

Abstract
Background
Long-term ingestion of alcohol produces marked alterations in hypothalamic-pituitary-adrenal axis activity. The authors engaged in a series of studies to determine the distinct role of the hypothalamus and the pituitary and adrenal glands in the disturbances observed in abstinent alcohol-dependent subjects. In this first of a two-part study, the authors report on (1) the basal secretory profile of corticotropin and cortisol from 2000 to 0800 hrs, (2) adrenocortical sensitivity in both the presence and absence of endogenous pituitary activation, and (3) pituitary glucocorticoid sensitivity to dexamethasone.
Methods
Eleven male, 4 to 6 weeks abstinent, alcohol-only–dependent subjects and 10 age-matched male healthy controls were studied. Basal circulating concentrations of corticotropin and cortisol were obtained from 2000 to 0800 hr. A submaximal dose of cosyntropin (0.01 μg/kg), a corticotropin analogue was then administered to assess adrenocortical sensitivity. In a separate session, cosyntropin was administered following high-dose dexamethasone (8 mg iv) to assess adrenocortical sensitivity in the relative absence of endogenous corticotropin. In addition, the corticotropin response to dexamethasone was measured to determine pituitary glucocorticoid responsiveness.
Results
Cortisol, but not corticotropin, pulse amplitude (p < 0.05) and mean concentration (p = 0.05) was significantly lower in alcohol-dependent subjects compared with controls. The cortisol response to cosyntropin was lower in alcohol-dependent subjects following endogenous corticotropin suppression by high-dose dexamethasone (p < 0.04) but not without dexamethasone pretreatment. Mean corticotropin (p < 0.004) and cortisol (p < 0.05) concentrations in response to dexamethasone were attenuated in the patients compared to controls. Basal concentrations of 11-deoxycortisol, the precursor to cortisol, were also decreased in alcohol-dependent subjects (p < 0.05).
Conclusion
Attenuated basal and stimulated adrenocortical concentrations in abstinent alcohol-dependent men are coupled with a nonhomeostatic increase in pituitary glucocorticoid inhibition. A decrease in stress-axis responsivity in alcohol dependence may have implications for treatment outcome.
Keywords: Adrenal Cortex, Alcoholism, Cosyntropin, Dexamethasone, Pituitary-Adrenal System
Disruption of Hypothalamic-Pituitary-Adrenal (HPA) axis activity is recognized as a pathophysiological alteration during the intoxicating, withdrawal, and abstinence phases of alcoholism. A number of studies have documented the increase in cortisol secretion during intoxication (Adinoff et al., 2003; Mendelson et al., 1971; Stokes, 1973) and withdrawal (Adinoff et al., 1991; Iranmanesh et al., 1989; Keedwell et al., 2001; Mendelson et al., 1971), occasionally resulting in a hypercortisolemic state mimicking the clinical and biochemical characteristics of Cushing syndrome (Jeffcoate, 1993; Veldman and Meinders, 1996). Concurrent with and after the resolution of this hypercortisolemic state, however, several investigators have observed an attenuation of the axis to pharmacological and psychological stressors. Although the increase in glucocorticoid secretion associated with intoxication and withdrawal resolves within a few days after the cessation of alcohol ingestion (Adinoff et al., 1991), suppression of HPA axis functioning seems to persist for at least the first several weeks of abstinence.
Attenuated responsiveness of the HPA axis has been revealed at each level of the axis in both drinking and abstinent alcohol-dependent patients. Central stimuli, including alcohol (Merry and Marks, 1972), operative trauma (Margraf et al., 1967), cold pressor (Errico et al., 1993), insulin-induced hypoglycemia (Chalmers et al., 1978; Costa et al., 1996), nicotine (Coiro and Vescovi, 1999), public speaking (Lovallo et al., 2000), m-chlorophenylpiperazine (Krystal et al., 1996), and hyperthermia (Vescovi et al., 1997) produce a blunted corticotropin and/or cortisol response in alcohol-dependent patients compared with healthy controls. Alcohol-dependent patients also have a muted corticotropin response to naloxone (Inder et al., 1995), an opioid antagonist that blocks the tonic inhibition of endogenous opioids on the hypothalamus. Pituitary stimulation by ovine or human CRH induces a blunted corticotropin and/or cortisol response for at least 3 weeks after drinking cessation (Adinoff et al., 1990; Costa et al., 1996; Ehrenreich et al., 1997). Finally, direct stimulation of the adrenal cortex by exogenously administered adrenocorticotropinα1–24 (cosyntropin) results in a markedly decreased cortisol response in alcohol-dependent individuals (Knudsen et al., 1987; Margraf et al., 1967; Wand and Dobs, 1991).
This impressive and consistent literature, however, offers only suggestive clues as to which specific levels of the axis are critical for the hyporesponsive state. Because of both positive and negative organizational loops, interactions between the various components of the HPA axis confound the interpretation of any specific stressor on a given hormonal unit. In addition, increases or decreases in endogenous hormone secretion could alter organ sensitivity to a given stimulus. Finally, many of the studies referenced above have significant confounders that make interpretation of their findings problematic. For example, chronic medical and/or psychiatric conditions (i.e., post–traumatic stress disorder [PTSD], depression) may interfere with HPA axis functioning.
To clarify the specific alterations in HPA axis functioning that occur after chronic and excessive use of alcohol, we designed a series of studies to explore basal pulsatile characteristics of corticotropin and cortisol, adrenocortical sensitivity to exogenous stimulation in the presence or absence of endogenous stimulation, and pituitary glucocorticoid sensitivity in 1-month-abstinent alcohol-dependent patients. Secretory characteristics of corticotropin and cortisol were determined by obtaining frequent sampling of plasma corticotropin and cortisol from 2000 to 0800 hr. Adrenocortical sensitivity to an exogenous stimulus was assessed by administering cosyntropin, a corticotropin analog (corticotropinα1–24). To avoid a maximal glucocorticoid response, or ceiling effect, that could obscure potential group differences, we assessed adrenocortical sensitivity with a very low dose (0.01 μg/kg) of cosyntropin. This is approximately 0.3% of the clinically recommended dose (250 μg) used in the cosyntropin stimulation test in the diagnosis of endocrine disorders. In a second test, we isolated the adrenal cortex from its major endogenous input (pituitary corticotropin) by first administering high-dose dexamethasone before the cosyntropin administration. We also determined hypothalamic-pituitary glucocorticoid sensitivity by measuring the corticotropin response to dexamethasone administration. Finally, to assess potential disruptions in adrenosteroid synthesis, we assessed glucocorticoid, minerocorticoid, and androgen hormones (in addition to cortisol) after cosyntropin administration. It was hypothesized that cortisol secretion in response to cosyntropin would be significantly attenuated in alcohol-dependent participants both with and without dexamethasone pretreatment.
MATERIALS AND METHODS
Alcohol-Dependent Patients
Eleven male alcohol-dependent participants (aged 42.8 ± 4.0 years [mean ± SD]; range, 38 –49 years) were recruited from patients requesting treatment for alcohol dependence at the Dallas VA Medical Center. Ten of these patients participated in both study sessions; one patient only participated in session B (see below). Patients reported an alcohol intake of at least 80 g of absolute alcohol on a daily basis for at least 2 weeks before the cessation of drinking and had at least a 10-year history of problematic drinking. Patients with a lifetime history of other DSM-IV (American Psychiatric Association, 1994) Axis I psychiatric disorders (such as anxiety, PTSD, or mood disorders) not associated with alcohol use, other substance use disorders (or weekly drug use) within the previous 12 months (excluding caffeine or nicotine use disorders), active medical disorders (i.e., hypertension, diabetes, chronic pain, or cardiac or pulmonary disorders), or a history of major head trauma were excluded from the study. Exclusion criteria also included use of any medications that may interfere with HPA axis functioning (i.e., psychotropics, antihypertensives, hypoglycemic agents) within 2 weeks of the study, Beck Depression Inventory (BDI; Beck et al., 1979) scores above 15 at the time of assessment, or an alanine aminotransferase (ALT) or aspartate aminotransferase (AST) 3.0 times greater than the clinical laboratory’s upper limit of normal. (See Table 1.)
Table 1.
Demographic and Clinical Characteristics of Alcohol-Dependent Participants and Healthy Controls
| Alcohol Dependent (n = 11) | Controls (n = 10) | t or χ2 | df | p Value | |
|---|---|---|---|---|---|
| Age (years) | 42.8 ± 4.0 | 39.6 ± 5.7 | 1.50 | 19 | NS |
| Race | 2.37 | 3 | NS | ||
| Asian | 0 | 1 | |||
| White | 6 | 7 | |||
| African-American | 3 | 1 | |||
| Hispanic | 2 | 1 | |||
| Marital status | 14.06 | 3 | 0.003 | ||
| Single | 0 | 2 | |||
| Married | 1 | 7 | |||
| Separated | 4 | 0 | |||
| Divorced | 6 | 1 | |||
| Employed | 3 | 10 | 11.75 | 1 | 0.001 |
| Education (years) | 13.0 ± 1.3 | 16.4 ± 2.9 | 3.39 | 17 | 0.003 |
| Homeless | 8 | 0 | 11.78 | 1 | 0.001 |
| Nicotine dependent | 9 | 1 | 10.83 | 1 | 0.001 |
| BDI | 8.0 ± 4.2 | 0.9 ± 1.7 | 4.71 | 18 | 0.000 |
| ALT (units/liter) | 59.0 ± 60.5 | 31.1 ± 10.4 | 1.44 | 19 | NS |
| AST (units/liter) | 57.1 ± 47.6 | 33.8 ± 27.0 | 1.36 | 19 | NS |
| DrInC | 37.6 ± 6.3 | 3.7 ± 3.5 | 14.97 | 19 | 0.000 |
| Years problem drinking | 20.8 ± 5.8 | ||||
| Drinks in past 30 days | 852.6 ± 577.5 | ||||
| Lifetime drinks | 131,129 ± 106,528 |
Data are mean ± SD. Comparisons between groups were by t test or χ2.
Healthy Controls
Eleven healthy men (aged 39.6 ± 5.7 years; range, 31–47 years) were individually age matched within a 5-year period with alcohol-dependent patients. Controls reported no lifetime history of any DSM-IV axis I disorder (except caffeine or nicotine use disorders), reported no medical disorders, and were taking no medications. Controls with a single first-degree relative or two second-degree relatives with an axis I disorder were excluded.
Clinical Assessment
All participants underwent a history and physical examination, routine clinical laboratory testing, electrocardiography, and urine drug screening. Psychiatric and substance use disorders were assessed using the SCID-Lifetime (First et al., 1996). Alcohol-dependent patients were detoxified from alcohol and then housed on a residential treatment until the studies were initiated. Urine drug screening results were obtained three times weekly, and Breathalyzer (Alco-Sensor III, Intoximeters, St. Louis, MO) test results were obtained whenever the patient left the unit unaccompanied by staff. Separate informed consents from both the University of Texas Southwestern Medical Center and the Dallas VA Medical Center were obtained after the study was fully explained, and participants were financially compensated for their participation. The Drinker Inventory of Consequences-Lifetime Consequences (DrInC-2L; Miller et al., 1995) was used to assess lifetime severity of alcohol-related problems. A timeline follow back (Sobell and Sobell, 1978) was used to assess 1-month, 1-year, and lifetime drinking history. (See Table 1.)
Procedure
All neuroendocrine studies were performed at the General Clinical Research Center (GCRC) at the University of Texas Southwestern Medical Center. The studies were performed in two separate sessions. Each session was separated by 1 week, and session order was balanced. Nicotine-dependent patients were given a nicotine patch appropriate to their level of cigarette use.
Session A
This study included a 24-hr urine collection for urinary free cortisol and 12-hr sampling for the measurement of plasma corticotropin and cortisol secretory dynamics and the cosyntropin stimulation test. Participants were brought to the GCRC at 1800 hr on day 1. A 24-hr urine collection was initiated at 2000 hr on day 1 for urinary free cortisol and continued until 2000 hr on day 2. An intravenous catheter was inserted in each arm at 1900 hr on day 2. Blood sampling was initiated for basal measures of corticotropin and cortisol 1 hr later (at 2000 hr) and continued through 0800 hr on day 3. After the 0800 hr blood draw on day 3, 0.01 μg/kg cosyntropin was administered intravenously over 1 min. [Cosyntropin was administered at 0800 hr to assure that in session B dexamethasone (administered 9 hr previously) would continue to maximally suppress corticotropin release throughout the cosyntropin paradigm.] Blood sampling was increased immediately after cosyntropin administration to every 5 min through 0900 and then decreased to every 10 min through 1000 hr. Intravenous lines were removed after the final 1000 hr blood draw. The blood sample collected just before cosyntropin administration was also used for the measurement of cortisol binding globulin (CBG). Blood samples at 0800, 0830, 0900, 0930, and 1000 hr were used for adrenosteroid steroid hormones other than cortisol (11-deoxycortisol, dehydroepiandrosterone, androstenedione, 17-hydroxyprogesterone, progesterone, and aldosterone).
Session B
This study included the pituitary and adrenocortical response to dexamethasone and the cosyntropin stimulation test after dexamethasone. Participants arrived at the GCRC at 2000 hr, and intravenous lines were placed into both arms at 2100 hr. Blood samples for the measurement of corticotropin and cortisol were obtained every 10 min from 2200 through 2300 hr. Dexamethasone, 8 mg, in 50 ml of dextrose 5% water (D5W) was administered intravenously from 2300 to 2330 hr. Blood sampling was continued from 2300 to 0500 hr and then restarted at 0700 to 0800 hr every 10 min. (Blood sampling was not obtained from 0500 to 0700 hr because of limitations on blood volume). The cosyntropin test was performed as described in session A.
Assays
Plasma concentrations of corticotropin were measured by immunoradiometric assay (IRMA), using reagents from DiaSorin (Stillwater, MN). This assay has a low-end sensitivity of 1.5 pg/ml, with an intra-assay coefficient of variation of 2.5–5.4% in the concentration range of 33 to 773 pg/ml. The interassay coefficient of variation is 3.2–5.7% in the concentration range of 8.7 to 257 pg/ml. Serum concentrations of cortisol were measured by radioimmunoassay (RIA) using reagents from DiaSorin. This assay has a low-end sensitivity of 0.21 μg/dl, with an intra-assay coefficient of variation of 6.6–7.7% in the concentration range of 2.9 to 47.1 μg/dl. The interassay coefficient of variation is 8.8–9.8% in the concentration range of 3.7 to 36.9 μg/dl. Serum concentrations of CBG were assayed by RIA using reagents from BioSource Europe S.A. (Nivelles, Belgium). This assay has a low-end sensitivity of 0.25 μg/ml, with an intra-assay coefficient of variation of 3.3 to 7.7 in the concentration range of 33 to 109 μg/ml. The interassay coefficient of variation is 4.5–5.4% in the concentration range of 32 to 105 μg/ml. Commercial RIA kits from Diagnostics Systems Laboratories, Inc. (Webster, TX) were used for the measurement of aldosterone, dihydroepiandrosterone, androstenedione, progesterone, and 17-OH-progesterone; RIA kits from ICN Pharmaceutical, Inc., Diagnostic Division (Costa Mesa, CA) were used for the measurement of 11-deoxycortisol; and RIA kits from DiaSorin were used for the measurement of urinary free cortisol.
Statistics
Data from one healthy control were deleted from the study because of occasional and unexplained pulses of corticotropin (up to 336 pg/ml) and multiple missing data points.
Demographics
Student’s t test (interval data) and χ2 contingency table analyses (nominal data) were used to compare demographic characteristics of the two groups. Items of interest included age, education, employment status, housing status, marital status, liver function, and smoking status. Descriptive statistics were used to quantify drinking characteristics of the alcohol-dependent group, including years of problem drinking, drinking days (30 days before drinking cessation and lifetime), standard drinks (30 days before drinking cessation and lifetime), and days abstinent at the time of testing. Student’s t test was used to compare mean scores on the BDI and the DrInC-2L.
Basal Corticotropin and Cortisol Pulsatile Characteristics
Because there is a circadian rhythm for the two hormones of interest, the assumption that all secretion was pulsatile leads to unreasonably long half-lives for corticotropin and cortisol, resulting in a poor fit of the calculated pulses to the actual data. Therefore, pulsatile characteristics were assessed by the Smoothing Baseline Pulse Pulses (SBPP) algorithm (Guo et al., 1999), which allows for a changing baseline. In general, missing values were not an issue in these data, but equally spaced observations are required for analysis in pulse-detection algorithms. Missing values were handled in the following way. If the concentration at the time point preceding the missing time point was lower than at the point after the missing time point, the missing concentration was replaced with the concentration value of the point preceding the missing time. This was done to avoid creating the possibility of a pseudopulse. Missing values that were part of a decreasing trend were linearly interpolated. For various reasons, some series had to be excluded from the secondary analysis, mainly because they did not show pulsatility. Pulsatile analysis was completed on nine alcohol-dependent patients and eight controls for corticotropin analyses, and seven patients and nine controls for the cortisol analyses. Student’s t test was used to compare group means. A brief description of some of the summary measures from SBPP is given below.
Total probability
The sum of the probabilities of there being a pulse at each point.
Adjusted input
The sum of input at each time point times the probability of input at each point. It is a weighted total input.
Number of inputs
The number of input locations with a probability greater than 0.5. SBPP models the probability of input at each time point, and we used the 0.5 cutoff to distinguish large input (where input is most likely occurring) from small input (unlikely) locations. This measure allows the comparison of input frequencies between the two groups but is not a measure of pulse number.
Average baseline
The mean of the baseline values estimated at each point. SBPP allows for a changing baseline, so baseline itself is no longer a parameter.
Mean amplitude
The net mean height of pulses after subtracting the changing baseline.
Time × Group Analysis
A repeated-measures ANOVA on the mean hormone level over four 3-hr blocks (i.e., 2000–2250, 2300–0150, 0200–0450, and 0500–0750 hr) was performed to incorporate all participants into the circadian analysis. Because the distribution of data was skewed, each block mean of both corticotropin and cortisol was log (base-e) transformed before analysis.
Pharmacologic Challenge Studies
Cortisol secretion after cosyntropin was compared between 0800 and 1000 hr by the area-under-the-curve method, or net integrated response, and by net peak cortisol for sessions A and B. Because the net integrated response was similar to the net peak response, only the former is reported. The net integrated response was calculated by taking the average hormone concentration between consecutive measurement points, multiplying by the time interval between the points, summing across time, and netting out basal hormone levels multiplied by the total time interval over which measurement occurred (i.e., 120 min). Basal hormone values were based on mean levels between the hours of 0730 and 0800. The response to dexamethasone was determined using mean corticotropin or cortisol response from 2330 to 0500 hr. Adrenosteroid steroids were measured after cosyntropin administration (without dexamethasone pretreatment). For each of these analyses, Student’s t test was used to compare group means. Missing values (six total) were estimated based on the mean of hormone measures taken directly before and after the missing values.
Correlation Analysis
The relation between drinking history (i.e., drinks in previous 30 days, lifetime drinks) and various neuroendocrine measures (basal cortisol concentration before cosyntropin administration [without dexamethasone], integrated cortisol response after cosyntropin, mean corticotropin response to dexamethasone, and integrated cortisol response after cosyntropin with dexamethasone pretreatment) was assessed by Pearson correlation.
RESULTS
Participant Characteristics
The 11 alcohol-dependent patients were more likely to be unemployed (p < 0.001) and homeless (p < 0.001), had fewer years of education (p < 0.003), and were more likely to be smokers (p < 0.001) relative to the healthy controls. The patient group also had higher BDI (p < 0.001) scores at the time of the first study compared with controls. Alcohol-dependent patients reported an onset of drinking problems at 21.1 ± 5.0 years (range, 14 –30 years) and had been drinking for an average of 20.8 ± 5.8 years (range, 12–28 years). These participants reported drinking 28.4 ± 3.6 days (range, 21–30 days) in the 30 days before abstinence and drank 853 ± 577 standard drinks (range, 384–2400 standard drinks) over this period (one standard drink = 1 oz spirits, 4 oz wine, or 12 oz beer). Alcohol-dependent patients were abstinent 31.0 ± 4.0 days (range, 24–39 days) at the time of the study. Total DrInC (Miller et al., 1995) scores in the alcohol-dependent group were 37.6 ± 6.3 compared with 3.7 ± 3.5 in the controls (p < 0.001). Despite their extensive drinking history, liver enzymes (ALT and AST) were not significantly different between alcohol-dependent and control groups, and levels of ALT and AST were not markedly higher than the clinical laboratory range for healthy individuals (ALT, 0 –40 units/liter; AST, 0 –37 units/liter) before the study. (See Table 1.)
Secretory Characteristics and Mean Basal Concentrations of Corticotropin and Cortisol
Secretory characteristics were determined by SBPP, an algorithm to assess pulsatility that allows for a changing baseline. Cortisol mean amplitude (net mean height of pulses after subtracting the changing baseline) was significantly lower (p < 0.05) in alcohol-dependent (1.9 ± 0.7 μg/dl; 95% confidence interval, 1.3~2.5) compared with healthy control participants (2.9 ± 1.0 μg/dl; 95% confidence interval, 2.2~3.6). Therefore, the mean amplitude in the alcohol-dependent group was approximately 65% that of the control group. There was also a statistical trend (p < 0.07) for an increase in the total probability (the sum of the probabilities of being a pulse at each point) of cortisol in the alcohol-dependent group relative to controls. Other corticotropin and cortisol pulsatile characteristics were not significantly different between the two groups. The time × group ANOVA analyses using all participants (10 in each group) found a significant group effect for mean cortisol concentrations, which were lower in alcohol-dependent patients compared with controls (p = 0.053). There was no significant group effect for corticotropin measures and no significant time × group effect for either hormone. As expected, a significant within-group time effect (p < 0.0001) was observed for both corticotropin and cortisol. (See Table 2 and Fig. 1.)
Table 2.
Corticotropin and Cortisol Pulsatile Characteristics as Determined by SBPP Analysis and Mean Corticotropin and Cortisol Concentrations Over 12 hr (2000–0800 hr) and in 3-hr Blocks
| Healthy Controls | Alcohol Dependent | t Stat | df | p Value | |
|---|---|---|---|---|---|
| Corticotropin pulsatile characteristics | |||||
| Total probability | 13.8 ± 7.0 | 15.2 ± 3.7 | 0.514 | 15 | NS |
| Adjusted input | 102.7 ± 47.9 | 119.6 ± 35.8 | 0.833 | 15 | NS |
| Number of inputs | 12 ± 5.9 | 12.7 ± 3.4 | 0.288 | 15 | NS |
| Average baseline | 17.7 ± 4.3 | 17.7 ± 3.2 | 0.001 | 15 | NS |
| Mean amplitude | 9.0 ± 5.1 | 8.0 ± 2.4 | 0.501 | 15 | NS |
| Mean corticotropin concentrations (pg/ml) | |||||
| 2000–0800 hr (total 12 hr) | 26.5 ± 7.6 | 26.0 ± 5.0 | 0.192 | 18 | NS |
| 2000–2250 hr | 19.9 ± 6.2 | 18.9 ± 6.8 | 0.349 | 18 | NS |
| 2300–0150 hr | 22.1 ± 9.0 | 18.2 ± 3.4 | 1.275 | 18 | NS |
| 0200–0450 hr | 28.8 ± 7.2 | 29.7 ± 9.8 | 0.218 | 18 | NS |
| 0500–0800 hr | 35.1 ± 10.9 | 37.2 ± 7.4 | 0.511 | 18 | NS |
| Cortisol pulsatile characteristics | |||||
| Total probability | 17.5 ± 5.0 | 22.2 ± 4.0 | 2.034 | 14 | 0.061 |
| Adjusted input | 49.2 ± 14.0 | 45.7 ± 13.2 | 0.508 | 14 | NS |
| Number of inputs | 16.1 ± 4.5 | 19.0 ± 3.9 | 1.343 | 14 | NS |
| Average baseline | 2.8 ± 0.8 | 2.3 ± 1.4 | 0.903 | 14 | NS |
| Mean amplitude | 2.9 ± 1.0 | 1.9 ± 0.7 | 2.224 | 14 | 0.043 |
| Mean cortisol concentrations (μg/dl) | |||||
| 2000–0800 hr (total 12 hr) | 6.9 ± 1.6 | 5.7 ± 1.4 | 2.071 | 18 | 0.053 |
| 2000–2250 hr | 3.4 ± 1.3 | 2.8 ± 2.3 | 0.647 | 18 | NS |
| 2300–0150 hr | 4.5 ± 2.5 | 2.9 ± 1.2 | 1.844 | 18 | 0.082 |
| 0200–0450 hr | 8.2 ± 3.2 | 6.6 ± 1.9 | 1.326 | 18 | NS |
| 0500–0800 hr | 11.9 ± 2.8 | 10.3 ± 2.0 | 1.525 | 18 | NS |
See text for description of SBPP measures.
Fig. 1.
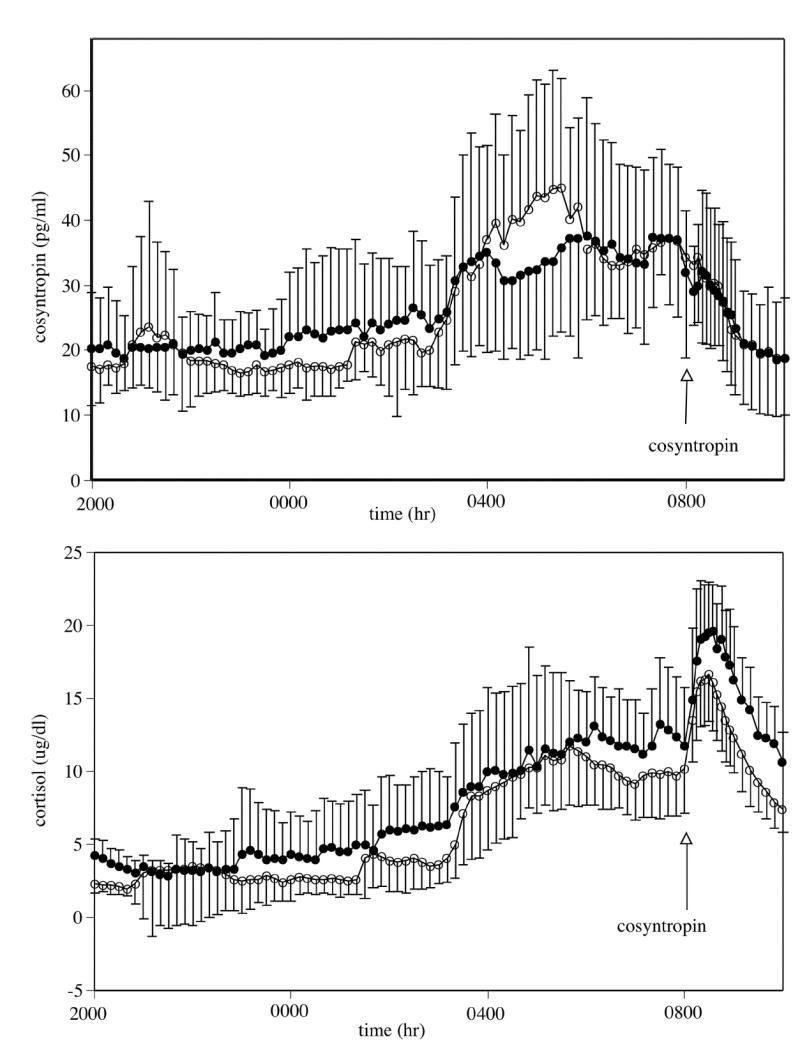
Mean ± SD of corticotrophin (ACTH) (top) and cortisol (bottom) concentrations. Measures between 2000 and 0800 hr reflect basal concentrations. Cosyntropin (0.01 μg/kg iv) was administered at 0800 hr. Mean basal cortisol concentrations were lower in the alcohol-dependent group compared with the healthy controls (p < 0.04). Closed circles represent healthy control participants; open circles represent alcohol-dependent participants.
Urinary Free Cortisol
There were no significant differences in urinary free cortisol in the alcohol-dependent and control groups (see Table 3.)
Table 3.
Urinary Free Cortisol, Basal and Stimulated Adrenosteroid Response to Cosyntropin (0.01 μg/kg iv), Pituitary-Adrenal Response to Dexamethasone (8 mg iv), and Basal and Adrenocortical Stimulated Response to Cosyntropin After Dexamethasone Pretreatment
| Healthy Controls | Alcohol Dependent | t Stat | df | p Value | |
|---|---|---|---|---|---|
| Urinary free cortisol (g/24 hr) | 61.4 ± 26.1 | 60.7 ± 31.9 | 0.058 | 18 | NS |
| Cosyntropin stimulation test without dexamethasone | NS | ||||
| Basal corticotropin (pg/ml) (0730–0800 hr) | 35.9 ± 10.5 | 36.9 ± 9.2 | 0.224 | 18 | NS |
| Basal cortisol (μg/dl) (0730–0800 hr) | 12.5 ± 4.1 | 9.9 ± 2.7 | 1.704 | 18 | NS |
| Cortisol binding globulin (μg/ml) (0800 hr) | 35.4 ± 8.0 | 38.2 ± 4.5 | 0.962 | 18 | NS |
| Basal free cortisol index (0730–0800 hr) | 0.5 ± 0.2 | 0.37 ± 0.1 | 2.414 | 18 | 0.027 |
| Basal aldosterone (0800 hr) (pg/ml) | 84.4 ± 56.3 | 108.0 ± 79.9 | 0.735 | 17 | NS |
| Basal androstenedione (0800 hr) (ng/ml) | 1.3 ± 0.5 | 1.6 ± 0.5 | 1.635 | 17 | NS |
| Basal dihydroepiandrosterone (0800 hr) (ng/ml) | 9.3 ± 3.3 | 8.5 ± 2.4 | 0.634 | 17 | NS |
| Basal 11-deoxycortisol (0800 hr) (ng/ml) | 1.5 ± 0.5 | 1.1 ± .3 | 2.326 | 17 | 0.033 |
| Basal progesterone (0800 hr) (ng/ml) | 0.7 ± 0.2 | 0.8 ± 0.1 | 0.786 | 17 | NS |
| Basal 17-OH-progesterone (0800 hr) (ng/ml) | 2.1 ± 1.1 | 2 ± 1.1 | 0.177 | 17 | NS |
| Net integrated cortisol response (μg/dl.2 hr) | 333 ± 378 | 242 ± 166 | 0.699 | 18 | |
| Net integrated aldosterone response (pg/ml.2 hr) | 4221 ± 3020 | 1383 ± 6187 | 1.116 | 15 | NS |
| Net integrated androstenedione response (ng/ml.2 hr) | 0.6 ± 0.4 | 0.6 ± 0.5 | 0.143 | 17 | NS |
| Net integrated dihydroepiandrosterone response (ng/ml.2 hr) | 39.8 ± 223.0 | −36.6 ± 184.5 | 0.817 | 17 | NS |
| Net integrated 11-deoxycortisol response (ng/ml.2 hr) | 29.0 ± 42.1 | 12.8 ± 14.1 | 1.153 | 17 | NS |
| Net integrated progesterone response (ng/ml.2 hr) | 20.3 ± 14.0 | 13.4 ± 20.0 | 0.872 | 17 | NS |
| Net integrated 17-OH-progesterone response (ng/ml.2 hr) | 59.7 ± 75.4 | 35.9 ± 63.0 | 0.750 | 17 | NS |
| Dexamethasone infusion | NS | ||||
| Mean basal corticotropin (pg/ml) (2200–2230 hr) | 18.3 ± 6.0 | 16.2 ± 3.6 | 0.988 | 19 | NS |
| Mean basal cortisol (μg/dl) (2200–2230 hr) | 3.1 ± 1.1 | 2.0 ± 1.0 | 2.367 | 19 | 0.03 |
| Mean ACTH response after dexamethasone (2330–0500 hr) (pg/ml) | 13.1 ± 2.5 | 9.6 ± 2.4 | 3.3 | 19 | 0.004 |
| Mean cortisol response after dexamethasone (μg/dl) (2330–0500 hr) | 1.9 ± 0.4 | 1.4 ± 0.6 | 2.219 | 19 | 0.04 |
| Cosyntropin stimulation test after dexamethasone | |||||
| Mean basal corticotropin (pg/ml) (0730–0800 hr) | 12.2 ± 2.1 | 8.6 ± 2.9 | 3.154 | 19 | 0.005 |
| Mean basal cortisol (μg/dl) (0730–0800 hr) | 1.2 ± 0.5 | 1.0 ± 0.5 | 1.030 | 19 | NS |
| Net integrated cortisol response (μg/dl.2 hr) | 1107 ± 249 | 804 ± 363 | 2.207 | 19 | 0.04 |
Data are mean ± SD.
Free Cortisol Concentrations
Measures of CBG were obtained at 0800 hr, just before cosyntropin infusion. There were no significant group differences between CBG levels. Calculated free cortisol index (le Roux et al., 2002; serum cortisol in μM/CBG in μM) for mean basal measures (0730 to 0800 hr) revealed that free cortisol in the patient group was significantly lower than in the control group (p < 0.05). (See Table 3.)
Cosyntropin Stimulation Test
The cortisol response to low-dose cosyntropin, as described by net integrated response (integrated response from 0800 to 1000 hr minus the mean basal concentration from 0730 to 0800 hr), was not significantly different between the two groups. (See Table 3 and Fig. 2.)
Fig. 2.
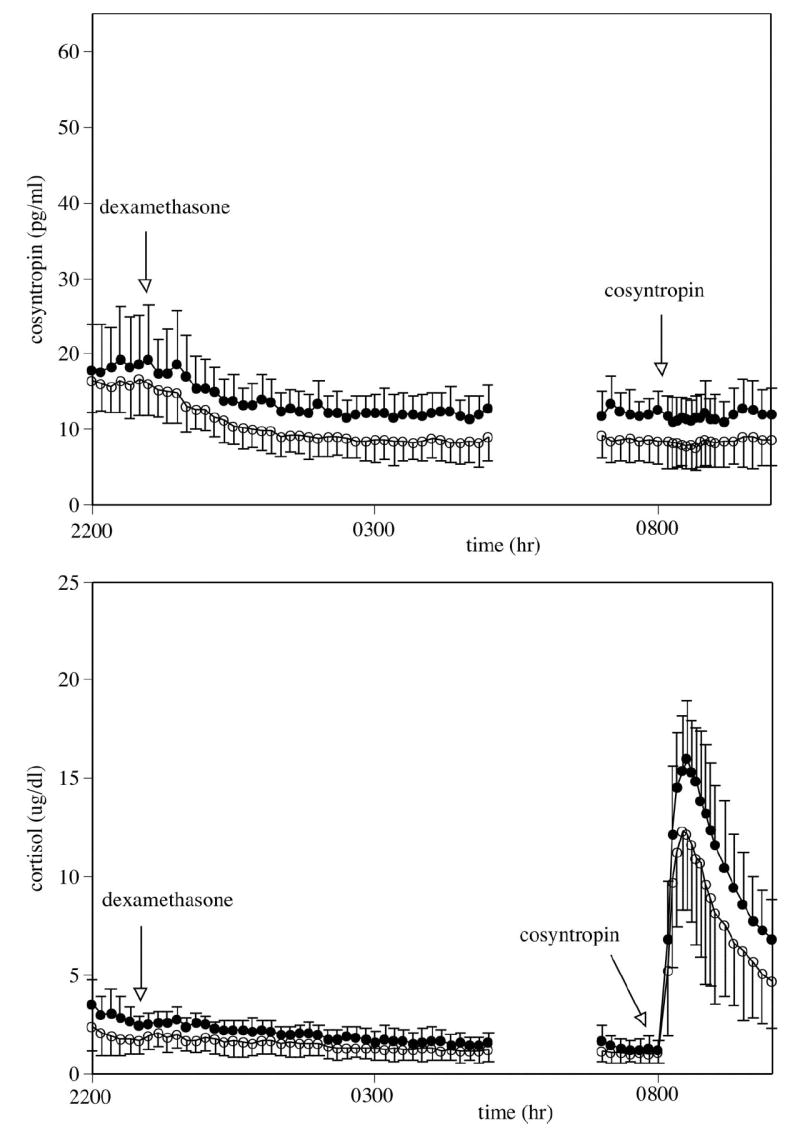
Mean ± SD of corticotrophin (ACTH) (top) and cortisol (bottom) concentrations. Measures between 2000 and 0800 hr reflect basal concentrations. Dexamethasone (8 mg iv) was administered at 2300 hr. Cosyntropin (0.01 μg/kg iv) was administered at 0800 hr. Abstinent alcohol-dependent participants suppressed corticotropin (p < 0.004) and cortisol (p < 0.05) after dexamethasone (2300 to 0500 hr) more than matched control participants. The cortisol response to cosyntropin was lower in the alcohol-dependent participants relative to the controls (p < 0.05). Closed circles represent healthy control participants; open circles represent alcohol-dependent participants.
Adrenosteroid Pathways
In a preliminary exploration of potential disruptions in glucocorticoid, minerocorticoid, and androgen steroid pathways that may underlie alterations in cortisol responsiveness, we assessed several adrenosteroid steroids after cosyntropin administration (without dexamethasone pretreatment). Basal concentrations of 11-deoxycortisol were significantly lower in the alcohol-dependent group relative to controls (p < 0.04), suggesting that the disruption in cortisol basal release occurred before the conversion of cortisol from 11-deoxycortisol. All other basal and stimulated adrenosteroid measures did not demonstrate significant differences between the two groups. (See Table 3.)
Dexamethasone Suppression of Corticotropin and Cortisol
Dexamethasone, 8 mg, was administered intravenously at 2300 hr, and the corticotropin and cortisol responses were assessed from 2300 to 0500 hr. Both corticotropin (p < 0.004) and cortisol (p < 0.04) mean concentrations had greater suppression in the alcohol-dependent group compared with controls. Both groups suppressed corticotropin concentrations to levels approximately 30% of those observed during peak hours, and mean cortisol concentrations were below 2 μg/dl from 0200 hr until the administration of cosyntropin at 0800 hr. (See Table 3 and Fig. 2.)
Dexamethasone Plus Cosyntropin Stimulation Test
Cosyntropin, 0.01 μg/kg, was administered after dexamethasone suppression of endogenous corticotropin. Just before cosyntropin administration, the mean corticotropin concentration (0730 to 0800 hr) remained significantly lower in the alcohol-dependent group compared with controls (p < 0.005). Mean cortisol concentrations (0730 to 0800 hr), however, were equally diminished in the alcohol-dependent and control participants. The net integrated cortisol response to the cosyntropin challenge at 0800 hr was lower in alcohol-dependent patients compared with controls (p < 0.04). Individual participant data revealed that 7 of the 11 alcohol-dependent patients who received cosyntropin after dexamethasone had a cortisol net integrated response lower than the lowest control response. (See Table 3 and Fig. 2.)
Correlations Between Neuroendocrine Measures and Drinking History
The relation between drinking history (i.e., drinks in previous 30 days, lifetime drinks) and neuroendocrine measures were considered. Neuroendocrine measures included mean basal cortisol concentration before cosyntropin administration (without dexamethasone), integrated cortisol response after cosyntropin, mean corticotropin response to dexamethasone, and integrated cortisol response after cosyntropin with dexamethasone pretreatment. There was a significant negative correlation between drinks in the previous 30 days with basal cortisol (r = −0.746, df = 10, p < 0.02) and lifetime drinks with the mean corticotropin response to dexamethasone (r = −0.755, df = 11, p < 0.007). Therefore, the more alcohol use there was, the greater the decrease in basal cortisol was and the more sensitive the pituitary corticotrophs were to dexamethasone suppression. Drinks in the previous 30 days and lifetime drinks were also highly correlated (r = 0.713, df = 11, p < 0.02)
DISCUSSION
Our findings reveal (1) decreased circulating basal concentrations of cortisol during the late night and early morning hours, coupled with a decreased cortisol pulse amplitude; (2) attenuated adrenocorticoid response to a submaximal dose of cosyntropin in the relative absence of pituitary corticotropin stimulation but not its presence; (3) heightened pituitary responsiveness to glucocorticoid feedback; and (4) preliminary evidence for a disruption in glucocorticoid synthesis occurring before the synthesis of 11-deoxycortisol. In summary, our findings suggest a significant decrease in both basal cortisol concentrations and stimulated cortisol responsiveness in 1-month alcohol-dependent men compared with healthy controls. The predicted homeostatic response to the attenuated cortisol concentrations would be a decrease in pituitary glucocorticoid receptor sensitivity, which would disinhibit cortisol feedback and allow a more robust pituitary-adrenal response. The observed exaggerated corticotropin response to glucocorticoid feedback therefore suggests a maladaptive response to the subdued cortisol basal concentrations and stimulated response.
There were several methodological strengths to our study. Our patient population was highly selective in terms of clinical characteristics and abstinence interval. The period of abstinence was controlled such that the study occurred within 4 to 6 weeks after the last drink. Because basal concentrations of cortisol are increased during the first few days of withdrawal and alterations in HPA axis responsiveness persist for at least several weeks after the cessation of drinking, the timing of these neuroendocrine measures are of critical importance. The coupling of dexamethasone pretreatment and cosyntropin stimulation has been previously described by others (Crowley et al., 1991; Krishnan et al., 1990). Our methodology was unique, however, in using an intravenous form of dexamethasone, thus avoiding intragroup and intergroup differences in absorption and first-pass effects, and in using a dose high enough to induce a near-maximal suppression of glucocorticoid secretion. In fact, 9 hr after dexamethasone administration, cortisol concentrations were less than 2 μg/dl in all participants during the circadian peak (0800 hr). In our study, all participants tolerated dexamethasone without subjective complaints. Although impaired liver function in the alcohol-dependent patients could have resulted in increased dexamethasone concentrations in this group, our alcohol-dependent population did not show clinical evidence of cirrhotic liver disease, liver enzymes were for the most part not markedly above the normal range, and CBG was similar in both groups. These findings suggest a relatively similar hepatic efficacy in both participant groups. It should be noted that our findings may not be generalized to alcohol-dependent females, and preliminary findings from our laboratory suggest that 4- to 8-week-abstinent females tested during the early follicular phase of the menstrual cycle do not show findings similar to those reported here in the males. In addition, our sample size is small and assesses only severely alcohol-dependent men. Although the patients were mostly smokers and the controls were not, Anthenelli et al. (2001) have previously reported that cigarette smoking did not alter HPA axis responses to a pharmacologic challenge in abstinent alcohol-dependent individuals.
Evidence of decreased basal circulating cortisol concentrations and cortisol pulse amplitude was unexpected. Previous studies assessing circadian and/or pulsatile measures of cortisol release during abstinence did not reveal differences between healthy controls and alcohol-dependent participants (Adinoff et al., 1991; Iranmanesh et al., 1989; Loosen et al., 1991). The SBPP modeling technique (Guo et al., 1999) may have uncovered group differences in cortisol amplitude not reported by others. However, mean basal concentrations of cortisol from 2000 to 0800 hr were also lower in alcohol-dependent participants relative to controls. Whereas Loosen et al. (1991) reported an attenuation of early morning corticotropin in abstinent alcohol-dependent patients, we found no differences between groups in either pulsatile or mean basal measures of corticotropin. The careful exclusion of patients with a lifetime history of other psychiatric disorders and/or a recent history of drug dependence, coupled with a circumscribed period of abstinence, may explain these differences. Using isolated measures of plasma cortisol, some other investigators have also reported lower resting AM cortisol concentrations in alcohol-dependent participants relative to controls (Anthenelli et al., 2001; Bailly et al., 1989; Kemper et al., 1990). Our findings showing decreased basal concentrations of 11-deoxycortisol in the alcohol-dependent group suggest that the disruption in cortisol secretion occurs before the conversion of cortisol from 11-deoxycortisol by 11-β hydroxylase.
After the near elimination of endogenous adrenocortical stimulation by dexamethasone, the cosyntropin challenge produced significantly less cortisol secretion in abstinent alcohol-dependent participants compared with healthy controls. In the absence of pituitary-adrenal suppression by pretreatment with dexamethasone, no group differences in the cortisol response to cosyntropin were observed. We hypothesize that the higher early morning concentrations of basal cortisol masked the group differences in adrenocortical sensitivity, possibly because of our use of a very low dose of cosyntropin. Visual inspection of the cortisol response to cosyntropin in Figs. 1 and 2 suggests that the group difference in the cortisol response to cosyntropin is similar both with and without dexamethasone, but the dexamethasone pretreatment removes the pre-existing differences in basal concentration. Therefore, the peak cortisol response to cosyntropin without dexamethasone pretreatment is approximately 8 μg/dl, or 50%, above the basal concentration, whereas cortisol increases approximately 15 μg/dl, or 700%, above basal concentrations in response to cosyntropin subsequent to dexamethasone pretreatment. The decreased glucocorticoid response in the alcohol-dependent group after cosyntropin with dexamethasone pretreatment may also reflect lower basal concentrations of corticotropin in this group. However, postdexamethasone corticotropin concentrations are quite low in both groups (approximately 30% of concentrations without dexamethasone pretreatment), and the differences in mean corticotropin concentrations between the two groups are relatively small, albeit significant.
Wand and Dobs (1991) also reported a blunted glucocorticoid response to cosyntropin in 1-day-abstinent alcohol-dependent participants, after both a high dose (250 μg) and a low dose (0.250 μg) of cosyntropin. The attenuated cortisol response to cosyntropin was apparent even in the presence of marked withdrawal-induced hypercortisolemia, although the adrenal response may have been heightened during the withdrawal state. Others have reported a decreased cortisol response to more indirect stressors. Blunted cortisol responses to ovine CRH (Bailly et al., 1989) and insulin (Knudsen et al., 1987) have been reported in 1-month-abstinent alcohol-dependent participants in the presence of a corticotropin response similar to healthy controls. Several other investigators (Coiro and Vescovi, 1999; Costa et al., 1996; Ehrenreich et al., 1997; Vescovi et al., 1997) have detected an attenuated cortisol response in tandem with corticotropin suppression, suggesting that the blunted glucocorticoid response may simply have been a consequence of the antecedent diminished corticotropin response. In other studies (Chalmers et al., 1978; Errico et al., 1993; Krystal et al., 1996; Lovallo et al., 2000; Merry and Marks, 1972), corticotropin was not assessed, such that it could not be determined whether the cortisol response was muted in an environment of normal or diminished corticotropin responsiveness.
The mechanistic etiology of the lower basal concentrations and the blunted glucocorticoid response remains speculative. The decrease in adrenocortical sensitivity could reflect a genetic vulnerability to alcohol dependence rather than a consequence. For example, Schuckit (1984) reported a decreased cortisol response in the sons of alcohol-dependent fathers after an alcohol challenge. However, Wand et al. (1999) demonstrated that the adrenocortical response to cosyntropin in the high-risk sons and daughters of alcohol-dependent parents is not significantly different from the response of low-risk young adults. In addition, the significant relation between recent and lifetime alcohol use and some of our dependent neuroendocrine measures suggest that the current alterations in HPA axis responsiveness are a result of previous alcohol use. Consistent with our findings is the work of the Guaza and Borrell (1984), who found that bathing the adrenals of rodents in ethanol decreases the corticosterone response to corticotropin in a dose-dependent manner. This work suggested a direct effect of alcohol on the adrenal cortex, similar to the inhibitory effect that ethanol exerts on testicular androgen synthesis (Badr et al., 1977; Cicero, 1981; Ellingboe and Varanelli, 1979). Surprisingly, the literature offers little guidance with respect to the pathology of the adrenal cortex in alcohol-dependent subjects.
Because both basal and stimulated cortisol responses seem to be decreased in alcohol-dependent patients, the expected adaptive response of the axis would be to attenuate pituitary glucocorticoid inhibitory feedback. In the relative absence of pituitary glucocorticoid inhibition, a given stimulus would then allow a more robust corticotropin response to a given stimulus, resulting in a heightened downstream cortisol release. In fact, the opposite is observed; increased pituitary glucocorticoid inhibition is observed despite attenuated cortisol sensitivity and basal secretion. [Although a number of previous studies have reported that the cortisol response to dexamethasone did not differ in alcohol-dependent participants and controls (Costa et al., 1996; Khan et al., 1984; Zern et al., 1986), these studies used oral, low-dose (1 mg) dexamethasone and did not assess plasma corticotropin concentrations.] Therefore, we hypothesize the following scenario: Two distinct allostatic changes (Schulkin et al., 1994) occur in the pituitary-adrenal axis in response to a physiological environment of persistent alcohol- and/or withdrawal-induced hypercortisolemia; first, a decrease in glucocorticoid sensitivity relative to corticotropin stimulation, and second, a heightened pituitary glucocorticoid inhibitory feedback to endogenous cortisol. Both altered set points would result in attenuated glucocorticoid responsiveness. We suggest that these adaptive responses persist for at least approximately 1 month after the resolution of alcohol- and withdrawal-induced hypercortisolemia. The adrenocortical hyposensitivity could be a result of altered adrenocortical corticotrophin receptor sensitivity, second messenger changes, or adjustments in the steroid metabolic pathway. Although assessment of androgen and minerocorticoid pathways in our study did not reveal group differences after cosyntropin without dexamethasone pretreatment, we also did not observe a difference in the cortisol response after this stimulus. The large variance in these steroids may have resulted in a type 2 error. A comparison of other steroid pathways may also have been more informative after the cosyntropin stimulus with dexamethasone pretreatment, as would an assessment of neurosteroids originating in the adrenal cortex. These compounds are active at central γ-aminobutyric acid–mediated receptor sites and have been implicated in the pathophysiology of alcohol dependence (Morrow et al., 2001).
Hypoadrenal states have been reported in a number of other disorders (for review, see Heim et al., 2000), including PTSD, chronic fatigue syndrome, somatoform disorders, rheumatoid arthritis, and asthma. The clinical consequences of these hypoadrenal states, if any, remain speculative, and it is unknown whether the findings reported here occur after any disorder evoking prolonged activation of the HPA axis or are specific to alcohol dependence. With respect to abstinent alcohol-dependent patients, low basal or stimulated cortisol may not appropriately prime or activate brain regions [i.e., amygdala (Barrot et al., 2000), ventral tegmental area (Saal et al., 2003), hippocampus (Erickson et al., 2003)] involved in stress regulation. Disruption in the HPA axis response to stress may subsequently heighten the likelihood of drinking in the abstinent patient after a relapse trigger (Sinha, 2001). A recent study by Kosten et al. (2002) found that inhibition of cortisol synthesis by ketoconazole increased cocaine use in methadone patients. A blunted cortisol response to the Trier Social Stimulation Test (Junghanns et al., 2003) and lower cortisol concentrations (Kiefer et al., 2002) predicted early relapse in alcohol-dependent participants, and cortisol concentrations and craving were inversely correlated after short-term naltrexone administration (O’Malley et al., 2002).
To understand the relevance of decreased glucocorticoid responsiveness to the clinical picture of alcoholism, future studies should explore the relation between psychiatric and physiological measures affected by both chronic alcohol use and the glucocorticoids, such as affective instability, episodic memory, impulsivity, immune functioning, and energy utilization. The persistence of HPA axis responsiveness should also be assessed because this physiological disruption may portend relapse risk in even long-term abstinent patients. The mechanistic underpinnings of this finding can be explored by assessing other steroid secretion in tandem with the glucocorticoids. Finally, adrenocortical hyposensitivity in alcohol-dependent patients may suggest that medications that replace glucocorticoids (i.e., prednisone) may be useful in the treatment of alcohol dependence. Alternately, medications that are beneficial in the treatment of alcohol dependence may do so as a consequence of their ability to activate the HPA axis [i.e., naltrexone (Farren et al., 1999; Hernandez-Avila et al., 2002; King et al., 2002), nalmefene (Schluger et al., 1998)].
Acknowledgments
The authors thank the Dallas VA Substance Abuse Team for assistance in the recruitment and clinical care of patients and the UT Southwestern GCRC staff for their excellent patient care and meticulous attention to research protocol. The authors also thank Ali Iranmanesh, MD, for his conceptual and instructional assistance and Dr. Joan Reisch and Alan C. Elliott, MAS, for their statistical expertise.
Supported by Grant AA1570 from the National Institute on Alcohol Abuse and Alcoholism and Grant M01-RR00633 from USPHS.
Footnotes
Presented at the 57th Annual Convention of the Society of Biological Psychiatry, May 18, 2002, Philadelphia, Pennsylvania; the 2003 Scientific Meeting of the Research Society on Alcoholism, June 22, 2003, Ft. Lauderdale, Florida; and the 12th World Congress on Biomedical Alcohol Research, October 1, 2004, Heidelberg/Mannheim, Germany.
References
- Adinoff B, Martin PR, Bone GHA, Eckardt MJ, Roehrich L, George DT, Moss HB, Eskay R, Linnoila M, Gold PW. Hypothalamic-pituitary-adrenal axis functioning and cerebrospinal fluid corticotropin releasing hormone and corticotropin levels in alcoholics after recent and long-term abstinence. Arch Gen Psychiatry. 1990;47:325–330. doi: 10.1001/archpsyc.1990.01810160025004. [DOI] [PubMed] [Google Scholar]
- Adinoff B, Risher-Flowers D, De Jong J, Ravitz B, Bone GHA, Nutt DJ, Roehrich L, Martin PR, Linnoila M. Disturbances of hypothalamic-pituitary-adrenal axis functioning during ethanol withdrawal in six men. Am J Psychiatry. 1991;148:1023–1025. doi: 10.1176/ajp.148.8.1023. [DOI] [PubMed] [Google Scholar]
- Adinoff B, Ruether K, Krebaum S, Iranmanesh A, Williams MJ. Increased salivary cortisol concentrations during chronic alcohol intoxication in a naturalistic clinical sample of men. Alcohol Clin Exp Res. 2003;27:1420–1427. doi: 10.1097/01.ALC.0000087581.13912.64. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. American Psychiatric Association; Washington, DC: 1994. [Google Scholar]
- Anthenelli RM, Maxwell RA, Geracioti TDJ, Hauger R. Stress hormone dysregulation at rest and after serotonergic stimulation among alcohol-dependent men with extended abstinence and controls. Alcohol Clin Exp Res. 2001;25:692–703. [PubMed] [Google Scholar]
- Badr FM, Bartke A, Dalterio S, Bulger W. Suppression of testosterone production by ethyl alcohol: Possible mode of action. Steroids. 1977;30:647–655. doi: 10.1016/0039-128x(77)90054-x. [DOI] [PubMed] [Google Scholar]
- Bailly D, Dewailly D, Beuscart R, Couplet G, Dumont P, Racadot A, Fossati P, Parquet PJ. Adrenocorticotropin and cortisol responses to ovine corticotropin-releasing factor in alcohol dependence disorder. Horm Res. 1989;31:72–75. doi: 10.1159/000181090. [DOI] [PubMed] [Google Scholar]
- Barrot M, Marinelli M, Abrous DN, Rouge-Pont F, Le Moal M, Piazza PV. The dopaminergic hyper-responsiveness of the shell of the nucleus accumbens is hormone-dependent. Eur J Neurosci. 2000;12:973–979. doi: 10.1046/j.1460-9568.2000.00996.x. [DOI] [PubMed] [Google Scholar]
- Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1979;4:561–571. doi: 10.1001/archpsyc.1961.01710120031004. [DOI] [PubMed] [Google Scholar]
- Chalmers RJ, Bennie EH, Johnson RH, Masterton G. Growth hormone, prolactin, and corticosteroid responses to insulin hypoglycaemia in alcoholics. BMJ. 1978;1:745–748. doi: 10.1136/bmj.1.6115.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicero TJ. Neuroendocrinological effects of alcohol. Ann Rev Med. 1981;32:123–142. doi: 10.1146/annurev.me.32.020181.001011. [DOI] [PubMed] [Google Scholar]
- Coiro V, Vescovi PP. Effect of cigarette smoking on ACTH/cortisol secretion in alcoholic after short- and medium-term abstinence. Alcohol Clin Exp Res. 1999;23:1515–1518. [PubMed] [Google Scholar]
- Costa A, Bono G, Martignoni E, Merlo P, Sances G, Nappi G. An assessment of hypothalamo-pituitary-adrenal axis functioning in non-depressed, early abstinent alcoholics. Psychoneuroendocrinology. 1996;21:263–275. doi: 10.1016/0306-4530(96)00001-7. [DOI] [PubMed] [Google Scholar]
- Crowley S, Hindmarsh PC, Holownia P, Honour JW, Brook CG. The use of low doses of ACTH in the investigation of adrenal function in man. J Endocrinol. 1991;130:475–479. doi: 10.1677/joe.0.1300475. [DOI] [PubMed] [Google Scholar]
- Ehrenreich H, Schuck J, Stender N, Pilz J, Gefeller O, Schilling L, Poser W, Kaw S. Endocrine and hemodynamic effects of stress versus systemic CRF in alcoholics during early and medium term abstinence. Alcohol Clin Exp Res. 1997;21:1285–1293. [PubMed] [Google Scholar]
- Ellingboe J, Varanelli CC. Ethanol inhibits testosterone biosynthesis by direct action on Leydig cells. Res Commun Chem Pathol Pharmacol. 1979;24:87–102. [PubMed] [Google Scholar]
- Erickson K, Drevets W, Schulkin J. Glucocorticoid regulation of diverse cognitive functions in normal and pathological emotional states. Neurosci Biobehav Rev. 2003;27:233–246. doi: 10.1016/s0149-7634(03)00033-2. [DOI] [PubMed] [Google Scholar]
- Errico AL, Parson OA, King AC, Lovallo WR. Attenuated cortisol response to biobehavioral stressors in sober alcoholics. J Stud Alcohol. 1993;54:393–398. doi: 10.15288/jsa.1993.54.393. [DOI] [PubMed] [Google Scholar]
- Farren CK, O’Malley S, Grebski G, Maniar S, Porter M, Kreek MJ. Variable dose naltrexone-induced hypothalamic-pituitary-adrenal stimulation in abstinent alcoholics: A preliminary study. Alcohol Clin Exp Res. 1999;23:502–508. [PubMed] [Google Scholar]
- First MH, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV Axis I Disorders, patient ed (SCID-I/P, version 2.0) Biometrics Research Department, New York State Psychiatric Institute; New York: 1996. [Google Scholar]
- Guaza C, Borrell J. Effect of ethanol on corticosterone production by dispersed adrenal cells of the rat. Life Sci. 1984;35:1191–1196. doi: 10.1016/0024-3205(84)90190-5. [DOI] [PubMed] [Google Scholar]
- Guo W, Wang Y, Brown MB. A signal extraction approach to modeling hormone time series with pulses and a changing baseline. J Am Stat Assoc. 1999;94:746–756. [Google Scholar]
- Heim C, Ehlert U, Hellhammer DH. The potential role of hypocortisolism in the pathophysiology of stress-related bodily disorders. Psychoneuroendocrinology. 2000;25:1–35. doi: 10.1016/s0306-4530(99)00035-9. [DOI] [PubMed] [Google Scholar]
- Hernandez-Avila CA, Oncken C, Van Kirk J, Wand G, Kranzler HR. Adrenocorticotropin and cortisol responses to a naloxone challenge and risk of alcoholism. Biol Psychiatry. 2002;51:652–658. doi: 10.1016/s0006-3223(01)01334-8. [DOI] [PubMed] [Google Scholar]
- Inder WJ, Joyce PR, Ellis MJ, Evans MJ, Livesey JH, Donald RA. The effects of alcoholism on the hypothalamic-pituitary-adrenal axis: Interaction with endogenous opioid peptides. Clin Endocrinol (Oxf) 1995;43:283–290. doi: 10.1111/j.1365-2265.1995.tb02033.x. [DOI] [PubMed] [Google Scholar]
- Iranmanesh A, Veldhuis JD, Johnson ML, Lizarralde G. 24-hour pulsatile and circadian patterns of cortisol secretion in alcoholic men. J Androl. 1989;10:54–63. doi: 10.1002/j.1939-4640.1989.tb00062.x. [DOI] [PubMed] [Google Scholar]
- Jeffcoate W. Alcohol-induced pseudo-Cushing’s syndrome. Lancet. 1993;341:676–677. doi: 10.1016/0140-6736(93)90434-i. [DOI] [PubMed] [Google Scholar]
- Junghanns K, Backhaus J, Tietz U, Lange W, Bernzen J, Wetterling T, Rink L, Driessen M. Impaired serum cortisol stress response is a predictor of early relapse. Alcohol Alcohol. 2003;38:189–193. doi: 10.1093/alcalc/agg052. [DOI] [PubMed] [Google Scholar]
- Keedwell PA, Poon L, Papadopoulos AS, Marshall EJ, Checkley SA. Salivary cortisol measurements during a medically assisted alcohol withdrawal. Addict Biol. 2001;6:247–256. doi: 10.1080/13556210120056580. [DOI] [PubMed] [Google Scholar]
- Kemper A, Koalick F, Thiele H, Retzow A, Rathsack R, Nickel B. Cortisol and beta-endorphin response in alcoholics and alcohol abusers following a high naloxone dosage. Drug Alcohol Depend. 1990;25:319–326. doi: 10.1016/0376-8716(90)90158-b. [DOI] [PubMed] [Google Scholar]
- Khan A, Ciraulo DA, Nelson WH, Becker JT, Nies A, Jaffe JH. Dexamethasone suppression test in recently detoxified alcoholics: Clinical implications. J Clin Psychopharmacol. 1984;4:94–97. doi: 10.1097/00004714-198404020-00006. [DOI] [PubMed] [Google Scholar]
- Kiefer F, Jahn H, Schick M, Wiedemann K. Alcohol self-administration, craving and HPA-axis activity: An intriguing relationship. Psychopharmacology (Berl) 2002;164:239–240. doi: 10.1007/s00213-002-1255-3. [DOI] [PubMed] [Google Scholar]
- King AC, Schluger J, Gunduz M, Borg L, Perret G, Ho A, Kreek MJ. Hypothalamic-pituitary-adrenocortical (HPA) axis response and biotransformation of oral naltrexone: Preliminary examination of relationship to family history of alcoholism. Neuropsychopharmacology. 2002;26:778–788. doi: 10.1016/S0893-133X(01)00416-X. [DOI] [PubMed] [Google Scholar]
- Knudsen GM, Christensen H, Benld D, Melgaard B, Kirkegaard C, Hasselbalch H. Discordance between the cortisol response to insulin-hypoglycemia and 30-minute ACTH stimulation test in chronic alcoholic men. Alcohol Clin Exp Res. 1987;11:323–325. doi: 10.1111/j.1530-0277.1987.tb01318.x. [DOI] [PubMed] [Google Scholar]
- Kosten TR, Oliveto A, Sevarino KA, Gonsai K, Feingold A. Ketoconazole increases cocaine and opioid use in methadone maintained patients. Drug Alcohol Depend. 2002;66:173–180. doi: 10.1016/s0376-8716(01)00198-3. [DOI] [PubMed] [Google Scholar]
- Krishnan KRR, Ritchie JC, Saunders WB, Nemeroff CB, Carroll BJ. Adrenocortical sensitivity to low-dose ACTH administration in depressed patients. Biol Psychiatry. 1990;27:930–933. doi: 10.1016/0006-3223(90)90476-i. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Webb E, Cooney NL, Kranzler HR, Southwick SW, Heninger GR, Charney DS. Serotonergic and noradrenergic dysregulation in alcoholism: m-Chlorophenylpiperazine and yohimbine effects in recently detoxified alcoholics and healthy comparison subjects. Am J Psychiatry. 1996;153:83–92. doi: 10.1176/ajp.153.1.83. [DOI] [PubMed] [Google Scholar]
- le Roux CW, Sivakumaran S, Alaghband-Zadeh J, Dhillo W, Kong WM, Wheeler MJ. Free cortisol index as a surrogate marker for serum free cortisol. Ann Clin Biochem. 2002;39:406–408. doi: 10.1258/000456302760042182. [DOI] [PubMed] [Google Scholar]
- Loosen PT, Chambliss B, Pavlou SS, Orth DN. Adrenal function in abstinent alcoholic men. Prog Neuropsychopharmacol Biol Psychiatry. 1991;15:771–780. doi: 10.1016/0278-5846(91)90006-m. [DOI] [PubMed] [Google Scholar]
- Lovallo WR, Dickensheets SL, Myers DA, Thomas TL, Nixon SJ. Blunted stress cortisol response in abstinent alcoholic and polysubstance-abusing men. Alcohol Clin Exp Res. 2000;24:651–658. [PubMed] [Google Scholar]
- Margraf HW, Moyer CA, Ashford LE, Lavalle LW. Adrenocortical function in alcoholics. J Surg Res. 1967;7:55–62. doi: 10.1016/0022-4804(67)90035-2. [DOI] [PubMed] [Google Scholar]
- Mendelson JH, Ogata M, Mello NK. Adrenal function and alcoholism: I. Serum cortisol. Psychosom Medicine. 1971;33:145–157. doi: 10.1097/00006842-197103000-00006. [DOI] [PubMed] [Google Scholar]
- Merry J, Marks V. The effect of alcohol, barbiturate, and diazepam on hypothalamic, pituitary/adrenal function in chronic alcoholics. Lancet. 1972;2:990–992. doi: 10.1016/s0140-6736(72)92403-8. [DOI] [PubMed] [Google Scholar]
- Miller WR, Tonigan JS, Longabaugh R. The Drinker Inventory of Consequences (DrlnC): An Instrument for Assessing Adverse Consequences of Alcohol Abuse. National Institutes of Health; Rockville, MD: 1995. [Google Scholar]
- Morrow AL, VanDoren MJ, Fleming R, Penland S. Ethanol and neurosteroid interactions in the brain. Int Rev Neurobiol. 2001;46:349–377. doi: 10.1016/s0074-7742(01)46068-5. [DOI] [PubMed] [Google Scholar]
- O’Malley SS, Krishnan-Sarin S, Farren C, Sinha R, Kreek J. Naltrexone decreases craving and alcohol self-administration in alcohol-dependent subjects and activates the hypothalamo-pituitary-adrenocortical axis. Psychopharmacology (Berl) 2002;160:19–29. doi: 10.1007/s002130100919. [DOI] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Schluger JH, Ho A, Borg L, Porter M, Maniar S, Gunduz M, Perret G, King A, Kreek MJ. Nalmefene causes greater hypothalamic-pituitary-adrenal axis activation than naloxone in normal volunteers: Implications for the treatment of alcoholism. Alcohol Clin Exp Res. 1998;22:1430–1436. doi: 10.1111/j.1530-0277.1998.tb03931.x. [DOI] [PubMed] [Google Scholar]
- Schuckit MA. Differences in plasma cortisol after ingestion of ethanol in relatives of alcoholics and controls: Preliminary results. J Clin Psychiatry. 1984;45:374–376. [PubMed] [Google Scholar]
- Schulkin J, McEwen BS, Gold PW. Allostasis, amygdala, and anticipatory angst. Neurosci Biobehav Rev. 1994;18:385–396. doi: 10.1016/0149-7634(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Sinha R. How does stress increase risk of drug abuse and relapse? Psychopharmacology (Berl) 2001;158:343–359. doi: 10.1007/s002130100917. [DOI] [PubMed] [Google Scholar]
- Sobell MB, Sobell LC. Behavioral Treatment of Alcohol Problems. Plenum Press; New York: 1978. [Google Scholar]
- Stokes E. Adrenocortical activation in alcoholics during chronic drinking. Ann NY Acad Sci. 1973;215:77–83. doi: 10.1111/j.1749-6632.1973.tb28251.x. [DOI] [PubMed] [Google Scholar]
- Veldman RG, Meinders AE. On the mechanism of alcohol-induced pseudo-Cushing’s syndrome. Endocr Rev. 1996;17:262–268. doi: 10.1210/edrv-17-3-262. [DOI] [PubMed] [Google Scholar]
- Vescovi PP, DiGennaro C, Coiro V. Hormonal (ACTH, cortisol, beta-endorphin, and met-enkephalin) and cardiovascular responses to hyperthermic stress in chronic alcoholics. Alcohol Clin Exp Res. 1997;21:1195–1198. [PubMed] [Google Scholar]
- Wand GS, Dobs AS. Alterations in the hypothalamic-pituitary-adrenal axis in actively drinking alcoholics. J Clin Endocrinol Metab. 1991;72:1290–1295. doi: 10.1210/jcem-72-6-1290. [DOI] [PubMed] [Google Scholar]
- Wand GS, Mangold D, Ali M, Giggey P. Adrenocortical responses and family history of alcoholism. Alcohol Clin Exp Res. 1999;23:1185–1190. [PubMed] [Google Scholar]
- Zern MA, Halbreich U, Bacon K, Galanter M, Kang B-J, Gasparini F. Relationship between serum cortisol, liver function, and depression in detoxified alcoholics. Alcohol Clin Exp Res. 1986;10:320–322. doi: 10.1111/j.1530-0277.1986.tb05097.x. [DOI] [PubMed] [Google Scholar]