

Abstract
A limiting factor to the clinical management of diabetes is iatrogenic hypoglycemia. With multiple hypoglycemic episodes, the collective neuroendocrine response that restores euglycemia is impaired. In our animal model of recurrent hypoglycemia (RH), neuroendocrine deficits are accompanied by a decrease in medial hypothalamic activation. Here we tested the hypothesis that the medial hypothalamus may exhibit unique changes in the expression of regulatory proteins in response to RH. We report that expression of the immediate early gene FosB is increased in medial hypothalamic nuclei, anterior hypothalamus, and posterior paraventricular nucleus of the thalamus (THPVN) of the thalamus following RH. We identified the hypothalamic PVN, a key autonomic output site, among the regions expressing FosB. To identify the subtype(s) of neuronal populations that express FosB, we screened candidate neuropeptides of the PVN for coexpression using dual fluorescence immunohistochemistry. Among the neuropeptides analyzed [including oxytocin, vasopressin, thyrotropin-releasing hormone, and corticotropin-releasing factor (CRF)], FosB was only identified in CRF-positive neurons. Inhibitory γ-aminobutyric acid-positive processes appear to impinge on these FosB-expressing neurons. Finally, we observed a significant decrease in the presynaptic marker synaptophysin within the PVN of RH-treated vs. saline-treated rats, suggesting that rapid alterations of synaptic morphology may occur in association with RH. Collectively, these data suggest that RH stress triggers cellular changes that support synaptic plasticity, in specific neuroanatomical sites, which may contribute to the development of hypoglycemia-associated autonomic failure.
Keywords: FosB, recurrent hypoglycemia, paraventricular nucleus, corticotropin-releasing factor, synaptophysin
the neuroendocrine counterregulatory response (CRR) that occurs to correct a single hypoglycemic (SH) episode becomes impaired when multiple episodes occur closely in time (recurrent hypoglycemia, RH), a situation often experienced by individuals with diabetes (18, 19). Although iatrogenic hypoglycemia represents a limitation to the clinical management of diabetes (49), the mechanisms underlying the failed neuroendocrine response are not completely understood.
The immediate early gene (IEG) and transcription factor, c-Fos, has served as a marker of neuronal activity in numerous in vivo and in vitro paradigms (36) and has been utilized to document activation of the central nervous system (CNS) as a result of SH in several laboratories (3, 23, 46). In a previous study, we used c-Fos to compare relative activation in the forebrain of rats receiving one or three bouts of hypoglycemia (23). That study revealed that three medial hypothalamic areas [the arcuate nucleus (ARC), the paraventricular nucleus (PVN), and the dorsomedial hypothalamus (DMH)] had decreased c-Fos expression in rats that had experienced three, vs. one, bout(s) of hypoglycemia. Furthermore, we determined that the PVN and DMH play key roles in mediating the hypothalamic-pituitary-adrenal axis (PVN and DMH) and sympathoadrenal (PVN) responses to hypoglycemia (24, 25). Accordingly, impaired activation of these medial hypothalamic areas would be expected to contribute substantially to the impaired CRR to RH. However, whether the decreased medial hypothalamic activation is the result of impaired afferent input [e.g., decreased input from glucosensing neurons (40, 53)], impaired excitability of neurons within the PVN and DMH, initiation of intracellular processes that decrease subsequent neuronal excitability, or a combination of these is still unknown.
Although c-Fos, because of its rapid activation and deactivation kinetics, functions as a marker for the acute activation induced by a single bout of hypoglycemia, absence of c-Fos expression cannot be used to identify the phenotype of neurons within the PVN and DMH that are impacted by RH. Within the PVN, such neurons might include preautonomic neurons or neuroendocrine cells expressing oxytocin (OT), vasopressin (VP), thyrotropin-releasing hormone (TRH), or corticotropin-releasing factor (CRF) that are implicated in stress responses and regulation of energy homeostasis (5, 6, 26, 35, 38, 42, 51), making them candidates for evaluation of their role in the response to hypoglycemia. In this study, we took advantage of the differential expression profile of another member of the “Fos” family of transcription factors (14, 15, 32) to begin to examine the delayed or longer-acting neuronal alterations that we hypothesize are associated with RH. Although the role of FosB isoforms has been studied most extensively in models of drug addiction, FosB has been implicated in local CNS responses to the pathophysiology of seizure (44) and Parkinson's disease (2, 13). As studied in the striatum by Nestler and colleagues (14, 15, 32), in the hours subsequent to the transient activation/deactivation time frame for c-Fos, alternative isoforms of Fos become expressed, notably FosB and delta FosB, which have longer half-lives, estimated to be in the range of days in vivo. This stability contributes to the potential accumulation of these isoforms following repeated stimulation and may serve to identify neurons that undergo delayed and longer-acting changes. These more stable IEGs can also control transcription of different sets of neuron-regulatory proteins, compared with c-Fos. Thus, with RH, transcription of alternative regulatory proteins that are not affected by SH might occur in the medial hypothalamus. To begin to evaluate this hypothesis, we measured the expression of FosB in the forebrain of rats following one or three bouts of hypoglycemia (or three control infusions). In addition to confirming that the medial hypothalamus is one of the brain regions that has increased FosB expression in association with RH, we used dual immunofluorescence to phenotype affected neurons in the PVN.
Various instances of neural plasticity, both behavioral and functional, are characterized by not only modified responses to repeated exposures of the same stimulus but changes in expression of key functional proteins in the neurons that are targeted by the stimulus. As such, we would predict that, in our model of RH, where repetitive exposure to the stress of hypoglycemia is associated with impaired neuroendocrine responsivity, there are underlying cellular mechanisms of neural adaptation. Thus transcription factors such as FosB could serve not only to simply identify the brain regions and neuronal phenotypes involved in mediating the impaired CRR to RH as discussed above but could represent a molecular step whereby persistent changes in gene expression can be induced and maintained. The functional implication of this for RH would be that neuronal activation is not merely decreased, but that neurons would essentially be reprogrammed.
As one additional index of change of function (as opposed to a simple decrease of activation) within the PVN, we evaluated expression of the synaptic marker synaptophysin (SYN) (41, 52, 61). Changes of SYN expression have been associated with the onset of plasticity in response to a variety of pathophysiological or adaptive changes (39) and challenges in the CNS, including hypoglycemia (52), although in that study the hypothalamus and individual brain regions were not evaluated. There are examples of early onset decreases of SYN and later-onset increases of SYN, which may reflect synaptic remodeling (33). We hypothesized that SYN expression would be altered in our model of acute RH.
EXPERIMENTAL PROCEDURES
Animals
Male Wistar rats (Simonsen, Gilroy, CA), 350–400 g, were maintained on ad libitum chow and water and a 12:12-h light-dark cycle (6:00 A.M. lights on; 6:00 P.M. lights off), and were studied during the light portion of the cycle. The study protocol was approved by the Institutional Animal Care and Use Committee of the Veterans Affairs Puget Sound Health Care System. After acclimation in the animal facility, rats were implanted with chronic venous catheters (jugular and submaxillary) according to our published methodology (23) to provide venous access for infusion and collection of blood samples (see Experimental protocol below). Rats recovered from the surgery and demonstrated a positive trajectory of weight gain before undergoing the experimental procedure.
Methods
Experimental protocol.
We utilized our published methodology (23) for inducing one or three bouts of hypoglycemia in a 2-day procedure. Rats were studied in the conscious, non-food-deprived, unstressed condition, with blood samples being collected remotely and volume replaced remotely with whole blood obtained from donor rats. Before the experiment, rats were brought into the experiment room and placed in acrylic testing chambers with clean bedding, for habituation to the testing conditions. On experimental days, rats were placed in acrylic chambers, and intravenous lines were hooked up. Rats were settled for ∼90 min before drawing a time 0 baseline sample, with subsequent infusion of insulin (0.25 U·100 g−1·2 h−1), or saline vehicle, at a rate of 1.146 ml/h using a microinfusion syringe pump (KD Scientific, Holliston, MA). On day 1, rats received a 2-h insulin (or saline) infusion in the late A.M. This was followed by a 60-min interval in which they were given chow and allowed to feed; we have determined that this feeding interval is sufficient to rescue plasma glucose levels back to within normoglycemia (80–85 mg/dl) by the start of the second infusion. A second 2-h insulin or saline infusion was then carried out, and rats were returned to home cages where they had ad libitum access to chow overnight. On day 2, at ∼9:00 A.M., rats were returned to their experimental chambers and, after baseline blood (time 0) collection, were subjected to either a third infusion of insulin or saline. Blood samples were then collected at 90 and/or 120 min thereafter for the measurement of glucose and the neuroendocrine CRR. Plasma samples obtained were stored at −80°C until assayed. Blood for the catecholamine assays was collected on EGTA-glutathione (2.3:1.5 mg/ml; Sigma). Tubes for glucagon assays contained 50 μl of 1 M benzamidine (Sigma) and 1 unit heparin. Blood for glucose, adrenocorticotropic hormone (ACTH), and corticosterone assays was collected on EDTA and aprotinin (1.7 tissue inhibitor unit; Sigma). The assays have been described previously. Briefly, a radioenzymatic method as described in Evans et al. (23) was used for determination of plasma epinephrine and norepinephrine. Plasma glucose was measured using the Beckman Glucose Analyzer. Glucagon was assayed by the Linco glucagon RIA kit (Linco Research, St. Charles, MO). Measurements of ACTH were made using the Nichols Institute Diagnostics immunoradiometric assay kit (Nichols Institute Diagnostics, San Juan Capistrano, CA). A radioimmunoassay procedure was used for plasma corticosterone measurement as described in van Dijk et al. (60).
Following the last infusion on day 2, rats were deeply anesthetized with either intraperitoneal pentobarbital sodium injection (50 mg·ml−1·kg−1) or isoflurane inhalation and perfused with 0.9% NaCl followed with cold 4% paraformaldehyde solution. One group of rats was killed 90 min following a single infusion of saline or insulin (SH). This corresponds to the timing of our previous determination of c-fos in response to a single bout of hypoglycemia (23). Note that essentially no FosB expression was observed following a single infusion of saline and, therefore, no quantitation was carried out for this group. The remaining groups were killed 90 min after the third infusion (saline or RH) on day 2. This time point should reflect an accumulation of FosB isoforms, as well as the c-fos response to a third bout of hypoglycemia. Brains were removed and postfixed overnight in the same fixative and subsequently placed in 30% sucrose-PBS solution. Brains were sectioned on a cryostat (Leica CM 3050S cryostat) for immunohistochemistry.
Immunohistochemistry.
Atlas-matched 40-μm sections were evaluated for comparison between insulin- and saline-treated rats, based upon Paxinos and Watson (47). Initial screen of forebrains revealed virtually no detectable FosB in most brain regions; therefore, we focused upon a limited number of areas for semiquantitation and for neuronal phenotyping (atlas figures 24–31, which include −1.4 to −3.30 mm posterior from bregma). Specifically, we focused upon hypothalamus [ventromedial hypothalamic nucleus (VMH), −2.12 to −3.14 mm; DMH, −2.56 to −3.14; PVN, −1.8 to −2.8; anterior hypothalamic area, −2.12 to −2.3]; posterior PVN of the thalamus (THPVP; −3.14 to −3.30), a region that is implicated in the habituation of the neuroendocrine response to other stressors (9), and is also activated in response to hypoglycemia (58); and anterior PVN of the thalamus (THPVA; −1.4 to −2.12) and central amygdala (−2.12 to −2.3) as control regions implicated in stress circuitry but not significantly responsive to hypoglycemia in our model.
FosB immunocytochemistry and quantitation.
The laboratory's established methodology for c-Fos quantitative immunocytochemistry (23) was adapted for quantitation of FosB. Sections were washed in PBS for 45 min; goat serum-Triton X-100 buffer (3% goat serum-0.3% Triton X-100 in PBS) for 30 min; gelatin blocking buffer (0.5% gelatin in goat serum-Triton X-100 buffer) for 60 min; and goat serum-Triton X-100 buffer for 30 min. Free-floating sections were then incubated with primary antibody (1:2,000) in goat serum-Triton X-100 buffer for 40–48 h at 4°C. Following incubation with primary antibody, tissue was washed at room temperature in PBS for 90 min, transferred to secondary antibody solution (5 μl secondary antibody/ml goat serum-Triton X-100 buffer) for 60 min, and then washed for 60 min in PBS. Sections were incubated with avidin-biotin solution at room temperature for 60 min, transferred to diaminobenzidine solution for hand mixing up to 2 min maximum, and washed in PBS for 30 min. Sections were mounted onto glass slides and coverslipped using Permount mounting medium. For quantitation (at ×40 magnification), atlas-matched sections from control or hypoglycemic brains were selected. ImagePro Plus software (Media Cybernetic) was utilized to capture an image of the desired area. An area was delineated for counting, and threshold for positive cell counts was established. The identical area and background (threshold) were utilized for control and hypoglycemic sections, and software counting of positive cells (quantitation) was carried out in the same session for control and hypoglycemic brain areas to prevent between-session changes in background setting. For statistical analysis, counts were taken from an individual rat only if complete sections through each area (as defined above) were available; data for a specific area were not taken from a rat if there was incomplete bilateral representation for that area.
Immunofluorescence.
Slide-mounted 12-μm whole brain coronal sections were washed three times in PBS (OXOID, Hampshire, UK). Sections were then blocked for 1 h at room temperature in PBS containing 5% normal goat serum. Sections were then washed multiple times in PBS and incubated overnight at 4°C in primary antibody solutions made up in PBS. Sections were washed three times in PBS and then incubated in the dark at room temperature in secondary antibody solution made up in PBS for 1 h. Included within each assay were control sections in which primary antibody was omitted (negative controls). As an additional control, sections were incubated in both monoclonal and polyclonal FosB antibodies together (see Materials), and it was confirmed that both antibodies colocalized to the same neurons. Sections were subsequently washed again in PBS and mounted and coverslipped in Vectashield hard set mounting medium (Vector, Burlingame, CA). In an effort to increase detection of CRF and TRH, the tyramide amplification system of immunofluorescence amplification was evaluated, but this did not improve the sensitivity over the regular protocol and, in fact, attenuated the observable signal; thus, those results are not included here. Digital Images of sections were acquired using a Nikon Eclipse E-800 fluorescence microscope connected to an Optiphot camera and using Image Pro Plus (Media Cybernetic) software.
Double-label immunofluorescence analysis.
Digital images (acquired as described above) taken at ×40 magnification, using atlas-matched sections from control or hypoglycemic brains, were selected for quantitation. An area was delineated for counting, and threshold for positive cell counts was established. The identical area and background (threshold) were used for sections from control and hypoglycemic rats, and manual counting of positive cells in each channel was conducted separately for FosB and CRF, and then as a merged image of both channels for colocalization. The colocalization was confirmed at higher (×60) magnification. Quantitation was carried out in the same session for control and hypoglycemic brain areas to prevent between-session changes in background setting.
Western blotting.
Brains were removed and snap-frozen in methylbutane. Thick (280 μm) cryosections of the tissue were prepared, and punches of the specific brain regions (see above for atlas coordinates) were taken and used to make homogenates using RIPA lysis buffer (Santa Cruz Kit) according to standard techniques (manufacturer's specifications). Protein estimates were performed using a BCA assay kit (Pierce, Rockford, IL). Samples were run on 10% Tris·HCl precast Ready Gels (Bio-Rad, Hercules, CA) and transferred to polyvinylidene difluoride membranes (Bio-Rad) for 2 h at 100 volts. Blots were blocked in Superblock Blocking Buffer (Pierce) made up in PBS, washed three times in Tris-buffered saline (TBS) containing 0.1% Tween 20 (Sigma, St. Louis, MO) (TBS-T), and incubated in primary antibody solution overnight at 4°C. Blots were washed again three times in TBS-T and incubated for 1–2 h at room temperature in secondary antibody solution. After multiple washes in TBS-T, blots were subjected to detection using the Supersignal West Femto ECL kit (Pierce) and developed on film (x-omat blueXB-1; Kodak). Digital images of blots were obtained using an Epson 836XL high-resolution document scanner, and the bands were quantitated using Bio-Rad's Quantity One analysis software.
Materials
Humulin R regular insulin, human USP rDNA origin (Elli Lilly, Indianapolis, IN), 0.25 U/100 g made up in saline, was used to induce hypoglycemia. Primary antibodies used included rabbit anti-FosB (1:500) (sc-48) (Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-FosB (1:500) (Abcam, Cambridge, MA), mouse anti-GAD (1:1,000) (Chemicon, Temecula, CA), mouse anti-OT (1:1,000) and mouse anti-VP (1:500) (generous gifts from Dr. Ann-Judith Silverman, Columbia University), rabbit anti-CRF (1:500) (#PBL rC70; generous gift from Dr. Wylie Vale, Salk Institute), mouse anti-SYN (1:1,000) (Chemicon), and rabbit anti-pre-pro-TRH (1:10,000) (generous gift from Dr. Eva Redei, Northwestern University). Secondary antibodies used included Cy3-conjugated goat anti-rabbit or anti-mouse (Jackson Immunoresearch, West Grove, PA), Alexa fluor 488 goat anti-mouse or anti-rabbit IgG (Molecular Probes, Eugene, OR), and biotinylated anti-rabbit IgG (Vector Laboratories). All secondary antibodies were diluted at 1:500.
RESULTS
Neuroendocrine Responses to SH and RH
Table 1 shows the metabolic and neuroendocrine response to three bouts of saline (control), SH, or RH in rats for which FosB was quantitated (Fig. 1). As expected, epinephrine and glucagon levels were elevated in hypoglycemic rats compared with the values of saline-infused control rats. The RH responses are comparable to our published data (23) and lower than those after SH. Table 2 shows plasma glucose and neuroendocrine data, in response to RH or three infusions of saline, in rats studied for the neural phenotype of Fos-B-expressing cells within the PVN (Figs. 2–5). Glucagon and epinephrine data are comparable to those reported in Table 1, consistent with a blunted response to RH. ACTH responses to RH are substantially lower than those we have reported previously as a response to SH [211 ± 22 (24); 193 ± 22 (25) pg/ml] and perhaps reflect the impaired CRR to RH, along with impaired glucagon and epinephrine. Corticosterone responses, as we have previously reported (23), are essentially normal after RH.
Table 1.
Plasma glucose, glucagon, and Epi responses to RH, SH, or saline (control) infusions
| Glucose, mg/dl | Glucagon, pg/ml | Epi, pg/ml | |
|---|---|---|---|
| Saline (n = 9) | |||
| Time 0 | 107±2 | 99±15 | 82±13 |
| Time 120 min | 101±3 | 101±3 | 52±3 |
| SH (n = 9) | |||
| Time 0 | 105±2 | 131±7 | 69±7 |
| Time 120 min | 31±2 | 574±94 | 4,081±511 |
| RH (n = 9) | |||
| Time 0 | 106±3 | 78±10 | 60±6 |
| Time 120 min | 31±2 | 211±24 | 2,358±422 |
Values are means ± SE; n, no. of rats. Epi, epinephrine; SH, single hypoglycemic; RH, recurrent hypoglycemia. Data were obtained from rats studied for FosB quantitation (Fig. 1).
Fig. 1.
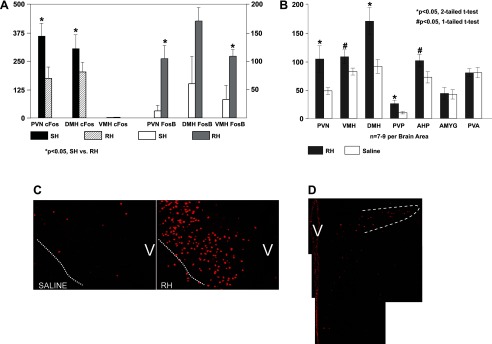
Recurrent hypoglycemia (RH) induces FosB expression. A: quantitative comparison of c-Fos (axes on left) and FosB (axes on right) cell counts following single hypoglycemic (SH) and RH in the paraventricular nucleus (PVN), ventromedial hypothalamic nucleus (VMH), and dorsomedial hypothalamic nucleus (DMH). B: FosB immunopositive neuron counts following three infusions of saline or insulin. Statistically significant increases in the number of FosB-positive cells in the RH-treated group compared with the saline control group were observed in all of the hypothalamic areas indicated and the posterior PVN of the thalamus (PVP). FosB expression was not increased in the other brain areas, as evaluated by either one- or two-tailed t-tests (hypothesis: increased FosB expression in RH- vs. saline-treated rats, one-tailed t-test; change in either direction of FosB expression in RH- vs. saline-treated rats, two-tailed t-test). C: immunofluorescence images taken at mid-PVN levels illustrating the expression of FosB in the PVN of a RH-treated rat (right) compared with a saline-treated rat (left). The dotted white line demarcates the ventral perimeter of the PVN, whereas “V” is placed over the region of the ventricle. D: immunofluorescence image of the caudal PVN; the dotted white line demarcates the lateral parvocellular subdivision.
Table 2.
Plasma glucose, glucagon, Epi, ACTH, and CORT responses to RH or saline (control) infusions
| Glucose, mg/dl | Glucagon, pg/ml | Epi, pg/ml | ACTH, pg/ml | CORT, μg/dl | |
|---|---|---|---|---|---|
| Saline (n = 7–8) | |||||
| Time 0 | 107±5 | 75±8 | 87±19 | 19±3 | 4.9±1.5 |
| Time 90 min | 119±2 | 84±9 | 67±25 | 17±3 | 5.5±2.0 |
| Time 120 min | 118±3 | 78±8 | 76±21 | 19±3 | 9.6±1.6 |
| Insulin (n = 7–8) | |||||
| Time 0 | 114±5 | 74±6 | 90±18 | 17±2 | 5.4±1.1 |
| Time 90 min | 46±5 | 220±32 | 1,658±555 | 93±13 | 26.9±2.1 |
| Time 120 min | 34±4 | 173±25 | 2,333±479 | 88±10 | 32.4±2.5 |
Fig. 2.
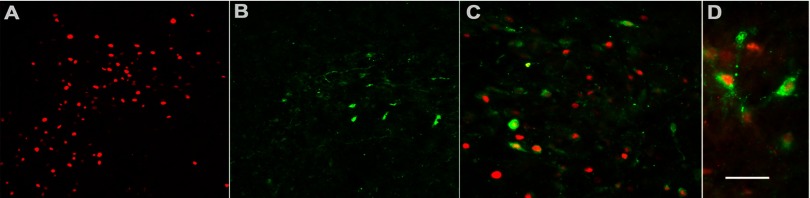
A subpopulation of FosB-positive neurons coexpresses corticotropin-releasing factor (CRF) in the PVN. A: immunofluorescence demonstrates RH-induced FosB expression (red). B: α-CRF antibody (green) identifies a subpopulation of neurons in the PVN and labels both cell bodies and fibers. C and D: successively higher (×40 and ×60) magnifications of merged images, respectively, of the same field double-labeled for both FosB and CRF exhibit colocalization of both antibodies in ∼30% of FosB-positive neurons. Scale bar represents 100 (A and B); 50 (C), and 25 (D) μm.
Fig. 5.
Glutamic acid decarboxylase (GAD) immunoreactivity, a marker of GABAergic transmission, does not colocalize with FosB in the PVN following RH. Immunofluorescence using α-GAD67 antibody revealed intense GAD (green) expression in the hypothalamus. A: GAD-positive staining in the PVN and peri-PVN. Note that the staining is almost exclusively of fibers within the PVN proper (outlined by dotted white line), whereas GAD-positive immunoreactive cell bodies are only observed in the area of the “peri-PVN” (white arrows). B: FosB-positive cells (red) in the PVN following RH. C: GAD immunoreactivity in the PVN and surrounding area in the same section. The neuropil in the immediate region of the PVN displayed intense GAD-positive fiber staining. D: merged images of the same field illustrating the intense GAD-positive neuropil with contacts appearing to impinge on FosB-positive cells. Scale bar: 100 μm.
C-Fos and FosB Responses to SH and RH
Figure 1A demonstrates c-Fos increases with SH compared with RH (Fig. 1A, axis on left). As we have reported previously, c-Fos is decreased in the PVN (P = 0.015) and DMH (P = 0.04) in response to RH vs. SH. There is no detectable c-Fos in the VMH in response to either SH or RH. In contrast, FosB (Fig. 1A, axis on right) levels are increased following RH (PVN, P = 0.04; VMH, P = 0.017; DMH, P = 0.063; overall medial hypothalamus, P < 0.001) compared with SH in the medial hypothalamus, consistent with a slower onset, and slower turnover, of expression. Figure 1B shows FosB-positive neuron counts in hypothalamic and limbic areas from rats after either a third bout of hypoglycemia (RH) or third saline infusion. There was an overall effect of treatment (RH vs. saline) in the medial hypothalamus (PVN, VMH, and DMH combined, P < 0.001), and post hoc one-tailed t-test (hypothesis: increased FosB expression in RH- vs. saline-treated rats) revealed statistically significant increases in all three individual regions (PVN, P = 0.02; VMH, P = 0.035; DMH, P = 0.005). There were also statistically significant increases in AHP (P = 0.04) and THPVP (P = 0.01), with no effect of insulin treatment on the THPVA or amygdala. As an example, Fig. 1, C and D, shows FosB expression within the PVN following RH. Figure 1C demonstrates robust FosB expression in the ventral and medial parvocellular subdivisions of the midlevel PVN, whereas Fig. 1D exhibits FosB expression in the medial and lateral pavocellular PVN subdivisions at a more caudal level.
Localization of FosB in PVN
Based on the FosB quantitation described above, we focused on the medial hypothalamus, specifically the PVN as a major autonomic center, to identify the phenotype(s) of neurons that express FosB following hypoglycemia, as vulnerable neuronal subtypes that might be implicated in the impaired CRR. To do this, we assayed coexpression of FosB with the candidate neuropeptides OT, VP, CRF, and TRH by means of double-label immunofluorescence in saline-treated and RH-treated rats.
Figure 2 shows representative images of sections from RH-treated animals. As in Fig. 1C, RH-induced FosB expression is observed (Fig. 2A). CRF expression in the hypothalamus was found to be limited to the parvocellular PVN region, and immunostaining of both cell bodies and fibers was observed (Fig. 2B). A proportion of FosB-positive neurons were found to coexpress CRF, as demonstrated by antibody colocalization (Fig. 2, C and D, at higher magnification). This subpopulation of neurons exhibited a central nuclear region expressing FosB surrounded by a cytoplasmic limbus of CRF immunoreaction product. Quantitation by means of cell counts revealed that ∼30% of FosB-positive cells express CRF. It is possible that this may be an underestimate of the FosB expression in CRF neurons, since rats were not colchicine pretreated to limit CRF transport out of the cell body (hence, lower cell body immunostaining).
Figure 3 illustrates FosB (Fig. 3A) and TRH (Fig. 3B) immunoexpression. The TRH labeling in the somata was seen to be predominantly cytoplasmic, whereas the fiber staining had a highly punctate appearance. In contrast to CRF, and within the limits of detection of TRH immunoreactivity, TRH-positive cell bodies did not appear to coexpress FosB, and clearly both immunoreactive products appeared to label separate neuronal populations (Fig. 3D). The lower-magnification image (Fig. 3C) confirms the presence of TRH-positive cells in the parvocellular region of the PVN, as expected.
Fig. 3.
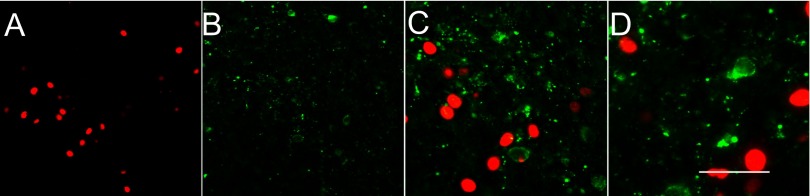
Thyrotropin-releasing hormone (TRH)-positive neurons in the PVN do not express FosB. A: immunofluorescence demonstrates RH-induced FosB expression (red) as observed in Figs 1 and 2. B: α-TRH antibody (green) identifies a subpopulation of neurons in the parvocellular PVN and labels both cell bodies and fibers. C and D: successively higher (×40 and ×60) magnification merged images, respectively, of the same field double-labeled for both FosB and TRH; no colocalization is observable. Scale bar represents 100 (A and B); 50 (C), and 25 (D) μm.
Analysis of VP expression revealed confinement of VP-expressing cell bodies to the magnocellular PVN region of the hypothalamus. Densely packed somata along with fiber staining were observed (Fig. 4B). Examination of OT expression in the hypothalamus again revealed a confinement to the magnocellular PVN region with clearly defined subpopulations of neurons displaying immunoreaction product in both cell bodies and fiber terminals (Fig. 4E). These OT-positive fibers displayed puncta-like varicosities along their lengths. However, double-label immunofluorescence revealed no colocalization of either VP or OT immunopositivity with FosB (Fig. 4, C and F, respectively), indicating that both of these neuropeptides, while robustly expressed in the hypothalamic PVN, are not the neuronal subpopulations in which FosB is being expressed.
Fig. 4.

Vasopressin (VP)- or oxytocin (OT)-positive neurons in the PVN do not express FosB. A and D: RH induces FosB expression (red) in the PVN. B and E: VP (B) cell bodies and fibers (green) and OT (E) cell bodies and fibers (green) are localized in the magnocellular region of the PVN. Images shown are of the same field as FosB. C and F: merged images of double-label immunofluorescence reveal distinct red (FosB-positive) and green [VP-positive (C) or OT-positive (F)] subpopulations of neurons with no coexpression. Scale bar represents 100 μm.
FosB expression within the parvocellular PVN compartments was observed at the mid-PVN levels in the medial and ventral parvocellular subdivisions and at the caudal PVN levels in the medial and lateral parvocellular subdivisions [Fig. 1, C and D; designations of the PVN subdivisions are based on the work of Lechan and Fekete (38)]. This finding is significant in that neurons in these parvocellular regions have been reported to send projections to preganglionic targets in the brain stem and spinal cord (12, 48, 54). The involvement of putative preautonomic projecting neurons is consistent with the impaired glucagon and epinephrine responses we measure in response to RH.
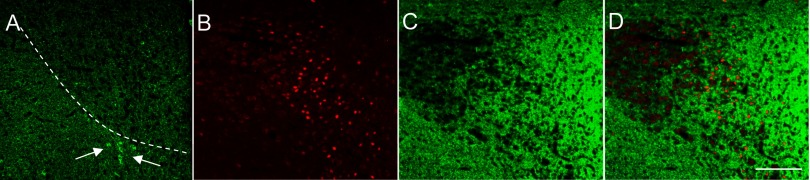
Finally, we evaluated expression of glutamic acid decarboxylase (GAD), the synthetic enzyme for the inhibitory neurotransmitter γ-aminobutyric acid (GABA). GABAergic innervation of the PVN is extensive and may be both local and extrinsic from regions such as the DMH (30, 31). Kovacs et al. (37) recently reported that, in the parvocellular PVN, 79% of GABAergic boutons terminate on CRF neurons and that these comprise 36% of the total synapses on CRF neurons. An increase in either GABA synthesis or signaling could be one mechanism contributing to the decreased PVN activation with RH. Immunohistochemistry revealed a high intensity of GAD immunoreactivity in the RH group (Fig. 5A) with very bright and widespread GAD-positive puncta, presumably reflecting synaptic terminals. A dense plexus of GAD-positive fiber staining was observed in the area of the PVN and the entire periventricular area, whereas GAD-positive somata were only observed in the area immediately adjacent to the PVN or the “peri-PVN” (see Fig. 5A). Double-label immunofluorescence was performed to examine the distribution and expression of GAD (Fig. 5C) relative to FosB (Fig. 5B). The GAD-positive staining did not appear to colocalize to the same cell bodies as FosB, but the GAD-positive fibers did appear to be impinging on the FosB-positive cells (Fig. 5D) and perhaps making synaptic contacts.
Measurement of Medial Hypothalamic SYN After RH
Having identified the CRF neuron as the predominant neuron within the PVN to express FosB, we then turned to examine expression of the putative presynaptic protein SYN as a marker of synaptic changes that might underlie the impaired CRR to RH. SYN expression can be altered (increased or decreased) in acute circumstances of stimulus-induced neuroplasticity (52, 61) (see Discussion). Expression of SYN in medial hypothalamic regions was analyzed by Western Blot (Fig. 6A) to investigate whether SYN changed in association with RH. Although no statistically significant changes were observed in the ARC or DMH/VMH, SYN expression was decreased significantly (P < 0.017) in the PVN with RH, as demonstrated by a decrease in the 37-kDa size band representing SYN in the PVN of RH rats compared with the PVN of saline controls (Fig. 6B).
Fig. 6.

Synaptophysin (SYN) is decreased in the PVN following RH. A: representative Western blot of SYN expression in the corresponding brain areas of interest (as identified in Fig. 1) from saline and RH-treated animals (n = 4/group). Top: blot probed with α-SYN antibody; bottom: blot probed with α-actin antibody as a control. Separator lines indicate where the image was cropped to include only the brain areas of interest. B: quantitation of Western blot SYN protein expression. *A statistically significant (P < 0.017) decrease in SYN expression is observed in the PVN following RH. Protein levels are corrected to actin expression. ARC, arcuate nucleus of the hypothalamus; D/V, combined DMH/VMH.
DISCUSSION
In the present study, we have begun to address the hypothesis that RH leads to a change in transcription of regulatory proteins that may decrease the responsiveness of key CNS regions that mediate the neuroendocrine response. We evaluated one aspect of regulatory protein transcription: changes in the expression of the IEG FosB following RH compared with SH or saline-infused controls. Here we present several novel findings. Relative to saline-infused controls, FosB expression is increased in the PVN, VMH, and DMH hypothalamic subnuclei of rats that experienced multiple bouts of hypoglycemia. We also observed significant FosB expression in the THPVP, a limbic structure that modulates the neuroendocrine response to recurrent or multiple stressors (e.g., see Ref. 9). This is consistent with its importance in modulating the CRR to hypoglycemia, as we and others have recently documented (1, 4). Whereas studies in models of seizure and Parkinson's disease (2, 13, 44) implicate a role for FosB expression in the amygdala and striatum, our finding represents the first report, to our knowledge, of expression of FosB isoforms in the PVN and in response to a metabolic stressor. Collectively, these studies suggest that FosB isoforms may play a role in the development of several neuropathologies, a commonality being substantial insult to the neuron with potential depletion of energy reserves.
Our observation of increased FosB expression in the PVN, a major efferent outflow site that receives multiple inputs from the entire neuraxis, then provided the rationale for examining FosB coexpression with specific neuropeptides, specifically, VP and OT, CRF, and TRH. Although robust expression of all of these neuropeptides in the PVN was observed as expected, our findings demonstrate that a significant proportion of FosB expression occurs within the hypothalamic PVN only in a subpopulation of neurons that express CRF but not VP, OT, or TRH. Furthermore, within the various subcompartments of the parvocellular PVN, we observed FosB expression to be included in ventral, medial, and lateral parvocellular compartments, regions that are reported to send preautonomic projections to the brain stem and spinal cord (12, 48, 54), thus potentially playing a major role in regulating the sympathoadrenal component of the CRR to hypoglycemia.
CRF is a neurotransmitter and hypothalamic neurohormone and functions as a major physiological regulator of the basal and stress-induced release of ACTH, β-endorphin, and other proopiomelanocortin-derived peptides from the pituitary (22, 59). Indeed, CRF appears to play a major role in both the activation and coordination of the stress response at the endocrine, behavioral, autonomic, and possibly even the immune levels (21). There is evidence that the repeated experience of certain stressors results in a process of adaptation such that subsequent exposure to the same stressor produces responses that become smaller and may even disappear. Consistent with this, Ma et al. (43) demonstrated desensitization and a loss in CRF mRNA response following repeated immobilization stress, concomitant with increased arginine VP expression in the parvocellular PVN. Somewhat surprisingly, we did not observe FosB coexpression with VP in our RH model. However, while not directly analogous to their recurrent stress model, the impaired neuroendocrine response to RH does include decreased ACTH release, implicating a change in the CRF-ACTH arm of the CNS response.
The ability of CRF neurons to have experience-specific (RH vs. SH) changes in the expression of a major transcription factor, FosB, is consistent with the reported modification of CRF output in a stressor-dependent manner (8) and, secondarily, with downstream actions of CRF in the CNS. In addition to mediating an acute response to stress, CRF has been shown to mediate long-term impacts of stress in several brain regions. For example, in the hippocampus, CRF signaling via the CRF1 receptor subtype has been reported to modulate dendritic length and branching (16, 55, 56). Thus it is conceivable that CRF-related plasticity may be one cellular mechanism that contributes to the impaired CRR observed in our model of RH.
We speculate that the decreased expression of the synaptic marker SYN specifically in the PVN reflects the initiation of structural changes in association with the impaired CRR response to RH. Functional plasticity in the brain is associated with structural remodeling of synapses (34) and with altered expression or regulation of specific synaptic proteins such as SYN (27). The decrease of SYN expression observed in the PVN supports the idea that initiation of synaptic reorganization there may accompany acute RH (and while we do not address the effect of SH on SYN expression, it is also possible that alterations in SYN expression can occur with SH) and is consistent with reports of decreases in SYN immunoreactivity observed with other models of stress (20, 50). Brief anoxic-hypoglycemic episodes have been reported to affect the structural and functional components of synaptic networks at both the pre- and postsynaptic levels (34). Clinically, hypoglycemia “unawareness” and the blunted epinephrine component of the defective CRR are reversed by 2–3 wk of scrupulous avoidance of hypoglycemia (18). Thus the impaired neuroendocrine CRR is a reversible phenomenon, and it is reasonable to propose that processes of reorganization and plasticity, rather than permanent synaptic or neural destruction per se (45), underlie the impaired CRR to RH.
Local inhibitory GABAergic and local excitatory glutamatergic synaptic inputs to hypothalamic neurosecretory neurons have been reported (10, 11, 28, 29, 30). Both neuroanatomical and electrophysiological studies have confirmed the presence of inhibitory GABAergic inputs to PVN parvocellular neurons (11, 57). Furthermore, in vitro studies have demonstrated that intrinsic GABAergic influences in the PVN impart a tonic inhibitory tone on parvocellular CRF neurons (7, 17, 37). These local networks play an essential role in communicating and integrating extrahypothalamic inputs. We did not observe GAD-positive cell bodies to coexpress FosB. Consistent with other reports in the literature, we found that GAD-positive cell bodies were not located in the PVN proper but appeared to be confined to the immediate perimeter, i.e., the peri-PVN. We observed an intense GAD-positive fiber plexus innervating the PVN and impinging on FosB-expressing cells. Our observation that GAD-positive terminals appear to impinge on FosB-positive cells is suggestive of a potential role for the inhibitory networks in regulating neurons that are engaged in response to hypoglycemia. Within the context of the present study, a potential role for GABAergic circuits projecting to the PVN may include limiting the stress response or modulating it as a function of physiological status. Decreased neuronal responsivity may serve a protective function in preventing the overactivation of specific circuitry or systems.
Taken together, our results suggest that the medial hypothalamic nuclei, particularly the PVN, may be targets for “reprogramming” in association with RH. We are currently exploring the functional significance of FosB expression on the neuroendocrine CRRs. Finally, our findings lay the groundwork for further studies aimed at the identification of specific proteins that decrease responsivity of PVN neurons, ultimately providing insight into the mechanism(s) underlying hypoglycemia-associated autonomic failure (HAAF).
Perspective
The impaired CRR to RH contributes to the syndrome of HAAF in diabetic individuals; it has been identified as a limiting problem in the use of intensive insulin therapy to minimize or prevent the chronic complications of diabetes. Identifying the CNS mechanisms that underlie the impaired CRR, which develops very acutely, is thus an important clinical goal. The current study extends our previous observations of the CNS efferent coordinator of neuroendocrine and autonomic responsivity, the PVN, as a target of RH. The finding of FosB, a conditionally expressed IEG that regulates transcription of a unique set of neuroregulatory proteins, in the PVN suggests that the PVN is not merely a “passive partner” in the CNS changes of RH. Rather, it implies that, within the PVN, adaptation to RH is an active process and may result in local changes of tone and responsivity to this metabolic stressor.
GRANTS
These studies were supported by the Department of Veterans Affairs (VA) Merit Review Program and Research Career Scientist (D. P. Figlewicz Lattemann), the VA Merit Review Entry Program (N. Sanders), and VA support to G. J. Taborsky and C. Wilkinson and by National Institute of Diabetes and Digestive and Kidney Diseases Grants R21-062446 and DK-40963 (S. Al-Noori) and 2 R01 DK-050154-09A1.
Acknowledgments
We thank Drs. Eva Redei, Ann-Judith Silverman, and Wylie Vale for generous gifts of antibodies; Dr. Patti Szot for helpful discussion of the manuscript; and Emily Haines, Libby Colasurdo, and Jen Bennett for excellent technical support with assays and surgical preparations.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Al-Noori S, Sanders NM, Taborsky GJ Jr, Wilkinson C, Figlewicz DP. Acute THPVP inactivation decreases the glucagon and sympathoadrenal responses to recurrent hypoglycemia. Brain Res 1194: 65–72, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersson M, Hilbertson A, Cenci MA. Striatal fosB expression is causally linked with l-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson's disease. Neurobiol Dis 6: 461–474, 1999. [DOI] [PubMed] [Google Scholar]
- 3.Ao Y, Wu S, Go VL, Toy N, Yang H. Maintaining euglycemia prevents insulin-induced Fos expression in brain autonomic regulatory circuits. Pancreas 31: 142–147, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Arbelaez AM, Powers WJ, Videen TO, Price JL, Cryer PE. Attenuation of counterregulatory responses to recurrent hypoglycemia by active thalamic inhibition: a mechanism for hypoglycemia-associated autonomic failure. Diabetes 57: 470–475, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armario A The hypothalamic-pituitary-adrenal axis: what can it tell us about stressors? CNS Neurol Disord Drug Targets 5: 485–501, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Arnhold MM, Wotus C, Engeland WC. Differential regulation of parvocellular neuronal activity in the paraventricular nucleus of the hypothalamus following single vs. repeated episodes of water restriction-induced drinking. Exp Neurol 206: 126–136, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bali B, Kovacs KJ. GABAergic control of neuropeptide gene expression in parvocellular neurons of the hypothalamic paraventricular nucleus. Eur J Neurosci 18: 1518–1526, 2003. [DOI] [PubMed] [Google Scholar]
- 8.Barakat Y, Pape JR, Boutahricht M, El Quezzani S, Alaoui A, Chaigniau M, Tramu G, Magoul R. Immunocytochemical detection of cholecystokinin and corticotrophin-releasing hormone neuropeptides in the hypothalamic paraventricular nucleus of the jerboa (Jaculus orientalis): modulation by immobilisation stress. J Neuroendocrinol 18: 767–775, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Bhatnagar S, Huber R, Nowak N, Trotter P. Lesions of the posterior paraventricular thalamus block habituation of hypothalamic-pituitary-adrenal responses to repeated restraint. J Neuroendocrinol 14: 403–410, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Boudaba C, Schrader LA, Tasker JG. Physiological evidence for local excitatory synaptic circuits in the rat hypothalamus. J Neurophysiol 77: 3396–400, 1997. [DOI] [PubMed] [Google Scholar]
- 11.Boudaba C, Szabo K, Tasker JG. Physiological mapping of local inhibitory inputs to the hypothalamic paraventricular nucleus. J Neurosci 16: 7151–7160, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carrasco M, Portillo F, Larsen PJ, Vallo JJ. Insulin and glucose administration stimulates Fos expression in neurones of the paraventricular nucleus that project to autonomic preganglionic structures. J Neuroendocrinol 13: 339–346, 2001. [DOI] [PubMed] [Google Scholar]
- 13.Cenci MA, Tranberg A, Andersson M, Hilbertson A. Changes in the regional and compartmental distribution of FosB- and JunB-like immunoreactivity induced in the dopamine-denervated rat striatum by acute of chronic l-dopa treatment. Neurosci 94: 515–527, 1999. [DOI] [PubMed] [Google Scholar]
- 14.Chen J, Kelz MB, Hope BT, Nakabeppu Y, Nestler EJ. Chronic Fos-related antigens: stable variants of deltaFosB induced in brain by chronic treatments. J Neurosci 17: 4933–4941, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, Nye HE, Kelz MB, Hiroi N, Nakabeppu Y, Hope BT, Nestler EJ. Regulation of delta FosB and FosB-like proteins by electroconvulsive seizure and cocaine treatments. Mol Pharmacol 48: 880–889, 1995. [PubMed] [Google Scholar]
- 16.Chen Y, Bender RA, Brunson KL, Pomper JK, Grigoriadis DE, Wurst W, Baram TZ. Modulation of dendritic differentiation by corticotropin-releasing factor in the developing hippocampus. Proc Natl Acad Sci USA 101: 15782–15787, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cole RL, Sawchenko PE. Neurotransmitter regulation of cellular activation and neuropeptide gene expression in the paraventricular nucleus of the hypothalamus. J Neurosci 22: 959–969, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cryer PE Mechanisms of sympathoadrenal failure and hypoglycemia in diabetes. J Clin Invest 116: 1470–1473, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cryer PE, Davis SN, Shamoon H. Hypoglycemia in diabetes. Diabetes Care 26: 1902–1912, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Cunha GM, Canas PM, Oliveira CR, Cunha RA. 2006 Increased density, and synapto-protective effect of adenosine A2A receptors upon sub-chronic restraint stress. Neuroscience 141: 1775–1781, 2003. [DOI] [PubMed] [Google Scholar]
- 21.De Souza EB Corticotropin releasing factor and interleukin-1 receptors in the brain-endocrine-immune axis. Role in stress response and infection. In: Corticotropin Releasing Factor and Cytokines: Role in the Stress Response, edited by Tache Y and Rivier C. New York, NY: Ann NY Acad Sci, 1993, p. 9. [DOI] [PubMed]
- 22.De Souza EB, Nemeroff CB. Corticotropin-Releasing Factor: Basic and Clinical Studies of a Neuropeptide. Boca Raton, FL: CRC, 1990.
- 23.Evans SB, Wilkinson CW, Bentson K, Gronbeck P, Zavosh A, Figlewicz DP. PVN activation is suppressed by repeated hypoglycemia but not antecedent corticosterone in the rat. Am J Physiol Regul Integr Comp Physiol 281: R1426–R1436, 2001. [DOI] [PubMed] [Google Scholar]
- 24.Evans SB, Wilkinson CW, Gronbeck P, Bennett JL, Taborsky GJ Jr, Figlewicz DP. Inactivation of the PVN during hypoglycemia partially simulates hypoglycemia-associated autonomic failure. Am J Physiol Regul Integr Comp Physiol 284: R57–R65, 2003. [DOI] [PubMed] [Google Scholar]
- 25.Evans SB, Wilkinson CW, Gronbeck P, Bennett JL, Zavosh A, Taborsky GJ Jr, Figlewicz DP. Inactivation of the DMH selectively inhibits the ACTH and corticosterone responses to hypoglycemia. Am J Physiol Regul Integr Comp Physiol 286: R123–R128, 2004. [DOI] [PubMed] [Google Scholar]
- 26.Fisher LA, Brown MR. Central regulation of stress responses: regulation of the autonomic nervous system and visceral function by corticotrophin releasing factor-41. Baillieres Clin Endocrinol Metab 5: 35–50, 1991. [DOI] [PubMed] [Google Scholar]
- 27.Grillo CA, Pirolli GG, Wood GE, Reznikov LR, McEwen BS, Reagan LP. Immunocytochemical analysis of synaptic proteins provides new insights into diabetes-mediated plasticity in the rat hippocampus. Neuroscience 136: 477–486, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Herman JP, Cullinan WE. Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci 20: 78–84, 1997. [DOI] [PubMed] [Google Scholar]
- 29.Herman JP, Cullinan WE, Morano MI, Akil H, Watson SJ. Contribution of the ventral subiculum to inhibitory regulation of the hypothalamo-pituitary-adrenocortical axis. J Neuroendocrinol 7: 475–482, 1995. [DOI] [PubMed] [Google Scholar]
- 30.Herman JP, Mueller NK, Figueiredo H. Role of GABA and glutamate circuitry in hypothalamo-pituitary-adrenocortical stress integration. Ann NY Acad Sci 1018: 35–45, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Herman JP, Tasker JG, Ziegler DR, Cullinan WE. Local circuit regulation of paraventricular nucleus stress integration: glutamate-GABA connections. Pharmacol Biochem Behav 71: 457–468, 2002. [DOI] [PubMed] [Google Scholar]
- 32.Hope BT, Nye HE, Kelz MB, Self DW, Iadarola MJ, Nakabeppu Y, Duman RS, Nestler EJ. Induction of a long-lasting AP-1 complex composed of altered Fos-like proteins in brain by chronic cocaine and other chronic treatments. Neuron 13: 1235–1244, 1994. [DOI] [PubMed] [Google Scholar]
- 33.Hou XE, Dahlstrom A. Synaptic vesicle proteins and neuronal plasticity in adrenergic neurons. Neurochem Res 25: 1275–1300, 2000. [DOI] [PubMed] [Google Scholar]
- 34.Jourdain P, Nikonenko I, Alberi S, Muller D. Remodeling of hippocampal synaptic networks by a brief anoxia-hypoglycemia. J Neurosci 22: 3108–116, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kiss A, Mikkelsen JD. Oxytocin–anatomy and functional assignments: a minireview. Endocr Regul 39: 97–105, 2005. [PubMed] [Google Scholar]
- 36.Kovacs KJ C-Fos as a transcription factor: a stressful (re)view from a functional map. Neurochem Int 33: 287–297, 1998. [DOI] [PubMed] [Google Scholar]
- 37.Kovacs KJ, Miklos IH, Bali B. GABAergic mechanisms constraining the activity of the hypothalamo-pituitary-adrenocortical axis. Ann NY Acad Sci 1018: 466–476, 2004. [DOI] [PubMed] [Google Scholar]
- 38.Lechan RM, Fekete C. The TRH neuron: a hypothalamic integrator of energy metabolism. Prog Brain Res 153: 209–235, 2006. [DOI] [PubMed] [Google Scholar]
- 39.Leuba G, Walzer C, Vernay A, Carnal B, Kraftsik R, Piotton F, Marin P, Bouras C, Savioz A. Postsynaptic density protein PSD-95 expression in Alzheimer's disease and okadaic acid induced neurite retraction. Neurobiol Dis 30: 408–419, 2008. [DOI] [PubMed] [Google Scholar]
- 40.Levin BE, Routh VH, Kang L, Sanders NM, Dunn-Meynell AA. Neuronal glucosensing: what do we know after 50 years? Diabetes 53: 2521–2528, 2004. [DOI] [PubMed] [Google Scholar]
- 41.Lu B, Chow A. Neurotrophins and hippocampal synaptic transmission and plasticity. J Neurosci Res 58: 76–87, 1999. [PubMed] [Google Scholar]
- 42.Lukic D, Haldar J. Isotonic and hypertonic saline act as stressful stimuli for oxytocinergic system of the pituitary, hypothalamus and spinal cord. Life Sci 53: 579–584, 1993. [DOI] [PubMed] [Google Scholar]
- 43.Ma XM, Levy A, Lightman SL. Emergence of an isolated arginine vasopressin (AVP) response to stress after repeated restraint: a study of both AVP and corticotropin-releasing hormone messenger ribonucleic acid (RNA) and heteronuclear RNA. Endocrinology 138: 4351–4357, 1997. [DOI] [PubMed] [Google Scholar]
- 44.Madsen TM, Bolwig TG, Mikkelsen JD. Differential regulation of c-Fos and FosB in the rat brain after amygdala kindling. Cell Mol Neurobiol 26: 87–100, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McEwen BS, Magarinos AM. Stress and hippocampal plasticity: implications for the pathophysiology of affective disorders. Hum Psychopharmacol 16: S7–S19, 2001. [DOI] [PubMed] [Google Scholar]
- 46.Niimi M, Sato M, Tamaki M, Yoshinaru W, Takahara J, Kawanishi K. Induction of Fos protein in the rat hypothalamus elicited by insulin-induced hypoglycemia. Neurosci Res 23: 361–364, 1995. [DOI] [PubMed] [Google Scholar]
- 47.Paxinos G, Watson C. Atlas of the Rat Brain in Stereotaxic Coordinates (4th ed.). San Diego, CA: Academic, 1998.
- 48.Portillo F, Carrasco M, Vallo JJ. Hypothalamic neuron projection to autonomic preganglionic levels related with glucose metabolism: a fluorescent labelling study in the rat. Neurosci Lett 210: 197–200, 1996. [DOI] [PubMed] [Google Scholar]
- 49.Raju B, Arbelaez AM, Breckenridge SM, Cryer PE. Nocturnal hypoglycemia in type 1 diabetes: an assessment of preventive bedtime treatments. J Clin Endocrinol Metab 91: 2087–2092, 2006. [DOI] [PubMed] [Google Scholar]
- 50.Sandi C, Davies HA, Cordero MI, Rodriguez JJ, Popov VI, Stewart MG. Rapid reversal of stress induced loss of synapses in CA3 of rat hippocampus following water maze training. Eur J Neurosci 17: 2447–2456, 2003. [DOI] [PubMed] [Google Scholar]
- 51.Shibata M, Fujihara H, Suzuki H, Ozawa H, Kawata M, Dayanithi G, Murphy D, Ueta Y. Physiological studies of stress responses in the hypothalamus of vasopressin-enhanced green fluorescent protein transgenic rat. J Neuroendocrinol 19: 285–292, 2007. [DOI] [PubMed] [Google Scholar]
- 52.Singh P, Heera PK, Kaur G. Expression of neuronal plasticity markers in hypoglycemia induced brain injury. Mol Cell Biochem 247: 69–74, 2003. [DOI] [PubMed] [Google Scholar]
- 53.Song Z, Routh VH. Recurrent hypoglycemia reduces the glucose sensitivity of glucose-inhibited neurons in the ventromedial hypothalamus nucleus. Am J Physiol Regul Integr Comp Physiol 291: R1283–R1287, 2006. [DOI] [PubMed] [Google Scholar]
- 54.Swanson LW, Sawchenko PE. Paraventricular nucleus: a site for the integration of neuroendocrine and autonomic mechanisms. Neuroendocrinol 31: 410–417, 1980. [DOI] [PubMed] [Google Scholar]
- 55.Swinny JD, Metzger F, Ijkema-Paassen J, Gounko NV, Gramsbergen A, van der Want JJ. Corticotropin-releasing factor and urocortin differentially modulate rat Purkinje cell dendritic outgrowth and differentiation in vitro. Eur J Neurosci 19: 1749–1758, 2004. [DOI] [PubMed] [Google Scholar]
- 56.Swinny JD, Valentino RJ. Corticotropin-releasing factor promotes growth of brain norepinephrine neuronal processes through Rho GTPase regulators of the actin cytoskeleton in rat. Eur J Neurosci 24: 2481–2490, 2006. [DOI] [PubMed] [Google Scholar]
- 57.Tasker JG, Dudek FE. Local inhibitory synaptic inputs to neurones of the paraventricular nucleus in slices of rat hypothalamus. J Physiol 469: 179–192, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teves D, Videen TO, Cryer PE, Powers WJ. Activation of human medial prefrontal cortex during autonomic responses to hypoglycemia. Proc Natl Acad Sci USA 101: 6217–6221, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science 213: 1394–1397, 1981. [DOI] [PubMed] [Google Scholar]
- 60.van Dijk G, Donahey JC, Thiele TE, Scheurink AJ, Steffens AB, Wilkinson CW, Tenenbaum R, Campfield LA, Burn P, Seeley RJ, Woods SC. Central leptin stimulates corticosterone secretion at the onset of the dark phase. Diabetes 46: 1911–1914, 1997. [DOI] [PubMed] [Google Scholar]
- 61.Varea E, Castillo-Gomez E, Gomez-Climent MA, Blasco-Ibanez JM, Crespo C, Martinez-Guijarro FJ, Nachr J. Chronic antidepressant treatment induces contrasting patterns of synaptophysin and PSA-NCAM expression in different regions of the adult rat telencephalon. Eur Neuropsychopharmacol 17: 546–557, 2007. [DOI] [PubMed] [Google Scholar]