|---|---|---|---|---|---|
| 6 |
c.879T>G |
p.L293L |
F5 |
N/A |
This study |
| 11 |
c.1935A>T |
p.T645T |
F5 |
N/A |
This study |
| 18 |
IVS17–8T>G |
F5 |
N/A |
[11] |
|
| 21 |
c.4457A>G |
p.K1486R |
F5 |
76/180 |
[11] |
| 34 |
c.6506T>C |
p.I2169T |
F3,F4,F5 |
27/100 |
[16] |
| 47 |
c.9340C>T |
p.P3114S |
F5 |
2/200 |
This study |
| 52 |
c.10232A>C |
p.E3411A |
F3,F4,F5 |
23/64* |
[17] |
| 53 |
IVS52–27T>C |
F3,F4,F5 |
N/A |
This study |
|
| 60 |
c.11602A>G |
p.M3868V |
F5 |
8/64* |
[17] |
| 63 |
c.12612A>G |
p.T4204T |
F3,F4,F5 |
N/A |
[17] |
| c.12666A>G |
p.T4222T |
F5 |
14/64* |
[17] |
|
| c.13191G>A | p.E4397E | F3,F4,F5 | 7/64* | [17] |
Abbreviations: N/A, data not available; the asterisk indicates that the allele frequency referred to patients sample.
Discussion
In this study, we examined three Chinese families with clinically diagnosed USH and screened the USH2A gene. We identified five novel mutations in the three families, which included three point mutations, one insertion mutation, and one deletion mutation. As shown in Figure 1, the three families carried different compound heterozygous mutations, which were consistent with a pattern of autosomal recessive inheritance of USH. Although all affected individuals experienced progressive hearing loss, the progression was slight, which was not as evident as it was in USH3. The clinical phenotype could be considered to be USH2.
As shown in Figure 6, USH2A isoform b consists of eight domains: N-terminal signal peptide, laminin G-like domain (Lam GL), laminin N-terminal (Lam NT), laminin-type EGF-like domain (EGF Lam), fibronectin type III (FN3), laminin G domains (LamG), transmembrane region (TM), and cytoplasmic domain with a PDZ-binding motif at its C-terminal end [7]. Four novel truncating mutations (p.R34fs, p.S2828fs, p.W3150X, and c.8559–2A>G), either directly or indirectly result in premature termination of USH2A translation or lead to deletion of more than one amino acid residue. This may cause significant changes in the primary protein structure. The c.99_100insT results in a frameshift at codon 34 and premature stop codon 8 amino acids downstream, which removes almost all protein except for the N-terminal signal peptide. The c.8483delC, located in the 14th FN3 repeat, causes a frameshift at codon 2828 and a premature stop codon 1 amino acid downstream. The nonsense mutation c.9450G>A leads to a premature stop codon at 3150. As the three premature stop codons are not located in the last exon of USH2A, there is a possibility that nonsense-mediated mRNA decay might be involved in abnormal RNA processing. Nonsense-mediated mRNA decay is a widespread cellular process that proofreads nascent mRNA transcripts and destroys those that bear premature termination codons before they are actually translated into truncated and potentially harmful proteins [14]. The splicing variant c.8559–2 A>G may result in the skipping of exon 43 of USH2A. The skipping of exon 43 doesn’t result in the interruption of the reading frame. However, it results in the deletion of 41 amino acids, which encode most regions of the 15th FN3 repeat.
Figure 6.
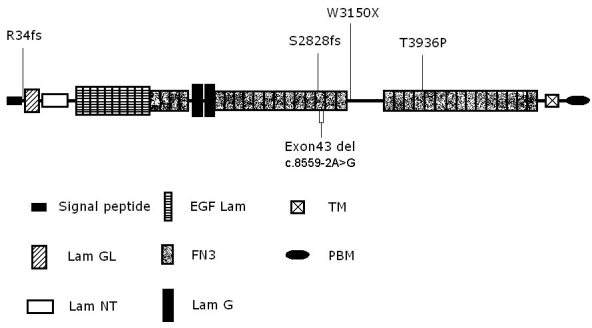
Schematic distribution shows the five novel mutations identified in the current study along the USH2A isoform b protein domains. Abbreviation: EGF-like domain (EGF Lam); Transmembrane region (TM); Laminin G-like domain (Lam GL); Fibronectin type III (FN3); PDZ-binding motif (PBM); Laminin N-terminal (Lam NT); Laminin G domains (Lam G).
Unlike the mutations that cause the loss of a large part of the protein, most of the amino acid substitutions occur at highly conserved sites and result in significant change in polarity or charge. The p.T3936 is located in the 21 FN3 repeat and is highly conserved in humans, mice, chicks, and dogs. The only missense mutation of p.T3936P (c.11806A>C) replaces a polar threonine with a nonpolar hydrophobic proline (Figure 7).
Figure 7.

Sequence alignment portion of the 21 FN3 domain spanning the novel missense mutation p.T3936P of human USH2A isoform b with other species. Identical residues are highlighted in purple. Arrow marks the identified mutation.
To date, more than 160 mutations have been reported in USH2A; however, mutation screening for Chinese patients was performed only once (the USH2A gene; Universal Mutation Database, UMD). Liu et al. [12] only screened the c.2299delG mutation in five Chinese families with USH2 and atypical USH and detected one homozygous and one heterozygous c.2299delG. This is the first report to describe mutation analysis of the complete coding sequence of USH2A in the Chinese population. The c.2299delG was the most common mutation with an allele frequency of 14–50% in patients from Europe and North America [6,11-13,15-19]. Dreyer et al. [20] found that c.2299delG was the result of an ancestral mutation that had spread throughout Europe and into the New World as a result of migration. The c.2299delG was only detected in single patients from South America, South Africa, and China [12,20]. It was not identified in the current study. As we only underwent mutation screening for three families, it is difficult to reach the conclusion that c.2299delG is a low frequency allele in the Chinese patients.
Of the five novel mutations detected in the current study, four are located in exons 22–72, which are specific to the long isoform of USH2A. Two families (F3 and F5) carrying compound heterozygous mutations (either two truncating or one truncating combined with one missense) in exons 22–72, presented with the USH2 phenotype. This is in accordance with Dreyer’s observation in a large unrelated Scandinavian patients study [21].
In conclusion, we have described five novel mutations in three Chinese families with USH2. Our findings expand the spectrum of UHS2A mutations in USH and disclose that the long isoform of USH2A may harbor even more mutations of the USH2A gene.
Acknowledgments
We thank the patients and their families for their participation in this study. We also thank the Beijing Tongren Hospital Joint Committee on Clinical Investigation for their kind support of this study.
Appendix 1. PCR primers used in this study
Oligonucleotide primers used for PCR amplification and sequencing of USH2A. To access the data, click or select the words “Appendix 1.” This will initiate the download of a pdf archive that contains the file.
References
- 1.Keats BJ, Corey DP. The usher syndromes. Am J Med Genet. 1999;89:158–66. [PubMed] [Google Scholar]
- 2.Smith RJ, Berlin Cl, Hejtmancik JF, Keays BJ, Kimberling WJ, Lewis RA, Moller CG, Pelias MZ, Tranebjaerg L. Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. Am J Med Genet. 1994;50:32–8. doi: 10.1002/ajmg.1320500107. [DOI] [PubMed] [Google Scholar]
- 3.Kremer H, van Wijk E, Märker T, Wolfrum U, Roepman R. Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum Mol Genet. 2006;15:R262–70. doi: 10.1093/hmg/ddl205. [DOI] [PubMed] [Google Scholar]
- 4.Ebermann I, Scholl HP, Charbel Issa P, Becirovic E, Lamprecht J, Jurklies B, Millán JM, Aller E, Mitter D, Bolz H. A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet. 2007;121:203–11. doi: 10.1007/s00439-006-0304-0. [DOI] [PubMed] [Google Scholar]
- 5.Ahmed Z, Riazuddin S, Khan S, Friedman P, Riazuddin S, Friedman T. USH1H, a novel locus for type I Usher syndrome, maps to chromosome 15q22-23. Clin Genet. 2008 doi: 10.1111/j.1399-0004.2008.01038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eudy JD, Weston MD, Yao S, Hoover DM, Rehm HL, Ma-Edmonds M, Yan D, Ahmad I, Cheng JJ, Ayuso C, Cremers C, Davenport S, Moller C, Talmadge CB, Beisel KW, Tamayo M, Morton CC, Swaroop A, Kimberling WJ, Sumegi J. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science. 1998;280:1753–7. doi: 10.1126/science.280.5370.1753. [DOI] [PubMed] [Google Scholar]
- 7.van Wijk E, Pennings RJ, te Brinke H, Claassen A, Yntema HG, Hoefsloot LH, Cremers FP, Cremers CW, Kremer H. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am J Hum Genet. 2004;74:738–44. doi: 10.1086/383096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reiners J, Nagel-Wolfrum K, Jürgens K, Märker T, Wolfrum U. Molecular basis of human Usher syndrome: Deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp Eye Res. 2006;83:97–119. doi: 10.1016/j.exer.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Adato A, Lefèvre G, Delprat B, Michel V, Michalski N, Chardenoux S, Weil D, El-Amraoui A, Petit C. Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum Mol Genet. 2005;14:3921–32. doi: 10.1093/hmg/ddi416. [DOI] [PubMed] [Google Scholar]
- 10.Maerker T, van Wijk E, Overlack N, Kersten FF, McGee J, Goldmann T, Sehn E, Roepman R, Walsh EJ, Kremer H, Wolfrum U. A novel Usher protein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum Mol Genet. 2008;17:71–86. doi: 10.1093/hmg/ddm285. [DOI] [PubMed] [Google Scholar]
- 11.Weston MD, Eudy JD, Fujita S, Yao S, Usami S, Cremers C, Greenberg J, Ramesar R, Martini A, Moller C, Smith RJ, Sumegi J, Kimberling WJ. Genomic structure and identification of novel mutations in usherin, the gene responsible for Usher syndrome type IIa. Am J Hum Genet. 2000;66:1199–210. doi: 10.1086/302855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu XZ, Hope C, Liang CY, Zou JM, Xu LR, Cole T, Mueller RF, Bundey S, Nance W, Steel KP, Brown SD. A mutation (2314delG) in the Usher syndrome type IIA gene: high prevalence and phenotypic variation. Am J Hum Genet. 1999;64:1221–5. doi: 10.1086/302332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernal S, Ayuso C, Antiñolo G, Gimenez A, Borrego S, Trujillo MJ, Marcos I, Calaf M, Del Rio E, Baiget M. Mutations in USH2A in Spanish patients with autosomal recessive retinitis pigmentosa: high prevalence and phenotypic variation. J Med Genet. 2003;40:e8. doi: 10.1136/jmg.40.1.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hentze MW, Kulozik AE. A perfect message: RNA surveillance and nonsense-mediated decay. Cell. 1999;96:307–10. doi: 10.1016/s0092-8674(00)80542-5. [DOI] [PubMed] [Google Scholar]
- 15.Seyedahmadi BJ, Rivolta C, Keene JA, Berson EL, Dryja TP. Comprehensive screening of the USH2A gene in Usher syndrome type II and non-syndromic recessive retinitis pigmentosa. Exp Eye Res. 2004;79:167–73. doi: 10.1016/j.exer.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 16.Kaiserman N, Obolensky A, Banin E, Sharon D. Novel USH2A mutations in Israeli patients with retinitis pigmentosa and Usher syndrome type 2. Arch Ophthalmol. 2007;125:219–24. doi: 10.1001/archopht.125.2.219. [DOI] [PubMed] [Google Scholar]
- 17.Aller E, Jaijo T, Beneyto M, Nájera C, Oltra S, Ayuso C, Baiget M, Carballo M, Antiñolo G, Valverde D, Moreno F, Vilela C, Collado D, Pérez-Garrigues H, Navea A, Millán JM. Identification of 14 novel mutations in the long isoform of USH2A in Spanish patients with Usher syndrome type II. J Med Genet. 2006;43:e55. doi: 10.1136/jmg.2006.041764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baux D, Larrieu L, Blanchet C, Hamel C, Ben Salah S, Vielle A, Gilbert-Dussardier B, Holder M, Calvas P, Philip N, Edery P, Bonneau D, Claustres M, Malcolm S, Roux AF. Molecular and in silico analyses of the full-length isoform of usherin identify new pathogenic alleles in Usher type II patients. Hum Mutat. 2007;28:781–9. doi: 10.1002/humu.20513. [DOI] [PubMed] [Google Scholar]
- 19.Bernal S, Medà C, Solans T, Ayuso C, Garcia-Sandoval B, Valverde D, Del Rio E, Baiget M. Clinical and genetic studies in Spanish patients with Usher syndrome type II: description of new mutations and evidence for a lack of genotype–phenotype correlation. Clin Genet. 2005;68:204–14. doi: 10.1111/j.1399-0004.2005.00481.x. [DOI] [PubMed] [Google Scholar]
- 20.Dreyer B, Brox V, Tranebjærg L, Rosenberg T, Sadeghi AM, Möller C, Nilssen Ø. Spectrum of USH2A mutations in Scandinavian patients with Usher syndrome type II. Hum Mutat. 2008;29:451. doi: 10.1002/humu.9524. [DOI] [PubMed] [Google Scholar]
- 21.Dreyer B, Tranebjærg L, Brox B. Rosenberg T, Möller C, Beneyto M, Weston MD, Kimberling WJ, Nilssen Ø. A common ancestral origin of the frequent and widespread 2299delG USH2A mutation. Am J Hum Genet. 2001;69:228–34. doi: 10.1086/321269. [DOI] [PMC free article] [PubMed] [Google Scholar]