

Abstract
Protein kinase C (PKC) signal transduction pathways have been implicated in mechanisms of synaptic plasticity and learning, however, the roles of the different PKC family isoforms remain to be clarified. Previous studies showed that NMDAR-mediated trafficking of GluR4-containing AMPARs supports conditioning and that the mitogen-activated protein kinases (MAPKs) have a central role in the synaptic delivery of GluR4 subunits. Here, an in vitro model of classical conditioning was used to assess the role of PKC isoforms in mechanisms underlying this form of learning. We show that the PKC antagonists chelerythrine and bisindolylmaleimide I attenuated conditioned response (CR) acquisition and expression, as did the PKCζ pseudosubstrate peptide inhibitor ZIP. Analysis of protein expression revealed that PKCζ is activated in early stages of conditioning followed shortly afterward by increased levels of PKCα/β and activation of ERK MAPK. Data also suggest that PKCζ is upstream from and activates ERK. Finally, protein localization studies using confocal imaging indicate that inhibitors of ERK, but not PKC, suppress colocalization of GluR1 with synaptophysin while inhibitors of PKC and ERK attenuate colocalization of GluR4 with synaptophysin. Together, these data suggest that acquisition of conditioning proceeds by two stages of AMPAR trafficking. The first is PKC-independent and ERK-dependent synaptic delivery of GluR1 subunits to activate silent synapses. This is followed by PKC-dependent and ERK-dependent synthesis and delivery of GluR4 subunits that supports the acquisition of CRs. Therefore, there is a selective role for PKC and MAPK signaling pathways in multistep AMPAR trafficking that mediates acquisition of classical conditioning.
Keywords: AMPAR trafficking, PKC, ERK, eyeblink conditioning, turtle
Introduction
Protein kinase C (PKC) is composed of a family of serine-threonine kinases that consist of three major classes and at least 11 different isoforms (Sossin, 2007). Conventional PKCs (α, βI, βII, and γ) are a major intracellular target of calcium and diacylglycerol (DAG) while novel PKCs (δ, ε, η, θ, and μ) are activated by DAG but not by calcium. Atypical PKCs (ζ, and λ), on the other hand, do not bind calcium or DAG but are instead regulated by the phosphoinositide 3 (PI3)-kinase signaling pathway. PKC functions in mechanisms underlying synaptic plasticity and learning but the exact nature of its role is not clearly defined. PKC is important in mechanisms of hippocampal LTP (Malinow et al., 1989; Klann et al., 1993; Wikstrom et al., 2003; Boehm et al., 2006) and is also involved in fear (Weeber et al., 2000) and eyeblink classical conditioning (Sunayashiki-Kusuzaki et al., 1993; Van der Zee et al., 1997). The PKCs appear to function both pre-and postsynaptically in synapse modification. For example, PKC rapidly phosphorylates Munc 18-1 upon depolarization which is necessary for synaptic vesicle release and short-term plasticity (Wierda et al., 2007). PKC signaling is also implicated in the postsynaptic trafficking and synaptic incorporation of AMPARs during hippocampal LTP and enhanced synaptic transmission by selective phosphorylation of AMPAR subunits (Boehm et al., 2006; Ling et al., 2006; Gomes et al., 2007). However, it is unknown if all PKC isoforms are activated during synaptic plasticity and whether they serve independent functions related to specific forms of plasticity. Evidence from Aplysia suggests that two different PKC isoforms selectively underlie short-term or intermediate-term forms of synaptic facilitation (Zhao et al., 2006).
Evidence using an in vitro model of eyeblink classical conditioning suggests that acquisition of conditioned responses (CRs) is associated with the synaptic insertion of AMPARs containing GluR4 subunits (Keifer, 2001; Mokin and Keifer, 2004; Mokin et al., 2006). In place of tone and airpuff stimuli as used in behaving animals, weak electrical stimulation of the auditory nerve (the “tone” conditioned stimulus; CS) is paired with strong stimulation of the trigeminal nerve (the “airpuff” unconditioned stimulus; US) and results in a neural correlate of conditioned eyeblink responses recorded from the abducens nerve (Keifer, 2003). Synaptic incorporation of GluR4-containing AMPARs during conditioning is NMDAR-dependent and regulated by the ERK and p38 mitogen-activated protein kinase (MAPK) signaling pathways (Keifer et al., 2007). Studies further show that synaptic incorporation of GluR1 subunits precede GluR4 to activate silent synapses in the initial stages of conditioning (Mokin et al., 2007). In addition, presynaptic alterations occur during conditioning involving enhanced expression of synaptophysin (Mokin and Keifer, 2004; Mokin et al., 2006). PKC, due in part to its high sensitivity to intracellular calcium concentration, is thought to be a major coordinator of synaptic modifications. Here, we examined the role of PKC isoforms PKCα/β and PKCζ in this in vitro form of associative learning. The results reveal coordinated regulation of PKC isoforms and ERK MAPK during the early stages of conditioning. Blockade of these pathways attenuates synaptic insertion of GluR4-containing AMPARs, the conditioning-related enhancement of synaptophysin, and acquisition of CRs. However, insertion of GluR1 subunits is unaffected by PKC inhibitors but is suppressed by inhibition of MEK-ERK signaling. These data indicate a selective role for PKC and MAPK signaling pathways in AMPAR trafficking and acquisition of classical conditioning.
EXPERIMENTAL PROCEDURES
Conditioning procedures
Freshwater pond turtles Pseudemys scripta elegans obtained from commercial suppliers were anesthetized by hypothermia until torpid and decapitated. Protocols involving the use of animals complied with the guidelines of the National Institutes of Health and the Institutional Animal Care and Use Committee. This model utilizes a brain stem preparation comprised of the pontine blink circuitry (Zhu and Keifer, 2004). The brain stem was transected at the levels of the trochlear and glossopharyngeal nerves and the cerebellum was removed as described previously (Anderson and Keifer, 1999). Therefore this preparation consisted only of the pons with the cerebellar circuitry removed. The brain stem was continuously bathed in physiological saline (2-4 ml/min) containing (in mM): 100 NaCl, 6 KCl, 40 NaHCO3, 2.6 CaCl2, 1.6 MgCl2 and 20 glucose, which was oxygenated with 95% O2/5%CO2 and maintained at room temperature (22-24° C) at pH 7.6 (Anderson and Keifer, 1999). Suction electrodes were used for stimulation and recording of cranial nerves. The US was an approximately two-times threshold single shock stimulus applied to the trigeminal nerve; the CS was a subthreshold 100 Hz, 1 sec train stimulus applied to the ipsilateral posterior root of the eighth nerve that was below the threshold amplitude required to produce activity in the abducens nerve (Keifer et al., 1995; Anderson and Keifer, 1999; Keifer, 2001). The latter nerve will be referred to as the auditory nerve as it carries predominantly auditory fibers. Neural activity was recorded from the ipsilateral abducens nerve that projects to the extraocular muscles controlling movements of the eye, nictitating membrane, and eyelid. The CS-US interval was 20 ms which is defined as the time between the offset of the CS and the onset of the US. This brief trace delay interval was found to be optimal for conditioning, however, conditioning is not supported using longer trace intervals (Keifer, 2001). The intertrial interval between the paired stimuli was 30 sec. A pairing session consisted of 50 CS-US presentations (taking 25 min in duration) followed by a 30 min rest period in which there was no stimulation (Keifer et al., 1995). Conditioned responses were defined as abducens nerve activity that occurred during the CS and exceeded an amplitude of double the baseline recording level. Conditioned preparations were those that received paired CS-US stimulation whereas pseudoconditioned control preparations received the same number of CS and US exposures that were explicitly unpaired using a CS-US interval randomly selected between 100 ms and 25 sec. Two stages of conditioning have been defined previously (Mokin et al., 2007) and are referred to here. The early stage of conditioning occurs during pairing sessions 1-2 when CRs undergo acquisition; the late stage of conditioning includes session 3 and those afterward when CR expression is at asymptotic levels.
Pharmacology
The selective and membrane-permeable PKC inhibitors chelerythrine chloride (Che; 35 μM), bisindolylmaleimide I (Bis, also known as GF109203X; 15 μM; Calbiochem, San Diego, CA), or the selective PKCζ pseudosubstrate peptide inhibitor ZIP (myristoylated, 5 μM; Biosource, Camarillo, CA) were dissolved in physiological saline and perfused through the bath to test for the function of PKC in conditioning. Additionally, the MEK inhibitor PD98059 which inhibits the ERK MAPK pathway (50 μM) or the p38 MAPK inhibitor SB203580 (200 nM; Calbiochem, San Diego, CA) were used. Finally, wortmannin, a cell-permeable inhibitor of PI3-kinase (400 nM to 200 nM, see Results; Calbiochem) was also used. In some experiments, drug application was performed prior to the beginning of conditioning and continued throughout the conditioning procedure to test for the effects of drug treatment on acquisition of abducens CRs. In other experiments, preparations underwent conditioning in normal physiological saline for two pairing sessions and were tested for drug effects on expression of CRs thereafter. Drug was then washed from the bath and conditioning resumed in normal saline to test for CR recovery. In experiments using ZIP or wortmannin, fresh compound was prepared and added to the bath after two sessions of testing for ZIP and every 30 min for wortmannin.
Western blot analysis and coimmunoprecipitation
Turtle brain stems were pseudoconditoned, conditioned, or treated with drug and frozen in liquid nitrogen immediately following the physiological experiments and stored at -70°C. Tissue was homogenized in lysis buffer (20 mM Tris pH 7.5, 1 mM EDTA, 1% NP40, 0.15 M NaCl, 10 mM Na4P2O7, 5% glycine) with a protease (Roche, Germany) and phosphatase inhibitor cocktail (Sigma), rotated at 4° C for 2 hr, centrifuged at 14,000 g for 20 min at 4°C, and the supernatants aliquoted and stored at -70° C. Protein sample concentrates were solubilized in 2 × SDS/β - mercaptoethanol and boiled for 3 min before separation by 10% SDS–PAGE. After electrophoresis, membranes were blocked with 5% nonfat dry milk in TBS/0.1% Tween-20 for 1 h at room temperature. The membranes were incubated with primary antibodies directed against protein for PKC isoforms (see Results below). We also used phosphorylation site-directed antibodies against p38 MAPK thr180/tyr182 (p-p38; Cell Signaling Technology, Danvers, MA) and ERK thr183/tyr185 (p-ERK; Promega, Madison, WI), or for total protein we used p38 (t-p38; Cell Signaling) and ERK (t-ERK; Chemicon). Membranes were incubated with primary antibodies overnight at 4° C, washed, and incubated with HRP-conjugated (1:10,000) or fluorescently tagged secondary antibodies (1:5,000) for 1 h at room temperature. Loading controls were performed using primary antibodies to actin (1:500, Chemicon). Proteins were detected using the ECL-Plus chemiluminescence system (Amersham Pharmacia, Piscataway, NJ) or the Odyessy infrared imaging system (Li-Cor Biotechnology, Lincoln, NE).
Immunoreactive signals were captured on Kodak X-omatic AR film and quantified by computer-assisted densitometry. Optical densities of the bands were determined relative to background levels. Quantification of total protein was determined relative to actin whereas phosphoprotein was determined relative to total protein for the same experiments. Ratios of total protein/actin or phosphoprotein/total protein were obtained for each experiment and averaged. Data are displayed as a percentage of normalized values from pseudoconditioned controls (n’s for each group, or number of preparations, are given in the text). For the coimmunoprecipitation experiments, brain stem preparations were homogenized in lysis buffer with a protease and phosphatase inhibitor cocktail. Protein G-Agarose beads slurry (Calbiochem) was added to cell lysates and incubated on ice for 2 hr. Samples were spun at 6,000 × g for 5 min at 4° C, the supernatant retained, and incubated with primary antibodies (same antibodies as used for western blotting) or nonspecific rabbit IgGs (Santa Cruz) at 4° C for 4 h. Agarose beads slurry was then added to the protein samples and incubated at 4°C overnight. After incubation, beads were washed three times with lysis buffer, solubilized in 2 × SDS-β-mercaptoethanol and boiled for 5 min to elute the proteins. Eluates were separated by 10% SDS-PAGE and then transferred to PVDF membranes. The membranes were blocked with 5% non-fat dry milk, incubated with primary antibodies overnight, washed and incubated in HRP-conjugated secondary antibodies. Immunoreactivity was visualized by chemiluminescence. These experiments were each repeated at least three times on different preparations.
Glutamate receptor localization, confocal imaging and data analysis
Immediately after the physiological experiments, brain stems were immersion fixed in cold 0.5% paraformaldehyde (Mokin and Keifer, 2004). Tissue sections were cut at 30 μm and preincubated in primary antibody overnight at 4° C with gentle shaking. The primary antibodies used were a polyclonal antibody raised in goat that recognizes the GluR4 subunit of AMPARs (1:100, Santa Cruz 7614), a polyclonal raised in rabbit that recognizes GluR1 (1:100, Chemicon 1504), and a monoclonal antibody raised in mouse that recognizes synaptophysin (1:1000, Sigma 5768, St. Louis, MO). The specificity of the antibodies was confirmed by Western blot. After the primary antibodies, sections were rinsed and incubated with secondary antibodies for 2 h using a concentration of 1:100 for GluR4 and GluR1, or 1:200 for synaptophysin. The secondary antibodies were a Cy3-conjugated goat anti-rabbit IgG for GluR1, a Cy3-conjugated rabbit anti-goat IgG for GluR4, and a Cy2-conjugated goat anti-mouse IgG for synaptophysin (Jackson ImmunoResearch, West Grove, PA) that were used to visualize the primary antibodies. After incubation in the secondary antibodies, sections were rinsed, mounted on slides and coverslipped. Images of labeled neurons in the principal or accessory abducens motor nuclei were obtained using an Olympus Fluoview 500 laser scanning confocal microscope. Tissue samples were scanned using a 60× 1.4 NA oil immersion objective with dual excitation using a 488 nm argon laser and a 543 nm HeNe laser. Quantification of punctate staining of at least twofold greater intensity above background was performed independently by two investigators using stereological procedures (Mokin and Keifer, 2006) with MetaMorph software (Universal Imaging Corp., Downington, PA). Images of two consecutive optical sections were taken using confocal microscopy. Protein puncta were counted in one optical section (sample section) if they were not present in the optical section immediately below the sample section (look-up section), and if they were within the inclusion boundaries of the unbiased counting frame. Colocalized staining indicating the presence of glutamate receptor subunits at synaptic sites was determined when red and green puncta were immediately adjacent to one another or if they were overlapping. Data were analyzed using StatView software (SAS, Cary, NC) by ANOVA.
RESULTS
Effects of PKC inhibitors on in vitro classical conditioning
The effect of selective PKC antagonists on the acquisition and expression of in vitro abducens classical conditioning was examined and these data are summarized in Figs. 1 and 2. Results using the PKC inhibitors Che and Bis are shown in Fig. 1. Representative abducens nerve recordings show an abducens nerve CR (Fig. 1A, arrow) followed by the UR recorded prior to drug application (Normal). Application of Che blocked the expression of CRs while leaving the UR unaffected (Fig. 1A; Che). Wash-out of drug resulted in recovery of CRs (Wash, arrow). Acquisition curves of the mean percent (± SD) of abducens CRs following drug treatment during the acquisition and expression phases of conditioning are shown (Fig. 1B,C). Application of Che prior to the beginning of conditioning resulted in no CR acquisition (Fig. 1B; n = 5). This compound also significantly attenuated CR expression to an average of 6 ± 6% CRs when applied for two pairing sessions after acquisition had been obtained (Fig. 1B; n = 7, P < 0.0001). The effect of Che was readily reversible. Note that in initial studies, application of 20 μM Che failed to significantly attenuate CRs once acquired (n = 3). Thereafter, the concentration of 35 μM was found to be effective for this preparation. Application of Bis, another selective PKC antagonist, similarly attenuated the acquisition and expression of abducens CRs. Treatment with Bis at the beginning of conditioning resulted in no CR acquisition (Fig. 1C; n = 5) and CR expression was significantly attenuated to an average of 13 ± 14% CRs after three pairing sessions (Fig. 1C; n = 8, P < 0.0001). This effect on CRs was also readily reversible. Additionally, we examined the function of PKCζ in conditioning. Application of the selective PKCζ pseudosubstrate inhibitor ZIP resulted in failure of CR acquisition in four sessions (Fig. 2B; n = 5). In experiments in which CRs had been recorded prior to drug application, ZIP also significantly attenuated CR expression (Fig. 2B; n = 5, P = 0.02, comparison of session 2 with 3-4), however, this effect reversed after prolonged exposure to ZIP. These findings are illustrated in the physiological recordings shown in Fig. 2A in which CRs recovered in the presence of ZIP later in the conditioning procedures. This effect was not likely due to degradation of the peptide inhibitor as it was changed to fresh compound after two sessions of application. Since PKCζ is activated downstream of PI3-kinase, we further tested the antagonist wortmannin in order to confirm the involvement of PKCζ in conditioning. Application of wortmannin completely inhibited acquisition of conditioning (200 nM; Fig. 2C; n = 3) and blocked CR expression (Fig. 2C; n = 3, P < 0.0001), an effect that was usually irreversible (i.e., 400 nM wortmannin failed to wash in one case and 200 nM washed in one of two cases). These pharmacological findings demonstrate that PKC supports both the acquisition and expression of in vitro abducens conditioning.
Fig. 1.

The PKC inhibitors Che and Bis attenuate acquisition and expression of conditioning. (A) Physiological records of abducens nerve recordings taken from one experiment in which the compound Che blocked expression of CRs. The traces show an abducens nerve CR (arrow) followed by the UR recorded in the second pairing session prior to drug application (Normal). Record obtained during application of Che in the fourth pairing session in which the expression of CRs was blocked but the UR was largely unaffected (Che). Wash-out of the drug in the seventh session resulted in re-expression of CRs (arrow, Wash). The CS and US are indicated at bottom. (B,C) Acquisition curves (means ± SD) of the percent of abducens CRs following Che or Bis treatment at the beginning of conditioning to test for acquisition (left panel) or after CRs had been obtained to test for CR expression (right panel). (B) Application of Che (35 μM) resulted in no acquisition and blocked expression of CRs. (C) Application of Bis (15 μM) also blocked acquisition and significantly attenuated CR expression. * indicate significant differences from session 2. P values are given in the text.
Fig. 2.

The selective PKCζ pseudosubstrate peptide inhibitor ZIP attenuates CR acquisition and expression, as does the PI3-kinase inhibitor wortmannin. (A) Physiological records of abducens nerve recordings prior to and during application of ZIP (5 μM) show suppressed expression of CRs (arrows) during early application. (B) Acquisition curves (means ± SD) show that ZIP significantly attenuated CR acquisition (left panel) and expression (right panel). However, the effect of ZIP on CR expression was reversed after prolonged exposure to the drug. (C) Bath application of wortmannin (200 nM; Wortm) also severly attenuated the acquisition and expression of CRs. * indicate significant differences from session 2. P values are given in the text.
PKC isoforms are differentially regulated during in vitro classical conditioning
The antibodies directed against total and phosphorylated PKC isoforms used in the present study were tested for their specificity by Western blot analysis of turtle and rat brain tissue (Fig. 3). The PKC antibody against total protein (t-PKC; Santa Cruz, 10800) recognizes all classical forms of PKC and identified a single band in both species at ~80 kDa. A phosphorylation site-directed antibody that recognizes PKCα/βII at thr638/641 (p-PKCα/β) resulted in a single band in rat and turtle at ~80 kDa (Cell Signaling, 9375). To examine the PKCζ isoform, an antibody to total protein (Zymed, 38-0100) was used that recognizes two bands in both rat and turtle, PKCζ at ~70 kDa (t-PKCζ, shown in Fig. 3) and PKMζ at ~55 kDa (not shown; Naik et al., 2000), although it is likely that this antibody also recognizes PKCλ. The data in the present study are from analysis of the t-PKCζ band since there were no conditioning-related changes in the t-PKMζ band (see below). Finally, a phosphorylation site-directed antibody that recognizes PKCζ/λ at thr410/403 (p-PKCζ/λ; Cell Signaling, 9378) demonstrated one band in both species at ~75 kDa. To further confirm the specificity of the p-PKCζ/λ antibody, the antigen also coimmunoprecipitated with t-PKCζ (Fig. 3B). Therefore, these antibodies demonstrated similar specificity in turtle brain compared to rat and were used for examination of in vitro classical conditioning.
Fig. 3.
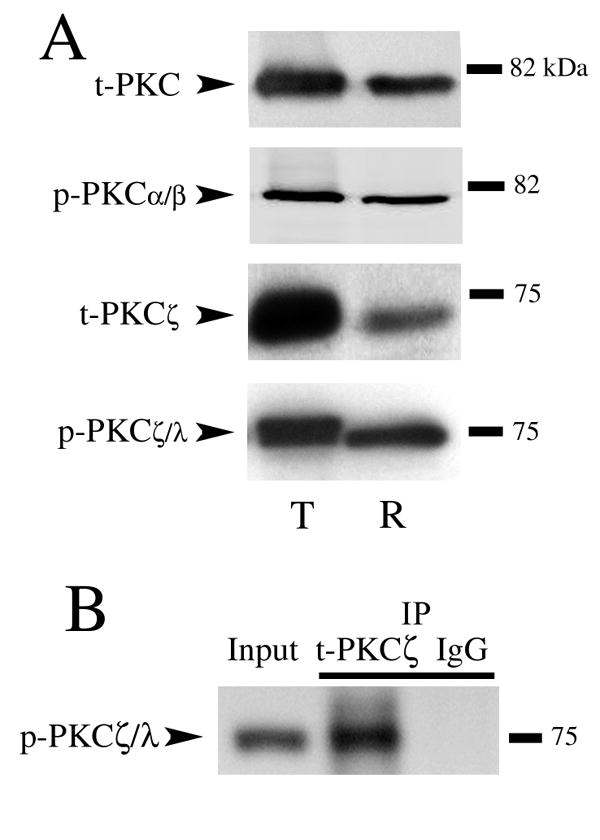
Western blots demonstrating specificity of the antibodies directed against PKC isoforms used in the present study. (A) Antibodies were tested on naive brain tissue from turtle (T) or rat (R). Bands appeared at the expected molecular weights in both species. See text for details. (B) t-PKCζ coimmunoprecipitated with p-PKCζ/λ supporting the specificity of the phosphoantibody.
Protein analysis of the α/β and ζ PKC isoforms following in vitro conditioning for one (C1), two (C2) or five (C5) pairing sessions and after pharmacological blockade of CRs using PKC inhibitors Che or Bis (for a total of five pairing sessions) was performed and these data are summarized in Fig. 4 (n = 4/group for A and B). The levels of both t-PKC and p-PKCα/β were significantly increased after conditioning for two and five pairing sessions above pseudoconditioned levels (Fig. 4A; t-PKC, P < 0.05; p-PKCα/β, P < 0.01). However, the ratio of phosphorylated to total protein across conditioning did not change (Fig. 4A; P = 0.78). Treatment of preparations by either Che or Bis, which blocked CR expression (to 4 ± 7% CRs and 8 ± 8% CRs, respectively), attenuated the conditioning-induced increases in both total and phosphorylated protein to pseudoconditioned levels (Che: t-PKC, P = 0.53, p-PKCα/β, P = 0.47; Bis: t-PKC, P = 0.79, p-PKCα/β, P = 0.11, differences from Ps5). Analysis of the PKCζ isoform showed a different pattern of conditioning-related changes in protein expression (Fig. 4B). While there was no change in the level of t-PKCζ protein across conditioning (Fig. 4B; P = 0.84), or in t-PKMζ (not shown; P = 0.85; phosphorylated PKMζ was not examined), there was a dramatic increase in p-PKCζ/λ, and therefore in the ratio of phosphorylated to total protein, after two pairing sessions of conditioning compared to pseudoconditioning (P < 0.0001). This was followed by significantly enhanced but slightly lower levels of protein phosphorylation later in conditioning after five sessions (P = 0.0002). Neither Che nor Bis significantly affected the enhanced phosphorylated state of PKCζ/λ protein even though CR expression was completely blocked (Che, p/t-PKCζ/λ: P = 0.72; Bis, p/t-PKCζ/λ: P = 0.52, differences from C5). These findings suggest that PKCα/β is regulated differently during the early stages of conditioning compared to PKCζ as both total and phosphorylated forms of PKCα/β are increased while only the phosphorylated form of PKCζ is enhanced. Moreover, PKCζ activation is not sufficient for conditioning as CRs are blocked during application of Che and Bis which do not affect PKCζ.
Fig. 4.

Differential regulation of PKCα/β and PKCζ protein during early and late stages of conditioning and effects of treatment by PKC inhibitors Che and Bis. Blots and quantitative data are shown for phospho-and total protein during one (C1), two (C2), or five (C5) pairing sessions of conditioning compared to pseudoconditioned controls (Ps). Western blots were also assessed following blockade of CRs by application of Che or Bis. (A) Data show significant increases in both p-PKCα/β and t-PKC after two and five sessions of conditioning. However, the ratio of phospho-to total protein did not change. The enhanced levels of PKC protein were attenuated by both Che and Bis. (B) There is a dramatic increase in the amount of p-PKCζ/λ, but not in t-PKCζ, after two pairing sessions of conditioning. These levels are maintained at significantly elevated values after five sessions, and are unaffected by the compounds Che or Bis. P values are given in the text.
Timing of PKC and MAPK protein phosphorylation during conditioning
To obtain a clearer picture of the timing of protein phosphorylation during conditioning we examined PKCα/β and PKCζ after one pairing session (or 25 minutes after conditioning stimulation onset), 15 min into the first rest period (C140; 40 min total), 30 min into the rest period (C155; 55 min), and after completion of two (80 min) or five pairing sessions (4 hr). Additionally, expression of the ERK and p38 MAPKs was determined as ERK has been previously shown to be activated early and p38 late in conditioning (Keifer et al., 2007). Among these groups there was significant CR expression only after two (mean of 86% CRs) and five (100%) pairing sessions. The Western blot analysis is summarized in Fig. 5 (n = 4/group in AD). Blots for total and phosphoprotein are shown while the ratio of protein is plotted. As described above, levels of both t-PKC and p-PKCα/β were increased significantly after two and five pairing sessions compared to pseudoconditioned levels, as can be seen in the blots shown in Fig. 5A (for C2 and C5). Protein ratio, however, did not change (P = 0.84). These findings suggest that levels of PKCα/β is increased at least 80 min after conditioning stimulation onset. Further scrutiny of the timing of protein phosphorylation consistently revealed increased p-PKCζ/λ after the C140 time point (40 min after stimulation) but not after C1 (Fig. 5B; P < 0.001). Examination of the MAPKs confirmed previous findings (Keifer et al., 2007) that p-ERK is maximal early in conditioning after C2 and declines (Fig. 5C; P = 0.02, C2 vs. Ps2) whereas p-p38 is activated late in conditioning after C5 (Fig. 5D; P = 0.005, C5 vs. Ps2). Therefore, these data reveal a pattern of protein expression during conditioning in which p-PKCζ/λ is activated early in conditioning about 40 minutes after stimulation onset, followed shortly thereafter by increases in p-PKCα/β and p-ERK. p-p38 MAPK is activated in the later stage of conditioning.
Fig. 5.

Characterization of the timing of PKC and MAPK protein expression during early conditioning revealed by Western blots. Blots for phospho-and total protein are shown while quantitative data for the ratio of phospho-to total protein is plotted. (A) Both p-PKCα/β and t-PKC increase at C2 and C5 but the ratio of protein expression is not enhanced. (B) Expression of p-PKCζ/λ increases dramatically at time point C140 (40 min). (C) Levels of p-ERK MAPK increase significantly at C2 and decline at C5. (D) Levels of phophorylated p38 MAPK increase late in conditioning at C5. C140, 15 min into the first rest period; C155, 30 min into the first rest period. P values are given in the text.
PKCζ activates ERK in early conditioning
It is recognized that PKC regulates MAPK activation in a variety of cell types. Evidence in our model system suggests that PKCζ regulates ERK early in conditioning and these data are shown in Fig. 6 (n = 4/group in A-C). Western blotting confirmed that application of the PKCζ inhibitor ZIP during two sessions of conditioning significantly dampened the activation of p-PKCζ/λ to pseudoconditioned levels (Fig. 6A; P = 0.25, ZIP vs. Ps2). Additionally, Che (or Bis) applied for two sessions had no significant effect on p-PKCζ/λ (Fig. 6A; P = 0.82, Che2 vs. C2). Our results consistently showed that activation of p-ERK after conditioning was significantly attenuated by ZIP (Fig. 6B; P = 0.36, ZIP2 vs. Ps2), however, Che, which inhibits p-PKCα/β, did not affect p-ERK (Fig. 6B; P = 0.001, Che2 vs. Ps2). Together, these data suggest that inhibition of p-PKCζ/λ, but not PKCα/β, suppresses p-ERK. There was no effect of ZIP or Che on expression of phosphorylated or total p-38 MAPK tested after five sessions of conditioning when p-38 is normally expressed (not shown; P = 0.11, ZIP5 vs. C5; P = 0.77, Che5 vs. C5). Moreover, application of the selective MEK-ERK and p38 MAPK inhibitors PD98059 or SB203580 failed to have an effect on the phosphorylation of PKCζ/λ (Fig. 6C; P = 0.60, PD and SB vs. C5) suggesting that ERK (or p38) MAPK does not activate PKCζ. Next, we performed coimmunoprecipitation experiments of PKC isoforms with ERK (Fig. 6D). The results showed that t-PKCζ, p-PKCζ/λ, as well as t-PKMζ strongly coimmunoprecipitated with t-ERK whereas t-PKC did not. Finally, PI3-kinase is an upstream activator of PKCζ. To confirm that PKCζ activates ERK, an inhibitor of PI3-kinase, wortmannin (200 nM), was tested (Fig. 7). The results from Western blot show that wortmannin application for two pairing sessions significantly attenuated the conditioning-related increase in phosphorylation of PKCζ/λ (Fig. 7A; n = 3, P = 0.0001, Wortm vs. C2), but did not affect p-PKCα/β (n = 3, P = 0.18; not shown). Furthermore, wortmannin also inhibited the activation of p-ERK (Fig. 7B; n = 3, P < 0.0001, Wortm vs. C2). Taken together, these data indicate that PI3-kinase-mediated phosphorylation of PKCζ is upstream from and activates p-ERK during conditioning in this preparation.
Fig. 6.

Evidence that PKCζ is upstream from and actives p-ERK during in vitro conditoning. (A) ZIP applied for two pairing sessions (ZIP2) at the beginning of conditioning suppresses expression of p-PKCζ/λ to pseudoconditioned levels compared to conditioning. (B) Application of ZIP also suppresses p-ERK expression, while the PKC antagonist Che does not. (C) The ERK and p38 MAPK antagonists PD98059 (PD) and SB203580 (SB), respectively, fail to affect levels of p-PKCζ/λ compared to conditioning. (D) Coimmunoprecipitation experiments reveal that t-PKCζ, p-PKCζ/λ, and PKMζ interact with t-ERK while t-PKC does not. * indicate significant differences from Ps. P values are given in the text.
Fig. 7.

The PI3-kinase inhibitor, wortmannin, inhibits phosphorylation of PKCζ/λ and ERK. (A) The conditioning-related increase seen in p-PKCζ/λ at C2 is signficantly attenuated by application of wortmannin (200 nM) for two sessions. (B) Application of wortmannin also significantly inhibits the phosphorylation of ERK. * Indicates significant differences from Ps2 (P = 0.0001); # indicates significant differences from C2 (P < 0.0001).
Presynaptic effects of PKCζ and ERK inhibitors
Protein localization studies of abducens motor neurons using immunocytochemistry were undertaken to determine pre-and/or postsynaptic effects of PKC and ERK inhibitors in the early stages of conditioning. In all of these immunocytochemical experiments (n = 3 preparations/group; 50-100 neurons for Syn, and 30-40 neurons each for GluR1 and GluR4 were analyzed), preparations treated with Che, ZIP or PD98059 for two pairing sessions showed no acquisition of CRs. Previous studies found that the presynaptic protein synaptophysin was significantly enhanced after in vitro conditioning (Mokin and Keifer, 2004; Mokin et al., 2006; Mokin et al., 2007). These findings were confirmed as shown in the confocal images from conditioned and pseudoconditioned preparations (Fig. 8A, compare C2 to Ps2) and in the quantitative data in Fig. 8B (Syn). Application of the general PKC inhibitor Che for two sessions failed to have an effect on the conditioning-related increase in synaptophysin (Fig. 8A,B; P = 0.20, Che2 vs. C2). On the other hand, application of the PKCζ inhibitor ZIP significantly attenuated this response compared to conditioned preparations (Fig. 8A,B; P < 0.0001, ZIP2 vs. C2). Similarly, the MEK-ERK antagonist PD98059 also significantly dampened synaptophysin expression (Fig. 8A,B; P < 0.0001, PD2 vs C2). These data suggest that PKCζ and its downstream target ERK are involved in presynaptic mechanisms of conditioning as assessed by expression of synaptophysin.
Fig. 8.

Colocalization studies show that synaptic incorporation of GluR4 AMPAR subunits is attenuated by inhibitors of PKC and ERK while GluR1 is attenuated by inhibitors of ERK. (A) Confocal images of selected abducens motor neurons from the different experimental groups showing punctate staining for synaptophysin (Syn), and colocalization of GluR4 subunits (GluR4 + Syn) and GluR1 subunits (GluR1 + Syn) with synaptophysin. Colocalization of AMPARs (red) with Syn (green) is defined by overlapping (yellow) or adjacent puncta and are indicated by the arrowheads. (B) Quantitative analysis of Syn, GluR4 + Syn, and GluR1 + Syn punctate staining for the different experimental groups is plotted. Preparations conditioned for two pairing sessions (C2) showed significantly greater punctate staining for Syn, GluR4 + Syn, and GluR1 + Syn compared to the pseudoconditioned group (Ps2). Treatment with the PKC inhibitor Che for two sessions (Che2) had no effect on the conditioning-induced increase in Syn or GluR1 + Syn, but significantly reduced GluR4 + Syn colocalization. Application of the PKCζ blocker ZIP for two sessions (ZIP2) significantly attenuated Syn and GluR4 + Syn, but not GluR1 + Syn. The MEK-ERK antagonist PD98059 (PD2) suppressed punctate staining for Syn, and colocalization of GluR4 + Syn and GluR1 + Syn. * indicate significant differences from Ps2. Scale bar = 2 μm.
PKC regulation of synaptic GluR4, but not GluR1, AMPAR subunits during acquisition
Conditioning in this preparation proceeds in two stages, first by synaptic insertion of GluR1-containing AMPARs followed by GluR4 subunits which are hypothesized to support CR acquisition (Mokin et al., 2007). Here, colocalization studies of GluR1 or GluR4 subunits with synaptophysin was performed to assess a role for PKC and ERK in early stages of AMPAR trafficking. Quantitative analysis of punctate staining showed that all three compounds tested during the first two pairing sessions, Che, ZIP and PD98059, significantly attenuated the colocalization of GluR4 subunits with synaptophysin compared to conditioned preparations as illustrated in the confocal images shown in Fig. 8A and summarized in the quantitative data in Fig. 8B (GluR4 + Syn, P < 0.0001, Che2, ZIP2 and PD2 vs. C2). This finding corresponded with the expression of 0% CRs in all three groups tested with drug. Additionally, the ratio of GluR4 in relation to synaptophysin puncta was unaltered in the ZIP and Che treated groups compared to conditioning (P = 0.96, ZIP2, and P = 0.59, Che2 vs. C2) and averaged about 1. However, the PD group was significantly lower (P = 0.002, PD2 vs C2) at about 0.5 having fewer GluR4 puncta. Analysis of the colocalization of GluR1 AMPAR subunits with synaptophysin showed that the enhanced levels of synaptic GluR1 subunits after early conditioning were not dampened by either of the PKC inhibitors Che or ZIP (Fig. 8A,B; P = 0.98, ZIP2, and P = 0.25, Che2 vs. C2) but was considerably reduced by the MEK-ERK antagonist PD98059 (P = 0.004, PD2 vs. C2). No significant changes in the ratio of GluR1 to synaptophysin puncta occured in the drug-treated groups compared to conditioning (P = 0.09). Therefore, these colocalization studies examining the early stages of conditioning indicate that PKC and ERK inhibitors attenuate the synaptic localization of GluR4-containing AMPARs, but only the MEK-ERK and not the PKC inhibitors affect the synaptic localization of GluR1 subunits.
DISCUSSION
Specificity of pharmacological compounds in turtle brain
Data suggest that the PKC antagonist Che inhibits all classes of PKC isoforms in mammalian brain, particularly at higher concentrations (Sossin, 2007). While Che does block the classical PKCs in this preparation, it fails to significantly inhibit protein phosphorylation levels of the atypical PKCζ/λ, at least as determined by antibodies that recognize this site. On the other hand, the compound Bis was found to inhibit p-PKCα/β and not the atypical PKCs as expected, and the pseudosubstrate peptide inhibitor ZIP also selectively blocked the atypical PKCs. Phosphorylation of PKCζ/λ at the site recognized by the antibodies used here is accomplished by 3-phosphoinositide-dependent protein-kinase-1 (PDK-1; Chou et al., 1998). Phosphorylation of this site in turtle is not inhibited by Che, but is blocked by ZIP, possibly by suppression of the upstream activator of PKCζ, PDK-1. Since PDK-1 can directly activate ERK through phosphorylation of MEK (Sato et al., 2004), this could also explain why ZIP resulted in inhibition of ERK. Our data showed that the PI3-kinase blocker wortmannin suppressed phosphorylation of PKCζ/λ and ERK, as well as conditioning. Therefore, the function of the PI3-kinase/PDK-1 signaling cascade in conditioning needs to be more clearly defined, as does the effect of Che and ZIP on PKCζ activity levels in this preparation.
Role of PKC in synaptic plasticity and conditioning
Our results show that PKC is required for an in vitro form of classical conditioning and conditioning-related AMPAR trafficking. Protein kinase inhibitors directed against PKC isoforms significantly attenuated both the acquisition and expression of CRs during conditioning. Evidence suggests that PKCζ is activated early in conditioning after the first pairing session followed shortly thereafter by increased levels of p-PKCα/β and p-ERK MAPK, and are maintained in that state throughout conditioning. Evidence further suggests that PKCζ activates ERK in this preparation while PKCα/β likely does not because ZIP and wortmannin, but not Che, inhibits phosphorylation of ERK. Synaptic incorporation of GluR4-containing AMPARs, which supports the acquisition and expression of conditioning (Keifer, 2001; Mokin and Keifer, 2004; Mokin et al., 2006), is also severely attenuated by PKC and MEK-ERK inhibitors. Thus, the coordinated actions of PKC and ERK MAPK (Keifer et al., 2007) have a major part in generating classical conditioning. Studies indicate that PKC has a significant role in the generation and maintenance of other forms of synaptic plasticity and learning. While PKC is clearly implicated in behavioral models of learning such as eyeblink (Sunayashiki-Kusuzaki et al., 1993; Van der Zee et al., 1997) and fear (Weeber et al., 2000) conditioning, comparatively little is known about the signaling pathways activated by this family of kinases in these forms of learning. In contrast, PKC has been shown to be important in LTP of hippocampal pyramidal neurons (Malinow et al., 1989; Klann et al., 1993; Wikstrom et al., 2003) and is a key factor in the synaptic incorporation of GluR1 AMPAR subunits during LTP by phosphorylation of ser818 (Boehm et al., 2006). Similar to the present findings on conditioning, PKC is activated shortly after tetanic stimulation and is maintained in that state with a time course similar to LTP (Klann et al., 1993). It is also recognized that a persistently activated form of PKCζ, PKMζ, contributes to maintenance of late-phase LTP (Serrano et al., 2005) at least in part by increasing the number of synaptic AMPARs (Ling et al., 2006). In Aplysia cell cultures, two different PKC isoforms that are either calcium-dependent or independent, Apl I and II, respectively, activate selective signaling pathways to mediate short-term and intermediate-term forms of synaptic plasticity (Zhao et al., 2006; Sossin, 2007). There also appears to be persistent activation of a PKC isoform in intermediate-term facilitation in Aplysia, Apl III, that may function similar to a PKM-like form of PKC (Sossin, 2007). Therefore, in addition to our own findings, studies indicate the importance of PKC isoforms in specific types of plasticity and learning, and in AMPAR subunit trafficking.
Our results using the atypical PKC inhibitor ZIP differ from those examining late-phase hippocampal LTP and long-term memory of conditioned taste aversion in rat cortex (Serrano et al., 2005; Shema et al., 2007). In those studies, ZIP had no effect on induction of early LTP or CR acquisition but irreversibly blocked late LTP and erased taste memory. However, in the present study, ZIP inhibited CR acquisition and had only transient effects on CR expression. Although ZIP blocks the atypical PKCs, we do not know if it inhibits activated PKMζ in this preparation as it does in mammalian tissue. However, lack of effect on PKMζ would not explain the inhibition of acquisition observed here. It is also likely that the reversible effects of ZIP on conditioning are related, at least in part, to involvement of multiple protein kinase signaling pathways (Wikstrom et al., 2003) that may have compensatory effects when the atypical PKCs are inhibited.
PKCζ activation of ERK MAPK
The PKCs regulate expression of the MAPKs in many cell types. PKCζ has been shown to activate ERK in several types of non-neuronal cells (e.g., Monick et al., 2000), however, there are very few studies of neurons. Using cultured embryonic hippocampal cell lines, Corbit et al. (2000) provided evidence that epidermal growth factor (EGF)-induced ERK MAPK activation is mediated by PKCζ through MEK. Evidence suggested that this function of PKCζ might be Raf-1-independent and act on MEK directly. Here, we show that the PKCζ pseudosubstrate peptide inhibitor ZIP, as well as wortmannin, an inhibitor of IP3-kinase, suppresses the phosphorylation of ERK when applied during two pairing sessions of conditioning. The general PKC antagonist Che, which does not significantly affect p-PKCζ/λ in our preparation, fails to attenuate p-ERK. Evidence also suggests that p-ERK does not feed back onto PKCζ because application of the MEK antagonist PD98059 does not affect phosphorylation of PKCζ/λ. Moreover, both t-PKCζ and p-PKCζ/λ coimmunoprecipitate with t-ERK while t-PKC, which recognizes conventional isoforms, does not. Together, these data support a signal transduction pathway during in vitro conditioning in which PKCζ activates MEK directly or through Ras/Raf to potentiate the function of p-ERK. We can not at this time, however, exclude the function of PI3-kinase/PDK-1 signaling in this process. The exact role of PKCζ in conditioning is currently unclear, especially since our data suggest it remains phosphorylated under conditions in which CRs are blocked (i.e., Che and Bis application). Other signaling pathways also activate ERK during conditioning in this preparation, such as PKA and BDNF, and therefore PKCζ is not required for this. Interestingly, the immunocytochemistry indicated that the atypical PKC inhibitor ZIP attenuated synaptophysin punctate staining as well as synaptic GluR4-containing AMPAR insertion, suggesting that PKCζ may have both pre- and postsynaptic effects in conditioning.
Signaling pathways for AMPAR trafficking during acquisition of conditioning
In a previous study (Mokin et al., 2007) we showed that acquisition of classical conditioning in this preparation proceeded in two stages of AMPAR trafficking. The first occurs very early in conditioning during the first pairing session, prior to CR acquisition, and involves synaptic insertion of GluR1 subunits in order to activate silent synapses. This phase is followed by replacement of GluR1 subunits with GluR4-containing AMPARs which parallels the acquisition of CRs. By examination of the early stages of CR acquisition here, we further show that synaptic incorporation of GluR1 subunits is attenuated by an inhibitor of the MEK-ERK pathway (PD98059), while GluR4-containing AMPARs are suppressed by inhibitors of both PKC (Che and ZIP) and MEK-ERK. Based on our findings, we propose a two stage model for the acquisition phase of in vitro classical conditioning. The first stage of acquisition (during pairing session 1) is initiated shortly after the onset of paired CS-US stimulation, prior to CR acquisition, and involves synaptic insertion of GluR1-containing AMPARs in order to activate silent synapses (Mokin et al., 2007). This process does not involve protein synthesis of GluR1 but is instead implemented by translocation of existing GluR1-containing AMPARs (Mokin et al., 2007). This stage does not require PKC but is ERK-dependent, as indicated by the inhibition of GluR1 synaptic localization by the MEK-ERK antagonist PD98059 (Fig. 8). However, since ZIP blocked ERK effectively and yet does not block GluR1 recruitment, there is some question as to whether synaptic GluR1 is exclusively ERK-dependent. There is a basal level of ERK expression in this preparation (Keifer et al., 2007), and data shown here indicate that low levels of p-ERK remain after both ZIP and wortmannin. One possibility is that there is conditioning-induced compartmentalization of Raf-MEK-ERK signaling to synapses conveying the CS that is not sensitive to inhibitors of PKC but is affected by MEK-ERK blockers (Shalin et al., 2006). Alternatively, an ERK-independent pathway may also be able to increase synaptic GluR1. This pathway may include PKA, CaMKII, or an interplay of both. Recent evidence suggests that the PKA inhibitor Rp-cAMPs suppresses recruitment of GluR1 (Zheng and Keifer, 2008). The second stage of acquisition (during pairing session 2 when CRs are expressed) involves NMDAR-dependent synaptic incorporation of GluR4-containing AMPARs which undergo protein synthesis (Keifer, 2001; Keifer and Clark, 2003; Mokin and Keifer, 2004). This stage is PKC-dependent as well as ERK-dependent. Since both Che and ZIP suppress synaptic localization of GluR4 subunits as well as CR acquisition, this process must utilize multiple PKC isoforms including the calcium-sensitive conventional and calcium-insensitive atypical PKCs. However, expression of PKCζ is not sufficient for conditioning as it may be activated in conditions in which CRs are not recorded, such as during Che or Bis inhibition. Other PKC isoforms not examined in the present study may also play a role in conditioning. For example, data suggest that PKCγ interacts directly with GluR4 subunits and functions in their surface expression (Correia et al., 2003; Gomes et al., 2007). Since the compounds Che and Bis both have strong effects in blocking conditioning it is likely that several of the conventional and novel PKC isoforms are involved. Parallel to the synthesis of GluR4 subunits, we have consistently observed increased synthesis of the presynaptic proteins synaptophysin and synapsin I (Mokin and Keifer, 2004; Mokin et al., 2006; Keifer et al., 2007). This indicates that there are coordinate presynaptic mechanisms in conditioning that may function in synaptogenesis or enhanced presynaptic release (Jovanovic et al., 2000; Antonova et al., 2001). Our immunocytochemical findings show that both ZIP and PD98059, but not Che, attenuate the conditioning-related increase in synaptophysin suggesting it may be mediated by PKCζ-MEK-ERK signaling pathways.
Neither GluR1 nor GluR4 AMPAR subunits are known to be directly phosphorylated by ERK MAPK. Each subunit, however, has sites for direct phosphorylation by PKA, PKC and CaMKII that can affect trafficking function (Carvalho et al., 1999; Derkach et al., 2007). What then are the mechanisms that incorporate them into synapses through ERK during conditioning? Data from Zhu et al. (2002) using hippocampal slices established that MEK-ERK mediated signaling pathways controlled through the GTPase Ras regulate the delivery of synaptic AMPARs. The exact nature of these pathways and the substrates for ERK are unknown but they are likely to be mediated in coordination with a number of other regulatory proteins. Control of receptor trafficking involves synaptic delivery, stabilization, and removal mechanisms (Derkach et al., 2007). We have interpreted the effects of PKC and ERK kinase antagonists on AMPAR localization to be the result of inhibition of receptor insertion. Alternatively, these observations could be viewed as facilitation of AMPAR withdrawal (Kim et al., 2005). Interestingly, and consistent with our model, bath application of BDNF alone activates ERK and results in synaptic insertion of both GluR1 and GluR4 AMPAR subunits (Li and Keifer, 2008). Among the primary differences between our findings and those from studies of LTP is that GluR1 synaptic insertion does not appear to be mediated by PKC. This is surprising as phosphorylation by PKC is strongly implicated in synaptic GluR1 delivery during LTP (Boehm et al., 2006). Our results emphasize that while the details of plasticity mechanisms may be specific to different systems or forms of learning, some general principles emerge. One of these is convergent evidence for multistep trafficking of AMPARs during plasticity and learning.
Acknowledgments
We thank Drs. Frances Day for assistance with the confocal microscopy and Brian Burrell for comments on the manuscript. Supported by National Institutes of Health Grants NS 051187 and P20 RR 015567 which is designated as a Center of Biomedical Research Excellence (COBRE) to J. K.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson CW, Keifer J. Properties of conditioned abducens nerve responses in a highly reduced in vitro brain stem preparation from the turtle. J Neurophysiol. 1999;81:1242–1250. doi: 10.1152/jn.1999.81.3.1242. [DOI] [PubMed] [Google Scholar]
- Antonova I, Arancio O, Trillat AC, Wang HG, Zablow L, Udo H, Kandel ER, Hawkins RD. Rapid increase in clusters of presynaptic proteins at onset of long-lasting potentiation. Science. 2001;294:1547–1550. doi: 10.1126/science.1066273. [DOI] [PubMed] [Google Scholar]
- Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Carvalho AL, Kameyama K, Huganir RL. Characterization of phosphorylation sites on the glutamate receptor 4 subunit of the AMPA receptors. J Neurosci. 1999;19:4748–4754. doi: 10.1523/JNEUROSCI.19-12-04748.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou MM, Hou W, Johnson J, Graham LK, Lee MH, Chen CS, Newton AC, Schaffhausen BS, Toker A. Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Curr Biol. 1998;8:1069–1077. doi: 10.1016/s0960-9822(98)70444-0. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Soh JW, Yoshido K, Eves EM, Weinstein IB, Rosner MR. Different protein kinase C isoforms determine growth factor specificity in neuronal cells. Mol Cell Biol. 2000;20:5392–5403. doi: 10.1128/mcb.20.15.5392-5403.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia SS, Duarte CB, Faro CJ, Pires EV, Carvalho AL. Protein kinase Cγ associates directly with the GluR4 α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor subunits. J Biol Chem. 2003;278:6307–6313. doi: 10.1074/jbc.M205587200. [DOI] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Gomes AR, Correia SS, Esteban JA, Duarte CB, Carvalho AL. PKC anchoring to GluR4 AMPA receptor subunit modulates PKC-driven receptor phosphorylation and surface expression. Traffic. 2007;8:259–269. doi: 10.1111/j.1600-0854.2006.00521.x. [DOI] [PubMed] [Google Scholar]
- Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci. 2000;3:323–329. doi: 10.1038/73888. [DOI] [PubMed] [Google Scholar]
- Keifer J. In vitro eye-blink classical conditioning is NMDA receptor dependent and involves redistribution of AMPA receptor subunit GluR4. J Neurosci. 2001;21:2434–2411. doi: 10.1523/JNEUROSCI.21-07-02434.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keifer J. In vitro classical conditioning of the turtle eyeblink reflex: approaching cellular mechanisms of acquisition. Cerebellum. 2003;2:55–61. doi: 10.1080/14734220310015610. [DOI] [PubMed] [Google Scholar]
- Keifer J, Armstrong KE, Houk JC. In vitro classical conditioning of abducens nerve discharge in turtles. J Neurosci. 1995;15:5036–5048. doi: 10.1523/JNEUROSCI.15-07-05036.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keifer J, Clark TG. Abducens conditioning in in vitro turtle brain stem without cerebellum requires NMDA receptors and involves upregulation of GluR4-containing AMPA receptors. Exp Brain Res. 2003;151:405–410. doi: 10.1007/s00221-003-1494-5. [DOI] [PubMed] [Google Scholar]
- Keifer J, Zheng Z, Zhu D. MAPK signaling pathways mediate AMPA receptor trafficking in an in vitro model of classical conditioning. J Neurophysiol. 2007;97:2067–2074. doi: 10.1152/jn.01154.2006. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A-and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Klann E, Chen SJ, Sweatt JD. Mechanism of protein kinase C activation during the induction and maintenance of long-term potentiation probed using a selective peptide substrate. Proc Natl Acad Sci. 1993;90:8337–8341. doi: 10.1073/pnas.90.18.8337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Keifer J. Coordinate action of pre- and postsynaptic BDNF is required for AMPAR trafficking and acquisition of in vitro classical conditioning. Neuroscience. 2008;155:686–697. doi: 10.1016/j.neuroscience.2008.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling DSF, Bernardo LS, Sacktor TC. Protein kinase Mζ enhances excitatory synaptic transmission by increasing the number of active postsynaptic AMPA receptors. Hippocampus. 2006;16:443–452. doi: 10.1002/hipo.20171. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Mokin M, Keifer J. Targeting of GluR4-containing AMPA receptors to synaptic sites during in vitro classical conditioning. Neurosci. 2004;128:219–28. doi: 10.1016/j.neuroscience.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Mokin M, Keifer J. Quantitative analysis of immunofluorescent punctate staining of synaptically localized proteins using confocal microscopy and stereology. J Neurosci Meth. 2006;157:218–224. doi: 10.1016/j.jneumeth.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Mokin M, Lindahl JS, Keifer J. Immediate-early gene-encoded protein Arc is associated with synaptic delivery of GluR4-containing AMPA receptors during in vitro classical conditioning. J Neurophysiol. 2006;95:215–224. doi: 10.1152/jn.00737.2005. [DOI] [PubMed] [Google Scholar]
- Mokin M, Zheng Z, Keifer J. Conversion of silent synapses into the active pool by selective GluR1-3 and GluR4 AMPAR trafficking during in vitro classical conditioning. J Neurophysiol. 2007;98:1278–1286. doi: 10.1152/jn.00212.2007. [DOI] [PubMed] [Google Scholar]
- Monick MM, Carter AB, Flaherty DM, Peterson MW, Hunninghake GW. Protein kinase C ζ plays a central role in activation of the p42/44 mitogen-activated protein kinase by endotoxin in alveolar macrophages. J Immunol. 2000;165:4632–4639. doi: 10.4049/jimmunol.165.8.4632. [DOI] [PubMed] [Google Scholar]
- Naik MU, Benedikz E, Hernandez I, Libien J, Hrabe J, Valsamis M, Dow-Edwards D, Osman M, Sactor TC. Distribution of protein kinase Mζ and the complete protein kinase C isoform family in rat brain. J Comp Neurol. 2000;426:243–258. doi: 10.1002/1096-9861(20001016)426:2<243::aid-cne6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T. Involvement of 3-phosphoinositide-dependent protein kinase-1 in the MEK/MAPK signal transduction pathway. J Biol Chem. 2004;279:33759–33767. doi: 10.1074/jbc.M402055200. [DOI] [PubMed] [Google Scholar]
- Serrano P, Yao Y, Sacktor TC. Persistent phosphorylation by protein kinase Mζ maintains late-phase long-term potentiation. J Neurosci. 2005;25:1979–1984. doi: 10.1523/JNEUROSCI.5132-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalin SC, Hernandez CM, Dougherty MK, Morrison DK, Sweatt JD. Kinase suppressor of Ras1 compartmentalizes hippocampal signal transduction and subserves synaptic plasticity and memory formation. Neuron. 2006;50:765–779. doi: 10.1016/j.neuron.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Shema R, Sacktor TC, Dudai Y. Rapid erasure of long-term memory associations in the cortex by an inhibitor of PKMζ. Science. 2007;317:951–953. doi: 10.1126/science.1144334. [DOI] [PubMed] [Google Scholar]
- Sossin WS. Isoform specificity of protein kinase Cs in synaptic plasticity. Learn & Mem. 2007;14:236–246. doi: 10.1101/lm.469707. [DOI] [PubMed] [Google Scholar]
- Sunayashiki-Kusuzaki K, Lester DS, Schreurs BG, Alkon DL. Associative learning potentiates protein kinase C activation in synaptosomes of the rabbit hippocampus. Proc Natl Acad Sci. 1993;90:4286–4289. doi: 10.1073/pnas.90.9.4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Zee EA, Kronforst-Collins MA, Maizels ET, Hunzicker-Dunn M, Disterhoft JF. Gamma isoform-selective changes in PKC immunoreactivity after trace eyeblink conditioning in the rabbit hippocampus. Hippocampus. 1997;7:271–285. doi: 10.1002/(SICI)1098-1063(1997)7:3<271::AID-HIPO3>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Atkins CM, Selcher JC, Varga AW, Mirnikjoo B, Paylor R, Leitges M, Sweatt JD. A role for the beta isoform of protein kinase C in fear conditioning. J Neurosci. 2000;20:5906–5914. doi: 10.1523/JNEUROSCI.20-16-05906.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierda KDB, Toonon RFG, de Wit H, Brussaard AB, Verhage M. Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron. 2007;54:275–290. doi: 10.1016/j.neuron.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Wikstrom MA, Matthews P, Roberts D, Collingridge GL, Bortolotto ZA. Parallel kinase cascades are involved in the induction of LTP at hippocampal CA1 synapses. Neuropharmacol. 2003;45:828–836. doi: 10.1016/s0028-3908(03)00336-8. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Leal K, Abi-Farah C, Mertin KC, Sossin WS, Klein M. Isoform specificity of PKC translocation in living Aplysia sensory neurons and a role for Ca 2+ -dependent PKC APL I in the induction of intermediate-term facilitation. J Neurosci. 2006;26:8847–8856. doi: 10.1523/JNEUROSCI.1919-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Keifer J. Abstract Viewer/Itenerary Planner. Washington, DC: Society for Neuroscience; 2008. PKA has a critical role in synaptic delivery of GluR1-containing AMPARs during the initial stages of acquisition of in vitro classical conditioning. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Keifer J. Pathways controlling trigeminal and auditory nerve-evoked abducens eyeblink reflexes in pond turtles. Brain Behav Evol. 2004;64:207–222. doi: 10.1159/000080242. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]