

Abstract
Epithelial sodium channels (ENaC) play an essential role in maintaining total body fluid and electrolyte homeostasis. As such, abnormal expression of ENaC at the cell surface is linked to several important human diseases. Although the stability of ENaC subunits has been extensively studied by protein biochemical analysis, the half-life of the functional channel in the apical membrane remains controversial. Because the functional stability of the multisubunit channel may be more physiologically relevant than the stability of individual subunit proteins, we performed studies of functional ENaC channels using A6 epithelial cells, a Xenopus laevis distal nephron cell line. We recorded single-channel activity in over 400 cells with the translation blockers cycloheximide (CHX) or puromycin, as well as the intracellular protein trafficking inhibitors brefeldin A (BFA) or nocodazole. Our cell-attached, single-channel recordings allow us to quantify the channel density in the apical membrane, as well as to determine channel open probability (Po) from control (untreated) cells and from cells at different times of drug treatment. The data suggest that the half-life of ENaC channels is ∼3.5 h following puromycin, BFA, and nocodazole treatment. Furthermore, these three drugs had no significant effect on the Po of ENaC for at least 6 h after exposure. A decrease in apical channel number and Po was observed following 2 h of CHX inhibition of protein synthesis, and the apparent channel half-life was closer to 1.5 h following CHX treatment. Treatment of cells with the translation inhibitors does not alter the expression of the protease furin, and therefore changes in protease activity cannot explain changes in ENaC Po. Confocal images show that BFA and nocodazole both disrupt most of the Golgi apparatus after 1-h exposure. In cells with the Golgi totally disrupted by overnight exposure to BFA, 20% of apical ENaC channels remained functional. This result suggests that ENaC is delivered to the apical membrane via a pathway that might bypass the Golgi vesicular trafficking pathway, or that there might be two pools of channels with markedly different half-lives in the apical membrane.
Keywords: cell-attached patch clamp, brefeldin-A, ENaC half-life
in mammals and vertebrates, the tight control of extracellular fluid homeostasis is mainly achieved by controlling salt and water reabsorption in the kidneys. Only 1 or 2% of the filtered load of sodium is reabsorbed in the distal nephron of the kidney. Nonetheless, the distal nephron is the major point for discretionary control of total body sodium balance. In the collecting tubule, the predominant pathway mediating sodium reabsorption is through highly selective epithelial sodium channels (ENaC) (11). Unlike voltage- and ligand-gated channels, epithelial sodium channels (ENaC) are constitutively active channels, and as such, one major mechanism for regulating channel activity is by controlling expression of the channels at the apical membrane.
The number of apical channels is determined by the balance between insertion of new channels into the membrane and the retrieval and degradation of channels from the membrane. The lifetime of ENaC in the apical membrane ultimately determines the rate of sodium reabsorption. Abnormal numbers of apical ENaC (i.e., abnormal membrane stability or lifetime) are related to human inherited diseases, such as Liddle's syndrome (32, 33, 37). This disease is caused by mutations in the β- or γ-ENaC subunit, which increase channel stability and thus result in an overall increase in ENaC-mediated sodium reabsorption.
Highly selective ENaC are composed of structurally conserved α-, β-, and γ-subunits and belong to the ENaC/degenerin family of proteins. It is clear that members of the ENaC/degenerin family share several structural features, such as two transmembrane-spanning domains (TM1 and TM2), intracellular NH2 and COOH termini, as well as a large extracellular domain (20). Recently, the crystal structure of ASIC, an Acid-Sensing Ion Channel member of the ENaC/degenerin family, has been described by Gouaux and colleagues (18) to be trimeric in the desensitized state. Since ENaC and ASICs belong to the same family of channel proteins, the crystal structure of ASIC suggests that ENaC may similarly be composed of three subunits. However, despite the structural composition of ENaC at the cell membrane, several studies have suggested that a mature and functional ENaC channel must undergo several posttranslational modifications, which include formation of intrasubunit disulfide bonds (7, 35), transformation of the N-glycan linkage on ENaC subunits from endoglycosidase H (endo-H)-sensitive to endo-H-insensitive forms (15, 17), as well as proteolytic cleavage of α- and γ-subunits. However, not every ENaC subunit expressed in the apical membrane is fully processed posttranslationally; both mature and immature forms of the sodium channels have been found to coexist in the plasma membrane (16). It has also been reported that the immature channels expressed in the plasma membrane have very low channel activity (5). Since both mature and immature channels have been described in the plasma membrane, perhaps the various different molecular weights for ENaC subunits that have been reported (using similar Western blotting techniques) are due to the differences in subunit maturity and posttranslational processing. Because there is uncertainty about the precise molecular weight of each subunit (based on standard Western blotting techniques), there remain issues related to ENaC processing at the membrane: 1) how posttranslational modifications or proteolytic cleavage can affect apparent subunit weight, 2) the precise stoichiometry of the subunits necessary to form a functional channel, and 3) the stability of membrane surface ENaC. Another reason it is difficult to correlate the biochemical data to the functional stability of ENaC is that several studies suggest that changes in α-, β-, and γ-ENaC subunit levels at the cell surface are not coordinately regulated (28, 40, 41), meaning that the half-life of each subunit may differ from one another. Therefore, it is difficult to accurately correlate the quantitative changes in the expression levels of ENaC (determined using biochemical approaches) to the actual functional stability of ENaC. However, determining the functional stability of ENaC is the most relevant physiological parameter determining distal nephron sodium reabsorption. In attempts to estimate the physiological contribution of the channels and to overcome the technical limitations of biochemical assays, a few groups have measured the decrease in amiloride-sensitive whole-cell current in oocytes or transepithelial current in cultured epithelial cells (9, 27, 36) under experimental conditions which prevent newly synthesized channels from being delivered to the plasma membrane. Posttranslational modifications and trafficking of ENaC in heterologous expression systems sometimes differ from channels in cells expressing endogenous ENaC (12, 41). Furthermore, amiloride may inhibit transporters and channels other than ENaC (10, 11), and therefore the half-life of ENaC observed based solely on the decline of amiloride-sensitive current may not actually reflect the half-life of individual ENaC. Additionally, protein biochemical data suggest that the half-life of apically expressed β-ENaC is much shorter than that of apically expressed α- and γ-ENaC subunits (41). The noncoordinated regulation of ENaC subunits may produce functional channels composed of α alone, or of α- and γ-channels, rather than channels composed of all three α-, β-, and γ-subunits (4, 8). Although the sodium channels composed of the α-subunit only or α- and γ-subunits are inhibited by amiloride, their characteristics are different from the highly Na+-selective ENaC that are believed to be composed of α-, β-, and γ-subunits (4). To overcome the technical limitations described above and to gain a better understanding of ENaC half-life at the apical membrane, we carefully measured single-channel (ENaC) activity using the cell-attached patch-clamp method and applied four different drugs to prevent either new synthesis of channels or intracellular trafficking to the apical membrane after synthesis. Single-channel measurements are particularly useful for this investigation since there can be no ambiguity about which channel we are measuring, and we can separate the effect of the drugs on channel density from those on open probability.
MATERIALS AND METHODS
A6 cell culture preparation.
A6 cells are distal nephron cells that are ideal for studying transepithelial Na transport because they express highly selective epithelial Na channels.
The 2F3 subclone of A6 cells was a gift from Dr. Dale Benos. They were originally derived in the laboratory of Dr. Bernard C. Rossier, University of Lausanne, Switzerland, by functionally selecting clonal A6 cells for a high transepithelial resistance monolayer and responsiveness to aldosterone (39). All of the A6 cells used in this study were this 2F3 subclone. A6 cells were maintained in plastic tissue culture flasks as described previously (1). For single-channel patch-clamp experiments, A6 cells were seeded onto permeable polyester inserts until they reached confluency. A6 cell culture medium was replaced 3 times/wk and consisted of 50/50 Dulbecco's MEM/Ham's F-12 base media (GIBCO, Grand Island, NY), 5% fetal bovine serum (GIBCO), 1.5 μM aldosterone, 1.0% streptomycin, and 0.6% penicillin (Irvine Scientific, Santa Ana, CA) at pH of 7.4. Patch-clamp experiments were carried out using A6 cells between passages 98 and 106.
Single-channel recordings.
The cell-attached configuration was used in all patch-clamp studies. Micropipettes were pulled from filamented borosilicate glass capillaries (TW-150F, World Precision Instruments) with a two-stage vertical puller (Narishige, Tokyo, Japan). The resistances of the pipettes were between 7 and 10 MΩ when filled with and immersed in patch solution containing (in mM) 96 NaCl, 3.4 KCl, 0.8 MgCl2, 0.8 CaCl2, and 10 HEPES, with pH adjusted to 7.4 by NaOH. Single-channel recordings were made from individual cells for ∼8–10 min at pipette holding potentials of 0 or 20 mV.
Data analysis.
Channel currents were recorded at 1 kHz with an Axopatch 1-D amplifier (Molecular Devices) with a low-pass, 100-Hz, eight-pole Bessel filter. Channel activity per patch was determined during an 8- to 10-min recording period. As a measure of epithelial sodium channel activity (NPo), we used pCLAMP 9 software (Molecular Devices). The channel NPo can be calculated from the single-channel record without any assumptions about the total number of channels in a patch or the Po of a single channel using the following relationship
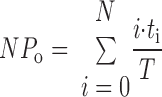 |
where T is the total recording time, i is the number of channels open, and ti is the time during the recording when there were i channels open. The channel density per patch, N, presented in this paper, is defined as the maximum number of unitary current transitions during 8–10 min of single-channel recording and was calculated for all patches including those patches without any observable active channels. We have previously shown that if channels open independently of one another, then the open probability of a single channel (Po) can be calculated by dividing NPo by the number of channels in a patch (26). The Po was calculated only for patches with active channels. For patches with no activity, it is unclear whether there are no channels or there are channels, but with zero open probability.
NPo.
N or Po values reported were averaged before and following various drug-treatment time points. Data are reported as means ± SE. Statistical analysis was performed with SigmaPlot and SigmaStat software (Jandel Scientific). Differences between groups were evaluated with one-way ANOVA with Holms-Sidak posttests, and results were considered significant if P < 0.05.
Drug treatments.
Cycloheximide (CHX; 20 μg/ml) and puromycin (50 μg/ml), drugs which interfere with normal protein synthesis, or brefeldin A (BFA; 300 nM) and nocodazole (20 μM), drugs which inhibit protein trafficking, were added to both the apical and basolateral sides of A6 cells during patch-clamp recordings. In general, cells were not exposed to patch-clamp solution for more than a total of 2 h. Drug treatments that were carried out for >2 h were done so in the presence of cell culture medium to maintain channel activity. All chemicals (except where explicitly mentioned in the paper) were purchased from Sigma.
Protein 35S labeling and analysis.
A6 cells were grown on 12-mm permeable insert supports for 10 days, after which cells were incubated with 0.5 ml cell culture medium containing 20 μCi/ml of 35S-labeled methionine and cysteine in the presence of 20 μg/ml CHX, or 50 and 75 μg/ml puromycin for 30 min. After labeling, the cells were washed three times with ice-cold phosphate-buffered saline (PBS) solution and then lysed with RIPA buffer (containing 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate in PBS). Protein was then precipitated with 10% trichloroacetic acid (TCA) and pelleted by centrifugation at 4°C. The pelleted protein was washed extensively (3 times) using ice-cold acetone and reconstituted in RIPA buffer. The radioactive 35S signal was quantified using liquid scintillation counting.
Western blot detection of furin.
A6 cells were grown on 24-mm permeable inserts for 10 days, after which cells were exposed to CHX or puromycin for 6 h and then homogenized using an Omni Tissue Homogenizer (Omni International, Warrenton, VA) in 20 mM Tris·HCl, 5 mM EGTA, and protease inhibitors, pH 7.4 buffer. Nuclei and intact cells were removed by gentle centrifugation at 1,000 g for 5 min, and the supernatant was centrifuged at 100,000 g for 1 h to obtain a crude membrane pellet, after which the pellet was reconstituted in RIPA buffer and subjected to standard Western blot analysis. Furin was detected by using 1:1,000 dilutions of rabbit anti-furin convertase polyclonal antibody (Abcam). A polyclonal anti-actin antibody used as a loading control (1:1,000) was purchased from Sigma. Western blot images were captured by Kodak Image station 2000MM (Eastman Kodak, Rochester, NY), and protein densities were calculated by Kodak MI SE software.
Golgi apparatus labeling.
After 10 days of culture on permeable supports, A6 cells were treated with 300 nM BFA or 20 μM nocodazole. At 1, 3.5, or 19 h after treatment, the cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. The Golgi apparatus was immunolabeled using a mouse monoclonal antibody against a Golgi marker protein, GM130, at a 1:100 dilution. The secondary antibody conjugated with Alexa Fluor 488, which detects murine IgG, was obtained from Santa Cruz Biotechnology and diluted 1:1,000. The nuclei of A6 cells were stained using Topro3 (Molecular Probes). Cells were mounted onto glass slides with ProLong antifade reagent (Molecular Probes) and imaged using a Zeiss LSM 510 NLO META laser-scanning confocal microscope.
RESULTS
Rate of loss of functional ENaC in A6 cells exposed to translation inhibitor CHX.
To examine the stability of functional ENaC in the apical membrane, we first recorded ENaC activity from A6 cells exposed to CHX for up to 6 h. CHX was added to the cell culture medium, as well as to the recording bath solution on both the apical and basolateral sides of cell-attached permeable supports. The single-channel activities were recorded at a membrane holding potential (−Vpip) of 0 or 20 mV, and the recording duration for each patch was ∼8–10 min. Although the resting potential of A6 cells can vary slightly and result in different single-channel current amplitudes at certain holding potentials (42), all data examined in our studies are of 4- to 5-pS, slowly gating, epithelial Na channels.
Figure 1A, top, shows a representative single-channel recording obtained from a patch on an A6 cell grown on a permeable support (untreated control). Figure 1A, bottom, shows a representative single-channel recording from a patch on an A6 cell treated with 20 μg/ml CHX for nearly 3.5 h. Figure 1B shows the average NPo values, as well as N and Po values, from cells that had been treated with 20 μg/ml CHX for between 1 and 6 h compared with untreated cells. Within the first hour of CHX exposure, the channel activity measured as NPo was 0.427 ± 0.083 (n = 35), which did not differ significantly from untreated cell values of 0.279 ± 0.042 (n = 68). After 1–2 h of CHX exposure, the channel activity substantially decreased to 0.096 ± 0.03 (n = 32), which was ∼30% of the untreated cells' NPo values. ENaC activity decreased further after 3–4 h of CHX treatment. In addition, we formed patches on another 11 cells exposed to CHX for 4–6 h and observed no ENaC activity in these cells. Since the calculation of channel activity (NPo) combines both channel open probability (Po) and channel number (N) in a patch, as previously described by Marunaka and Eaton (26), NPo can be separated into N and Po with the degree of certainty dependent mostly on the total length of the recording period and, to a lesser extent, Po. In general, if the Po is between 0.1 and 0.9, and the recording period is near 10 min, N and Po can be determined with >95% confidence. During 1–2 h of CHX treatment, the N values (1.31 ± 0.231, n = 35) were significantly lower than control cells (2.47 ± 0.16, n = 68) by ∼50% (P = 0.001). The N values decreased further as the drug exposure time increased up to 4 h (Fig. 1C). After treatment for >4 h, ENaC activity was not observed (Fig. 1B). Cells that were exposed to CHX for >1 h also showed changes in Po. The Po of untreated cells was 0.091 ± 0.008 (n = 62), whereas the Po after 1–2 h of CHX exposure decreased significantly to 0.057 ± 0.013 (n = 21, P = 0.03). Po values continued to decrease with longer CHX treatment (Fig. 1D). Data from Fig. 1 show that CHX effectively inhibits both ENaC N and Po values 1–6 h following treatment. In the interpretation of Fig. 1, it may also be important to take into account that the determination of N depends, to an extent, on the value of Po; hence, reduction in the Po may cause an underestimation of the number of channels per patch as well as the mean half-life (see discussion).
Fig. 1.

Effect of cyclohexamide (CHX) on the inhibition of epithelial Na channel (ENaC) activity. A: representative single-channel recordings obtained from A6 cells (control and CHX treatment). The closed state of ENaC is indicated with a “c,” and channel opening(s) are marked with an “o.” As shown in the top panel, there are 3 levels of ENaC openings in Ctrl, while in the bottom panel there is only 1 level of open channel (in 3-h, 20-min CHX exposure). B: average ENaC NPo values (see C and D) from untreated control and CHX-exposed cells between 1 and 6 h. The number heading each column indicates the number of total observations. *P < 0.05, **P < 0.02, ***P < 0.01 compared with control. C and D: average number (N) of channels within a patch and average channel open probability (Po) are independently scrutinized by first determining N and then dividing NPo by the N within each patch for the Po values. The number heading each column in D indicates patches with active channels.
Rate of loss of functional ENaC in A6 cells exposed to the translation inhibitor puromycin.
To show that the observed reduction in ENaC activity following CHX treatment is not attributable to other effects of CHX unrelated to protein synthesis, we next tested the stability of apical ENaC with another protein translation inhibitor, puromycin. The recording and timing of drug exposures analyzed were the same as that described for CHX. In contrast to the effect of CHX, ENaC activity in puromycin-treated cells decreased gradually so that the decrease could only be observed in the cells exposed to puromycin after 4–6 h. After 4–6 h of treatment, ENaC NPo values decreased significantly from initial NPo values of 0.279 ± 0.043 (n = 68) to 0.06 ± 0.02 (n = 18, P = 0.011) (Fig. 2A). Calculation of N and Po show that puromycin decreases ENaC N values without significantly altering the Po following 4–6 h of treatment (Fig. 2, B and C). Our results show that the channel number decreased as the exposure time increased. After 3–4 h of exposure, the number of channels (N) significantly decreased to 0.928 ± 0.438 (n = 14) from control values of 2.471 ± 0.196 (n = 68, P = 0.002). Interestingly, the Po stayed the same in all experiments (maximum: 0.138 ± 0.026; minimum: 0.090 ± 0.021) and did not significantly differ from control values of 0.09 ± 0.01(n = 62) shown in Fig. 2C.
Fig. 2.

Effect of 50 μg/m puromycin on ENaC activity. A–C: ENaC NPo, N, and Po, respectively, following 0–6 h of puromycin treatment. See text for details.
Estimating ENaC half-lives in cells treated with CHX or puromycin.
Since we used similar treatment protocols for CHX and puromycin, we could compare the half-lives of sodium channels in the apical membrane following each drug treatment. To do so, the channel number (N) at different times after each drug treatment was fit to a simple decreasing exponential function. The data points in Fig. 3A are mean values for N at each time point, with the best-fit curve determined from all individual data points. The average half-life of ENaC in CHX-treated cells is 1.45 ± 0.24 h, with an r2 value of 0.81 (Fig. 3A). The average half-life of ENaC in puromycin-treated cells is 3.28 ± 0.89 h, and the r2 value is 0.86 (Fig. 3B). The fitted data suggest that CHX and puromycin affect ENaC turnover time differently, with ENaC having a shorter half-life in CHX-treated cells than in puromycin-treated cells. To be certain that both compounds are effective in inhibiting cellular translational processes, we used 35S-labeled methionine and cysteine incorporation into A6 cells in the presence of CHX and puromycin as an indicator of the efficacy of the protein translation inhibitors (Fig. 4). CHX decreased radioactive incorporation (which is a measure of protein synthesis) at all time points. In the presence of CHX, radioactive incorporation was ∼10% of control (untreated A6 cells) for all cells at times greater than 30 min of radioactive labeling. This is similar to that reported by Butterworth et al. (3) in epithelial MPKCCD cells. The radioactive incorporation of puromycin-treated cells (both 50 or 75 μg/ml) was only ∼50% of control. Figure 4 provides a side-by-side comparison of CHX and puromycin efficacy in A6 epithelia, which has not been previously reported. The results in Fig. 4 seemingly indicate that puromycin is not as effective an inhibitor of translation as CHX. However, these results may simply reflect the fact that these two compounds inhibit protein translation through different mechanisms. Since CHX blocks protein translation initiation, there should be little, if any, radioactively labeled amino acid incorporation into nascent protein compared with puromycin-treated cells. Since puromycin prevents complete protein synthesis by blocking elongation, and thus allows generation of short (yet incomplete) polypeptides that would incorporate 35S, puromycin may be just as effective as CHX in blocking complete protein synthesis (particularly for large proteins like ENaC).
Fig. 3.

Estimation of ENaC half-life based on observed channel N in cell-attached patches following translational inhibition. The channel N observed at various time of drug treatment was fit to a simple exponential decay equation, and the fitted curves were superimposed on the averaged N values shown in Figs. 1C and 2B. A: ENaC half-life from cells exposed to CHX (t1/2 = 1.45 ± 0.24 h). B: ENaC half-life from cells exposed to puromycin (t1/2 = 3.28 ± 0.89 h).
Fig. 4.

Effects of CHX and puromycin on A6 cell global protein synthesis. Confluent A6 cells were fed with 35S-labeled methionine and cysteine in the presence of 20 μg/ml of CHX (•) or 2 different concentrations of puromycin [50 μg/ml (▵); 75 μg/m (○)]. At the indicated time of 35S labeling, protein obtained from control, CHX-, and puromycin-treated cells was precipitated and radioactive 35S incorporation was counted by liquid scintillation. A: relative intensity of radioactive signal from cells sampled at different times of drug treatment. B: enlarged view of the effect of CHX and puromycin on protein synthesis at 5, 10, and 30 min of drug treatment.
Acute inhibition of protein translation does not affect furin expression levels in A6 cells.
Proteases such as furin and prostasin have established roles as ENaC regulators (2, 13, 29, 34). Therefore, protein synthesis inhibitors, like CHX and puromycin, may be altering the level of furin expression in A6 cells and thus altering the protease-mediated level of ENaC activity. This is an important point to consider, since the rapid turnover of a protease could also contribute to the decrease in ENaC NPo observed following CHX or puromycin treatment described above. To address this possibility, we performed Western blot analysis on endogenous furin protein levels in A6 cells following 20 μg/ml CHX or 50 μg/ml puromycin for 6 h. Figure 5A shows that the amount of furin in A6 crude membrane preparations did not change significantly following treatment with protein synthesis inhibitors. The same membrane was stripped and reprobed using an anti-β actin antibody to demonstrate that equal amounts of protein were loaded. Western blot images were captured by Kodak Image station 2000MM (Eastman Kodak), and protein densities were calculated by Kodak MI SE software. This result suggests furin expression levels in A6 cells are not impacted by CHX or puromycin treatment. Therefore, the decrease in ENaC half-life and activity reported is unlikely to be due to changes in proteolytic activation of the channel by furin.
Fig. 5.

Effect of CHX and puromycin on furin expression. A6 cells were grown on permeable supports for 10 days, after which cells were exposed to 20 μg/ml CHX or 50 μg/ml puromycin for 6 h and then homogenized. The crude membrane was collected and reconstituted in RIPA buffer. A: proteins were subjected to standard SDS-PAGE and then transferred onto nitrocellulose membrane and probed with anti-furin (top) and anti-actin (bottom) antibody. B: quantification of normalized furin protein levels (n = 3).
Estimating ENaC half-lives in cells of treated with protein trafficking inhibitors.
We also recorded single-channel activity at the apical membrane while we inhibited newly synthesized protein delivery to the plasma membrane using either BFA, a fungal metabolite which prevents vesicular anterograde transport from the endoplasmic reticulum (ER) to the cis-Golgi, or nocodazole, a specific microtubule inhibitor that prevents microtubular-dependent vesicular trafficking. The decrease in single-channel activity was determined at times up to 22 h after BFA treatment. We observed that the channel activity decreased gradually after BFA exposure (Fig. 6A). Within 2–4 h of BFA treatment, the average channel NPo values had already appeared to decrease to 0.149 ± 0.038 (n = 23) compared with the initial (untreated control) NPo value of 0.279 ± 0.042, (n = 68). A significant difference in channel activity can be seen after 6 h of BFA exposure, and the channel activity decreased by ∼80% after overnight (16–22 h) exposure to BFA. Next, N and Po were determined separately (Fig. 6, B and C). ENaC N values (Fig. 6B) significantly decrease after only 1 h of BFA treatment (and this decrease in N continues for up to 22 h of BFA treatment). Figure 6B shows that the N value in cells treated with BFA for 1–2 h is 1.53 ± 0.43 (n = 15), which is significantly lower than the control N value that started out at 2.47 ± 0.20 (n = 68, P = 0.046). Surprisingly, 20% of channel activity persists even after overnight treatment. Although BFA treatment also affects Po, it did not do so within the first 6 h of treatment when N was decreasing significantly (Fig. 6C). Only after 6–22 h of BFA treatment, channel Po values begin to decrease significantly (Fig. 6C). This implies that the only effect of BFA in the first 6 h is to reduce the number of channels which reach the apical membrane rather than any posttranslational modification that affects ENC Po.
Fig. 6.

Effect of brefeldin A (BFA) on ENaC activity. A–C: NPo, N, and Po of control cells, respectively, following up to 22 h of BFA treatment. All data were calculated in a manner similar to that shown in Fig. 1.
The effect of nocodazole on ENaC activity was similar to the effect of BFA. Nocodazole is a specific microtubule inhibitor which prevents protein delivery from the ER to the plasma membrane by disrupting microtubular structures, and thereby prevents microtubular-dependent vesicle trafficking and causes disassembly of the Golgi apparatus. Thus nocodazole, like BFA, is another useful compound in testing the half-life of functional ENaC. Figure 7A (top) shows a representative recording of ENaC activity in the presence of vehicle alone (0.2% DMSO) and vehicle plus nocodazole (Fig. 7A, bottom). A summary of all observations from DMSO-treated or nocodazole plus DMSO-treated cells on ENaC NPo is shown in Fig. 7, B–D. Figure 7B shows that there was no significant change in ENaC NPo during the initial 0- to 6-h period of nocodazole treatment. There was, however, large inherent variability in ENaC Po. However, independent analysis of changes in average N (Fig. 7C) and average Po (Fig. 7D) values shows significant changes following nocodazole treatment. The average N value of single-channel measurements performed in control cells decreased from 2.542 ± 0.371 (n = 24) to 1.38 ± 0.40 (n = 13), ∼50% of control, after 3–4 h of nocodazole treatment. The number of channels observed in the apical membrane of cells continued to decrease after 4–6 h of nocodazole treatment. In contrast to the gradual reduction of N, Po values showed no significant change among control or cells exposed to nocodazole for any length of time up to 6 h. In Fig. 8, we determined the half-life of ENaC in cells treated with BFA and nocodazole in the same manner as we had previously done in Fig. 3. Again, the data points are mean values for each time point; however, the best-fit curve was determined from all individual data points obtained within the first 6 h of nocodazole treatment. Although we observed the effect of BFA up to 22 h, the exponential fitting of N values was done with data up to 6 h of drug treatment due to significant decreases in ENaC Po following long-term exposure (shown in Fig. 6C). The estimated ENaC half-life in BFA exposed cells was 3.36 ± 0.97 h, with r2 = 0.86 (Fig. 8A). In nocodazole-exposed cells, the half-life of ENaC was 3.77 ± 1.17 h (r2 = 0.97) (Fig. 8B).
Fig. 7.

Effect of nocodazole on ENaC activity. A: representative traces of single-channel activity from a control (DMSO-treated) A6 cell and a 2-h, 55-min nicodazole-exposed cell. B–D: NPo, N, and Po, respectively, of control cells and different times of nocodazole treatment.
Fig. 8.

ENaC channel N, indicative of channel half-life, from cells exposed to BFA and nocodazole. A: ENaC half-life from cells exposed to BFA. The average N presented here is the same N value presented in Fig. 5B (t1/2 = 3.36 ± 0.96 h). B: ENaC half-life from cells exposed to nocodazole. The N value is the same as in Fig. 6C (t1/2 = 3.77 ± 1.17 h).
Effect of BFA and nocodazole on Golgi apparatus.
It has been previously reported that the Golgi apparatus in epithelial Madin-Darby canine kidney (MDCK) cells is resistant to low concentrations of BFA (24), but whether the Golgi of A6 cells is also resistant to BFA is unclear. This question was especially relevant, since, as pointed out above, even after 21 h of BFA treatment, 20% of ENaC channels were still active. Therefore, we labeled Golgi in A6 cells with an antibody to GM 130, a Golgi matrix protein, to compare the effectiveness of BFA and nocodazole in disrupting the A6 Golgi complex. In Fig. 9, the fluorescent green signal is anti-GM 130 binding to the Golgi apparatus with Topro3 (magenta) labeling of the nuclei. Figure 9A is a control and shows that the anti-Golgi antibody recognizes cisternae structures of the Golgi apparatus (green fluorescent structures encircling the Topro-labeled nuclei). Figure 9B shows that there is no nonspecific binding of the secondary anti-murine IgG antibody. Golgi labeling is still visible in most cells after BFA and nocodazole exposure for half an hour (data not shown), but, after 1-h BFA treatment (Fig. 9C) or 1-h nocodazole treatment (Fig. 9D), there are Golgi complexes in some cells, although most Golgi have been disrupted. Our immunohistochemical labeling of the Golgi complex indicates that either 1-h BFA or nocodazole treatment disrupts Golgi complexes to a similar extent. We also examined disruption of Golgi cisternae following 19.5 h of BFA exposure, since our single-channel recordings indicated that ∼20% of channels remained functional even after cells were treated with BFA overnight. Despite this observation, Fig. 9E shows that, in fact, the Golgi apparatus is completely disrupted following overnight BFA treatment.
Fig. 9.

Confocal image of Golgi apparatus affected by BFA and nocodazole: A: Golgi apparatus was labeled using GM130 (primary) mouse monoclonal antibody labeling and subsequent anti-mouse IgG secondary antibody conjugated to Alexa Fluor 488. B: untreated A6 cells were stained with Alexa Fluor 488 conjugated with anti-mouse IgG antibody (2nd antibody control). The nucleus was stained using Topro3. C: disrupted Golgi cisternae structure in A6 epithelial cells following 1-h exposure to 300 nM BFA. D: 1-h exposure to 50 μM nocodazole. E: 19-h exposure to 300 nM BFA.
DISCUSSION
The reported values for the half-life of ENaC in the plasma membrane vary widely from as short as 15 min (6) to over 24 h (41). We have pointed out in the introduction that problems with knowing exactly which posttranslational form of the different ENaC subunits actually forms a functional channel precludes determining the stability of functional ENaC in the apical membrane of native epithelial cells. However, determining the functional stability of ENaC is the most relevant physiological parameter in determining distal nephron sodium reabsorption. In attempts to estimate the physiological contribution of the channels and to overcome the technical limitations of biochemical assays, a few groups have measured the decrease in amiloride-sensitive current under experimental conditions which prevent newly synthesized channels from being delivered to the plasma membrane with either BFA or CHX (9, 27, 36). However, such approaches rely on the ability to pharmacologically separate ENaC from all other transporters and channels. This point is particularly relevant since there have been several reports that amiloride-sensitive channels can be formed from α-subunits alone or α- and γ-subunits. These difficulties can be overcome by measuring the half-life of single 4-pS ENaC in the apical membrane. Single-channel measurements imply that we only examine the half-life of highly-selective, 4-pS channels (and no other channel) and that, unlike measurements of short-circuit current, we can determine the effect of the four different drugs on the density of channels and generally exclude the effect of changes in Po on apical sodium current. For these reasons, in this study, we examined the half-life of functional ENaC in A6 cells by examining the density of single ENaC after exposing cells to four different drugs which either prevented protein synthesis or the trafficking of newly synthesized channels to the plasma membrane. By counting the channel number in patches at different times after drug exposure, each of the four sets of experiments suggest that the half-life of ENaC in A6 cells range between 1.5 and 3.5 h. We observed that the channel number decreased by 50% in ∼3.5 h in BFA-, nocodazole-, and puromycin-treated cells compared with untreated control cells. The 1.5-h half-life of channels after CHX treatment appears to be slightly less than that of the other three drugs. Additionally, CHX is the only drug of the four that has a significant effect on Po. One possible explanation may be related to the estimation of N depending on Po and low values of Po requiring longer recording periods to estimate N accurately. For example, if the recording period is similar among each compound tested (it was in our experiments), then N values in patches containing channels with low Po will possibly be underestimated. Therefore, N in CHX-treated patches will be underestimated as Po decreases (i.e., at longer times after CHX treatment). Thus N will appear to decrease more rapidly after CHX than for the other three agents, where there is no decrease in Po within the first 6 h. The issue of underestimating N has been considered on a probability basis by Kemendy et al. (21). Using their methods, the low Po (0.057 ± 0.013) after 4 h of CHX treatment with an 8-min recording period implies that, even if we only observe one level of channel opening, there is more than a 76% chance of there being two or more channels in the patch. If the number of channels present in patches treated with CHX for >2 h increased from 1.5 to 2 times, the half-life would increase to >3 h. The determination of CHX half-life is likely underestimated in the concurrent change in N and Po; therefore, the half-lives for all the agents are likely to be close to 3 h.
Nonetheless, the question remains of why CHX should reduce Po while the other agents do not. The CHX-induced reduction in Po could result from inhibiting translation of ENaC regulatory proteins. One possibility for such regulatory proteins is proteases implicated in activating ENaC. Proteases such as furin and prostasin have been reported to increase ENaC Po by converting immature channels with low activity into mature highly active channels through proteolytic cleavage of ENaC on α- and γ-subunits in overexpression systems (15, 34). However, in A6 cells, trypsin increases transepithelial current only after a 3- or 4-h pretreatment with aprotinin (data not show). The above observation implies that most, if not all, apical membrane ENaC in A6 cells are proteolytically processed. We examined the amount of furin, a protease endogenously expressed in A6 cells, in untreated cells and following 6 h of CHX or puromycin treatment to determine whether proteolytic processing of ENaC in A6 cells may be altered following inhibition of protein synthesis. We found no change in the furin protein expression level following each treatment, and therefore we speculate that another ENaC regulatory protein could be important in CHX-mediated effects. In considering what sort of regulatory protein might be involved, we need to recognize that CHX reduces Po and that puromycin, another translation inhibitor, does not. The difference between CHX and puromycin is that CHX blocks translation initiation while puromycin blocks elongation. Thus CHX prevents translation of all protein, but puromycin may allow translation of small proteins. Therefore, if the ENaC regulatory protein had a low molecular weight, then there might still be sufficient protein to maintain ENaC activity in the presence of puromycin but not CHX. Examples of such small regulatory proteins are the small G proteins, several of which have been implicated in ENaC regulation (14, 30, 38). The estimated half-life of ENaC can also be affected by drug latency and efficacy. CHX and BFA are more commonly used reagents than puromycin or nocodazole, and their latency and efficacy have been well documented. CHX prevents protein synthesis following just a few minutes of exposure (31); consistent with these reports, we indeed observed effects within 5 min. The inhibitory efficiency of CHX was judged by protein translation assays that reached ∼90% of control cells exposed to CHX for 30 min. The effect of BFA on Golgi in normal rat kidney and human fibroblast M1 cells is also fast; Lippincott-Schwartz (22) reported that the perinuclear cisternal structure of the Golgi disappears within minutes in cells exposed to BFA at 37°C. Although the Golgi of MDCK epithelial cells were reported to be resistant to 0.5 μg/ml BFA, protein secretion at the apical surface of this cell was inhibited (24). In our results (Fig. 9), the effect of BFA on Golgi cisternae formation in A6 cells is very clear; however, the latency we observed is longer than its effect on Golgi of normal rat kidney and M1 cells. The slow effect of BFA on Golgi in A6 cells might result from the lower temperature (26°C) at which the cells are grown; reduced temperatures inhibit retrograde transport of Golgi membrane into the ER in BFA-treated cells (22). However, we cannot exclude the possibility that BFA latency is longer and its efficiency is lower than that of CHX on ENaC in A6 cells. In our experiments, we showed that nocodazole and BFA disrupt the Golgi with similar time courses, and ENaC half-life estimated from cells treated with both drugs is also similar, which implies that both drugs reduce plasma-membrane-protein trafficking to the same extent even though they disrupt different cellular machinery.
Kabra et al. (19) biotin labeled plasma membrane proteins and found the half-life of proteolytically cleaved α-ENaC, which formed a functional channel, at the plasma membrane is ∼15 min. Although they studied ENaC in an overexpression system, the large difference between their report and our observation of a much longer ENaC half-life may be mainly due to the fact that they measured the half-life of ENaC at the surface membrane, whereas we measured the half-life of ENaC that underwent endocytosis and exocytosis processes. The half-life of ENaC that has been delivered to the apical membrane and the half-life of ENaC at the apical membrane are two different concepts; the half-life of the former may much longer than the latter because channels may be presented to the apical membrane several times via endocytosis and exocytosis processes (3, 25). The channel activity we have recorded in any particular patch reflects the steady-state balance of endocytosis, exocytosis recycling and degradation of ENaC in the apical membrane. Although BFA and nocodazole are vesicle trafficking inhibitors, they should have minor or negligible effects on ENaC recycling between the apical membrane and early endosome. Butterworth et al. (3) reported that 5 μg/ml of BFA had no obvious effect on ENaC's response to cAMP-induced recycling of the channel for at least 2 h. The BFA concentration that we used was 300 nM (∼0.1 μg/ml), and at such a low concentration, the toxic effect of BFA on ENaC recycling for the first 6 h of drug exposure should be negligible. Another drug, nocodazole, which disrupts microtubule filament formation, is also unlikely to significantly alter ENaC recycling. Lu et al. (25) observed that a large pool of ENaC recycles between the plasma membrane and just underneath the plasma membrane, and the major cytoskeleton component underneath the plasma membrane is the actin filament. Our suggestion that BFA and nocodazole have negligible effects on ENaC recycling is also supported by the results observed from puromycin-treated cells. Puromycin prevents ENaC trafficking to the apical membrane through a different mechanism compared with BFA or nocodazole treatment; however, the half-life determined for all three experiments do not differ.
Two populations of ENaC, a rapidly decaying pool and a stable pool, were observed in MDCK1 cells overexpressing all three mENaC subunits, and the half-life of the rapid pool was 2.6 h in cells exposed to CHX (27). The ENaC half-lives in this paper were calculated from cells exposed to drugs for 6 h, making our results likely equivalent to the half-life of rapid pools observed by others. We cannot easily determine whether, in addition to this rapidly processed pool, there may be longer-lived, stable pools of ENaC in A6 cells, because it is extremely difficult to form tight seals on cells that have been exposed to CHX, puromycin, or nocodazole for >6 h. We have recorded ENaC activity from BFA-treated cells for up to 22 h, and even though ENaC half-life following BFA treatment is 3.6 h, there is still ∼20% of channel activity remaining at the end of this period. However, we are not certain whether the residual channels are from the stable pool. Since both mature and immature ENaC appear to reside in the apical membrane, Hughey et al. (16) suggested that there might be a trafficking pathway that bypasses the Golgi to deliver ENaC directly from the ER to the plasma membrane.
An alternative explanation of the action of BFA requires that BFA interferes with the endocytic machinery. If the degradative pathway is disrupted before all of the apical ENaC is degraded, then some apical ENaC would persist as a long-lived pool. BFA does cause endosomes and lysosomes to form tubular structures (23), but it is unclear whether the morphological change implies a loss of function.
In summary, we have shown, using several different pharmacological inhibitors, that the half-life of functional ENaC in the apical membrane is ∼3 h. This implies that the three ENaC subunits remain together for at least this long. However, since the half-lives of individual subunits determined by biochemical analysis is, in general, longer, our results imply that channels become nonfunctional before leaving the membrane or that subunits separate from one another before being degraded.
GRANTS
This work was supported by National Institutes of Health Grants HL-071621 and DK-37963 awarded to D. C. Eaton. This research was also made possible by support from Emory University through Digestive Disease Research Development Center Grant DK-064399.
Acknowledgments
The authors thank Dr. Janet Klein and Chris Martin for help in the successful immunolabeling of the Golgi apparatus in A6 cells. Billie Jean Duke maintained A6 cells in culture for the duration of all experiments.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Becchetti A, Kemendy AE, Stockand JD, Sariban-Sohraby S, Eaton DC. Methylation increases the open probability of the epithelial sodium channel in A6 epithelia. J Biol Chem 275: 16550–16559, 2000. [DOI] [PubMed] [Google Scholar]
- 2.Bruns JB, Carattino MD, Sheng SH, Maarouf AB, Weisz OA, Pilewski JM, Hughey RP, Kleyman TR. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the gamma-subunit. J Biol Chem 282: 6153–6160, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Butterworth MB, Edinger RS, Johnson JP, Frizzell RA. Acute ENaC stimulation by cAMP in a kidney cell line is mediated by exocytic insertion from a recycling channel pool. J Gen Physiol 125: 81–101, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of 3 homologous subunits. Nature 367: 463–467, 1994. [DOI] [PubMed] [Google Scholar]
- 5.Carattino MD, Sheng SH, Bruns JB, Pilewski JM, Hughey RP, Kleyman TR. The epithelial Na+ channel is inhibited by a peptide derived from proteolytic processing of its alpha subunit. J Biol Chem 281: 18901–18907, 2006. [DOI] [PubMed] [Google Scholar]
- 6.de la Rosa DA, Li H, Canessa CM. Effects of aldosterone on biosynthesis, traffic, and functional expression of epithelial sodium channels in A6 cells. J Gen Physiol 119: 427–442, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Firsov D, Robert-Nicoud M, Gruender S, Schild L, Rossier BC. Mutational analysis of cysteine-rich domains of the epithelium sodium channel (ENaC)—identification of cysteines essential for channel expression at the cell surface. J Biol Chem 274: 2743–2749, 1999. [DOI] [PubMed] [Google Scholar]
- 8.Firsov D, Schild L, Gautschi I, Merillat AM, Schneeberger E, Rossier BC. Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: a quantitative approach. Proc Natl Acad Sci USA 93: 15370–15375, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher RS, Grillo FG, SaribanSohraby S. Brefeldin A inhibition of apical Na+ channels in epithelia. Am J Physiol Cell Physiol 270: C138–C147, 1996. [DOI] [PubMed] [Google Scholar]
- 10.Frelin C, Barbry P, Vigne P, Chassande O, Cragoe EJ, Lazdunski M. Amiloride and its analogs as tools to inhibit Na+ transport via the Na+ channel, the Na+/H+ antiport and the Na+/Ca2+ exchanger. Biochimie 70: 1285–1290, 1988. [DOI] [PubMed] [Google Scholar]
- 11.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev 77: 359–396, 1997. [DOI] [PubMed] [Google Scholar]
- 12.Hanwell D, Ishikawa T, Saleki R, Rotin D. Trafficking and cell surface stability of the epithelial Na+ channel expressed in epithelial Madin-Darby canine kidney cells. J Biol Chem 277: 9772–9779, 2002. [DOI] [PubMed] [Google Scholar]
- 13.Harris M, Garcia-Caballero A, Stutts MJ, Firsov D, Rossier BC. Preferential assembly of epithelial sodium channel (ENaC) subunits in Xenopus oocytes—role of furin-mediated endogenous proteolysis. J Biol Chem 283: 7455–7463, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helms MN, Chen XJ, Ramosevac S, Eaton DC, Jain L. Dopamine regulation of amiloride-sensitive sodium channels in lung cells. Am J Physiol Lung Cell Mol Physiol 290: L710–L722, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong QS, Carattino MD, Johnson JP, Stockand JC, Kleyman TR. Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem 279: 18111–18114, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Hughey RP, Bruns JB, Kinlough CL, Kleyman TR. Distinct pools of epithelial sodium channels are expressed at the plasma membrane. J Biol Chem 279: 48491–48494, 2004. [DOI] [PubMed] [Google Scholar]
- 17.Hughey RP, Mueller GM, Bruns JB, Kinlough CL, Poland PA, Harkleroad KL, Carattino MD, Kleyman TR. Maturation of the epithelial Na+ channel involves proteolytic processing of the alpha- and gamma-subunits. J Biol Chem 278: 37073–37082, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 19A resolution and low pH. Nature 449: 316-+, 2007. [DOI] [PubMed]
- 19.Kabra R, Knight KK, Zhou R, Snyder PM. Nedd4-2 induces endocytosis and degradation of proteolytically cleaved epithelial Na+ channels. J Biol Chem 283: 6033–6039, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev 82: 735–767, 2002. [DOI] [PubMed] [Google Scholar]
- 21.Kemendy AE, Kleyman TR, Eaton DC. Aldosterone alters the open probability of amiloride-blockable sodium-channels in A6 epithelia. Am J Physiol Cell Physiol 263: C825–C837, 1992. [DOI] [PubMed] [Google Scholar]
- 22.Lippincott-Schwartz J, Donaldson JG, Schweizer A, Berger EG, Hauri HP, Yuan LC, Klausner RD. Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin a suggests an ER recycling pathway. Cell 60: 821, 1990. [DOI] [PubMed] [Google Scholar]
- 23.Lippincottschwartz J, Yuan L, Tipper C, Amherdt M, Orci L, Klausner RD. Brefeldin-A's effects on endosomes, lysosomes, and the Tgn suggest a general mechanism for regulating organelle structure and membrane traffic. Cell 67: 601–616, 1991. [DOI] [PubMed] [Google Scholar]
- 24.Low SH, Wong SH, Tang BL, Tan P, Subramaniam VN, Hong WJ. Inhibition by brefeldin-A of protein secretion from the apical cell surface of Madin-Darby canine kidney cells. J Biol Chem 266: 17729–17732, 1991. [PubMed] [Google Scholar]
- 25.Lu C, Pribanic S, Debonneville A, Jiang C, Rotin D. The PY motif of ENaC, mutated in Liddle syndrome, regulates channel internalization, sorting and mobilization from subapical pool. Traffic 8: 1246–1264, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Marunaka Y, Eaton DC. Effects of vasopressin and camp on single amiloride-blockable Na channels. Am J Physiol Cell Physiol 260: C1071–C1084, 1991. [DOI] [PubMed] [Google Scholar]
- 27.Mohan S, Bruns JR, Weixel KM, Edinger RS, Bruns JB, Kleyman TR, Johnson JP, Weisz OA. Differential current decay profiles of epithelial sodium channel subunit combinations in polarized renal epithelial cells. J Biol Chem 279: 32071–32078, 2004. [DOI] [PubMed] [Google Scholar]
- 28.Planes C, Blot-Chabaud M, Matthay MA, Couette S, Uchida T, Clerici C. Hypoxia and beta2-agonists regulate cell surface expression of the epithelial sodium channel in native alveolar epithelial cells. J Biol Chem 277: 47318–47324, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Planes C, Leyvraz L, Uchida T, Angelova MA, Vuagniaux G, Hummler E, Matthay M, Clerici C, Rossier B. In vitro and in vivo regulation of transepithelial lung alveolar sodium transport by serine proteases. Am J Physiol Lung Cell Mol Physiol 288: L1099–L1109, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Pochynyuk O, Stockand JD, Staruschenko A. Ion channel regulation by Ras, Rho, and Rab small GTPases. Exp Biol Med 232: 1258–1265, 2007. [DOI] [PubMed] [Google Scholar]
- 31.Robert F, Gao HQ, Donia M, Merrick WC, Hamann MT, Pelletier J. Chlorolissoclimides: new inhibitors of eukaryotic protein synthesis. RNA 12: 717–724, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schild L The ENaC channel as the primary determinant of two human diseases: Liddle syndrome and pseudohypoaldosteronism. Nephrologie 17: 395–400, 1996. [PubMed] [Google Scholar]
- 33.Schild L, Canessa CM, Shimkets RA, Gautschi I, Lifton RP, Rossier BC. A mutation in the epithelial sodium-channel causing Liddle disease increases channel activity in the Xenopus laevis oocyte expression system. Proc Natl Acad Sci USA 92: 5699–5703, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheng S, Carattino MD, Bruns JB, Hughey RP, Kleyman TR. Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am J Physiol Renal Physiol 290: F1488–F1496, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Sheng SH, Maarouf AB, Bruns JB, Hughey RP, Kleyman TR. Functional role of extracellular loop cysteine residues of the epithelial Na+ channel in Na+ self-inhibition. J Biol Chem 282: 20180–20190, 2007. [DOI] [PubMed] [Google Scholar]
- 36.Shimkets RA, Lifton RP, Canessa CM. The activity of the epithelial sodium channel is regulated by clathrin-mediated endocytosis. J Biol Chem 272: 25537–25541, 1997. [DOI] [PubMed] [Google Scholar]
- 37.Shimkets RA, Warnock DG, Bositis CM, Nelsonwilliams C, Hansson JH, Schambelan M, Gill JR, Ulick S, Milora RV, Findling JW, Canessa CM, Rossier BC, Lifton RP. Liddles syndrome—heritable human hypertension caused by mutations in the beta-subunit of the epithelial sodium channel. Cell 79: 407–414, 1994. [DOI] [PubMed] [Google Scholar]
- 38.Staruschenko A, Patel P, Tong QS, Medina JL, Stockand JD. Ras activates the epithelial Na+ channel through phosphoinositide 3-OH kinase signaling. J Biol Chem 279: 37771–37778, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Verrey F, Schaerer E, Zoerkler P, Paccolat MP, Geering K, Kraehenbuhl JP, Rossier BC. Regulation by aldosterone of Na+, K+-ATPase messenger RNAs, protein synthesis, and sodium transport in cultured kidney cells. J Cell Biol 104: 1231–1237, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weisz OA, Johnson JP. Noncoordinate regulation of ENaC: paradigm lost? Am J Physiol Renal Physiol 285: F833–F842, 2003. [DOI] [PubMed] [Google Scholar]
- 41.Weisz OA, Wang JM, Edinger RS, Johnson JP. Non-coordinate regulation of endogenous epithelial sodium channel (ENaC) subunit expression at the apical membrane of A6 cells in response to various transporting conditions. J Biol Chem 275: 39886–39893, 2000. [DOI] [PubMed] [Google Scholar]
- 42.Yu L, Eaton DC, Helms MN. Effect of divalent heavy metals on epithelial Na+ channels in A6 cells. Am J Physiol Renal Physiol 293: F236–F244, 2007. [DOI] [PubMed] [Google Scholar]