

Abstract
In addition to providing a pathway for intercellular communication, the gap junction-forming proteins, connexins, can serve a growth-suppressive function that is both connexin and cell-type specific. To assess its potential growth-suppressive function, we stably introduced connexin 37 (Cx37) into connexin-deficient, tumorigenic rat insulinoma (Rin) cells under the control of an inducible promoter. Proliferation of these iRin37 cells, when induced to express Cx37, was profoundly slowed: cell cycle time increased from 2 to 9 days. Proliferation and cell cycle time of Rin cells expressing Cx40 or Cx43 did not differ from Cx-deficient Rin cells. Cx37 suppressed Rin cell proliferation irrespective of cell density at the time of induced expression and without causing apoptosis. All phases of the cell cycle were prolonged by Cx37 expression, and progression through the G1/S checkpoint was delayed, resulting in accumulation of cells at this point. Serum deprivation augmented the effect of Cx37 to accumulate cells in late G1. Cx43 expression also affected cell cycle progression of Rin cells, but its effects were opposite to Cx37, with decreases in G1 and increases in S-phase cells. These effects of Cx43 were also augmented by serum deprivation. Cx-deficient Rin cells were unaffected by serum deprivation. Our results indicate that Cx37 expression suppresses cell proliferation by significantly increasing cell cycle time by extending all phases of the cell cycle and accumulating cells at the G1/S checkpoint.
Keywords: gap junction, tumor, cancer, angiogenesis
gap junctions have long been recognized as serving growth regulatory and tumor suppressor functions (40), but the underlying mechanism(s) remain uncertain. With the discovery that gap junctions are composed of one or more of the 21 members of the connexin gene family (56), the possibility of connexin-specific roles for these proteins in supporting tissue function seemed increasingly likely (29). Since nearly all cells express multiple connexin isoforms, identification of their unique contributions to cell and tissue function has been difficult. Nevertheless, connexin-specific deletion and replacement studies (32, 49, 54) emphasize the importance of specific connexins to development and tissue function (1, 44, 46, 60). The specific properties of particular connexins that render them irreplaceable in the animal remain ill-defined. However, from expression studies it is clear that connexin-specific properties include channel permselectivity, regulation by intracellular signaling cascades, and protein-protein interactions (29, 30).
Connexins are thought to contribute to coordinated tissue function and growth suppression in (at least) three ways. First, through formation of intercellular channels and gap junctions, their best documented role, connexins mediate direct intercellular exchange of small molecules and metabolites that support tissue homeostasis and electrical and chemical signaling. This intercellular signaling contributes to coordinated tissue responses such as contraction/relaxation and secretion, as well as controlled growth, cell proliferation, and cell migration (14, 20, 21, 48). Second, through formation of functional hemichannels, connexins participate in transmembrane signaling, mediating autocrine and paracrine responses especially during injury, ischemia, and inflammation (18, 28, 62). Finally, through selective protein-protein interactions, connexins may contribute to controlled growth, proliferation, apoptosis, and possibly other cell and tissue behaviors (27, 29).
Numerous studies indicate that connexins serve a tumor suppressor function. In mice with targeted deletions of connexin 43 (Cx43) or Cx32, susceptibility to radiation-induced tumorigenesis is significantly increased (30). Typically, tumorigenesis is linked to decreased gap junctional intercellular communication, reduced cell-cell contact, and enhanced cell proliferation (4, 25, 52). Transfection of connexin-encoding genes (e.g., for Cx43, Gja1) into some tumorigenic cell lines slows cell proliferation rates and tumor growth (25, 29), but underlying mechanisms of growth suppression remain uncertain. However, given the variable efficacy of a specific connexin in suppressing growth of diverse tumor types, it appears that the cellular milieu likely modulates the efficacy of growth-suppression by specific connexins. Thus it is currently unclear whether connexin-mediated growth suppression is unique to certain connexins or whether it is a shared feature of all connexins (with possible mechanistic differences).
With regard to Cx37, there are no published data concerning its growth-suppressive potential in tumorigenic or normal cells and tissues. However, data from studies of the response of the vascular endothelium to injury and other proliferation-inducing stimuli suggest such a role for Cx37. For example, whereas the endothelial cells of mature blood vessels with normal laminar blood flow are quiescent (39), in regions of chronic high shear or turbulent flow, endothelial cell proliferation rates increase (37). In comparably stimulated regions, Cx37 expression is decreased, Cx43 expression is increased, and Cx40 expression is largely unchanged (15). These studies and others (26, 55) indicate that these connexins likely make unique contributions to regulated cell proliferation in vivo.
The goal of the current study was to determine whether Cx37 may serve a growth-suppressive function and, if so, to begin identifying the underlying mechanisms. Using an inducible expression system, we show that expressed Cx37 forms functional gap junction channels. However, unlike Cx40 or Cx43, Cx37 expression drastically slowed the proliferation of rat insulinoma (Rin) cells, significantly increased their cell cycle duration by extending all phases therein, and accumulated these cells at the G1/S checkpoint. These cell cycle effects of Cx37 were augmented by serum deprivation.
MATERIALS AND METHODS
Antibodies and reagents.
All general chemicals, unless otherwise noted, were purchased from Sigma-Aldrich Chemical. Two Cx37 antibodies [either αCx37-18264 (53) or αCx37-ADI (catalog no: Cx37A11-A; Alpha-Diagnostics)] were used for both immunoblots and immunocytochemistry. Cx43 antibody was from Sigma (C6219); Cx40 antibody is an affinity-purified, rabbit anti-rat Cx40 (residues 234-332)-glutathione S-transferase fusion protein; Cy3-conjugated anti-rabbit IgG (Jackson Immunoresearch) was used for immunocytochemistry; horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence strategies were used in immunoblotting [SuperSignal West Pico System (catalog no. 32106, Pierce) or SuperSignal West Femto System (catalog no. 34095, Pierce), as appropriate].
Cell culture and expression vectors.
Communication-deficient Rin cells [Rin1046-38 (6, 16)] were obtained from R. Lynch (University of Arizona). Cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS; Gemini Bioproducts, Sacramento, CA) and antibiotics (300 μg/ml penicillin and 500 μg/ml streptomycin) at 37°C in a humidified, 5% CO2 incubator. pTET-ON (Clontech, Mountain View, CA) was transfected into Rin cells using Lipofectamine (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Cells were selected in 300 μg/ml of G418 (GIBCO-Invitrogen), subcloned, and screened for inducible expression of transiently transfected luciferase according to the manufacturer's instructions. The clone exhibiting the highest luciferase expression, iRin, was selected for further use. Mouse Cx37 (mCx37) was subcloned from a pCIneo-Cx37 construct (provided by Karen Hirschi, Baylor College of Medicine, Houston, TX) into pTRE2-hygro (Clontech) using the BamH I [carried from pBS-SK+ when subcloned into pCIneo's NheI and XhoI sites (61)] and NotI. The iRin cells were transfected with the resulting pTRE2-mCx37 plasmid using Lipofectamine (Invitrogen) according to the manufacturer's instructions. Stably transfected cells were selected in 100 μg/ml hygromycin and dilution subcloned.
Immunoblotting.
Whole cell protein was prepared as follows. Cells were washed three times with PBS and harvested by scraping. Cells were pelleted, lysed in sample buffer [100 mM Tris, 4% SDS, 10% glycerol, 5 mM NaF, 0.25 mM Na3VO4, 2 mM PMSF, and 0.02% bromophenol blue with added protease inhibitor cocktail (catalog no. 11836153001, Roche); pH 6.8], sonicated briefly, and debris was removed by centrifugation at 16,000 g for 10 min. Protein content of the supernatant was measured using the BCA assay (Pierce Chemical, Rockford, IL). Samples (5–50 μg total protein) were electrophoresed on 12% SDS-PAGE gels (Bio-Rad, Hercules, CA) and transferred to nitrocellulose for detection by αCx37-18264 or αCx37-ADI.
The Triton X-100-insoluble fraction was isolated as follows. Cells were rinsed with PBS and scraped from the dish after addition of lysis buffer [1% Triton X-100, 5 mM NaF, 0.25 mM Na3VO4, and 2 mM PMSF with added protease inhibitor cocktail (Roche)]. This lysate was homogenized (4 passes through a 26-gauge needle), and the Triton X-100-insoluble fraction was pelleted by centrifugation (16,000 g, 10 min). Sample buffer was added to solubilize the Triton X-100-insoluble pellet, and after brief sonication, protein content was analyzed as described above.
Immunocytochemistry.
Cells plated on glass coverslips for a minimum of 24 h were fixed in 100% methanol (−20°C, 5 min) or 70% ethanol (4°C, overnight). For immunofluorescent detection of connexin, cells were treated with 0.2% Triton X-100 and 0.5 M NH4Cl (with preceding and intervening PBS washes) and incubated in blocking reagent (10 min; 4% fish skin gelatin, 1% normal goat serum, and 0.25% Triton X-100 in PBS) before incubation in primary and then secondary antibody (both for 2–3 h at room temperature with intervening washes with blocking reagent). Connexin proteins were detected using the specified antibodies and Cy3-conjugated secondary antibody (diluted 1:200 in blocking reagent). Proliferating cell nuclear antigen (PCNA) was visualized with a commercially available PCNA staining kit (Zymed 93-1143) used according to the manufacturer's directions.
Proliferation.
Proliferation assays were performed on cells plated in six-well plates, with an initial seeding density of 7.2, 10, or 30 × 103 cells/well (approximately 750–3,125 cells/cm2; most experiments were performed at the highest density). Twenty-four hours after plating (day 0 of proliferation assays), doxycycline (2 μg/ml, unless otherwise indicated) was added to three of six wells devoted to each time point. Cells were fed every 48 h with or without added doxycycline, as appropriate. Every 3 days for a total of 21 days, the cells in each well devoted to a time point were harvested (using trypsin), and the number of cells per well was determined using a hemocytometer.
Cell cycle analysis.
Subconfluent plates of cells in complete medium containing 10% FBS were induced or not to express Cx37 for 2 or 5 days, as indicated; Rin40 and Rin43 cells were cultured similarly but were analyzed 48 h after plating. All cells in the dish were then harvested for analysis of cell cycle position as follows. Cells were trypsinized, pelleted, and resuspended in 1 ml medium, and cell number was determined (hemocytometer). The cells were then pelleted, and the pellets were fixed in 1 ml ice-cold 70% ethanol with vigorous vortexing. After repelleting, the ethanol was removed and the cells were resuspended (at a density of 2 × 106 cells/ml) in cold PBS to which 50 μg RNase A total and 50 μg/ml propidium iodide were added. After incubation at 37°C for 30 min, the cells were transferred to ice and analyzed by fluorescence-activated cell sorting (FACS). Because Cx37 has been reported to reduce adhesivity in some cell types (62) and to induce apoptosis in others (51), the position of unattached cells in the cell cycle was also evaluated.
In some experiments, the impact of serum starvation (0% FBS) with or without Cx37 expression on cell cycle progression was evaluated. Rin1046 cells or iRin37-F cells were plated in 100-mm plates at a density of approximately 12–38 × 103 cells/cm2 in 10% FBS. Twenty-four hours later, culture medium was replaced with medium containing either 10% or 0% FBS, and after 48 h, doxycycline was added (or not) to each plate for 24 h. After this induction period, medium containing 10% FBS (with or without doxycycline, as appropriate) was restored to all plates, and cells were harvested immediately or 4–96 h later.
Junctional conductance.
Confluent cells were trypsinized and replated at low density onto glass coverslips, with doxycycline added at the time of replating. After 24 h, coverslips were mounted in a custom-made chamber, and an Olympus inverted (IMT2) microscope with phase contrast optics was used to identify pairs of cells in the dish (typically only two or three pairs of cells were found on any given 25-mm coverslip). Cells were bathed in external solution containing (in mM) 142.5 NaCl, 4 KCl, 1 MgCl2, 5 glucose, 2 sodium pyruvate, 10 HEPES, 15 CsCl, 10 TEA-Cl, 1 BaCl2, and 1 CaCl2, pH 7.2 and osmolarity of 330 mosM. Junctional conductance was determined on all pairs found within 30 min (the window of time to which all our electrophysiology experiments are typically restricted) using dual whole cell voltage-clamp techniques as previously described (34). The pipette solution contained (in mM) 124 KCl, 14 CsCl, 9 HEPES, 9 EGTA, 0.5 CaCl2, 5 glucose, 9 TEA-Cl, 3 MgCl2, and 5 disodium ATP, pH 7.2 and osmolarity of 326 mosM. Junctional conductance was evaluated with 10-mV transjunctional pulses, and channel conductance was evaluated with a 25-mV transjunctional voltage difference.
Statistical analysis.
Statistical comparisons of junctional conductance and cell cycle data were conducted using unpaired, two-tailed Student's t-tests. Significant differences are indicated by P < 0.05. Data from serum starvation experiments were subjected to single-factor ANOVA, and where significance was indicated, multiple comparisons were performed using Tukey's test for unequal sample sizes (63).
RESULTS
Cx37-expressing Rin cells.
Our initial attempts to isolate a cell line that constitutively expressed Cx37 were unsuccessful. Stable introduction of Cx37 into Rin and 23-3 [Cx43−/− fibroblasts (41)] cell lines was attempted using pCIneo, expression driven by the cytomegalovirus promoter, or pHβApr-4 (19, 45), expression driven by the β-actin promoter. Cx37 was detected in the transfected cells 48 h after transfection, but it could not be detected in several neomycin-resistant clonal cell populations derived from these transfectants (see Fig. 1S in supplemental data, available in the online version of this article at the American Journal of Physiology-Cell Physiology website). Since both of these cell lines have been used successfully to express other connexin proteins (22, 36, 41), we hypothesized that cells expressing Cx37 failed to proliferate and, consequently, during dilution cloning, were overgrown by non-Cx37-expressing, but neomycin-resistant cells.
To test this hypothesis, we transfected the Rin cells with the pTET-ON vector and isolated a cell line, iRin, into which the gene encoding Cx37 was introduced using the pTRE2-hygromycin vector (for doxycycline-inducible expression of Cx37). Several hygromycin-resistant clones of iRin-Cx37 (iRin37) cells were isolated, two (the F and H clones; see supplemental data Fig. 1S-B) were selected for further study. Cx37 expression by these clones was dose and time dependent, with maximal expression occurring at 24 h and 2 μg/ml doxycycline (see supplemental data Fig. 2S). Immunofluorescence techniques were used to determine Cx37 localization in doxycycline-induced and -noninduced cells. As shown in Fig. 1, Cx37 was readily detected in iRin37-F cells induced with 0.25 and 2 μg/ml doxycycline (Fig. 1, B and C) but not evident in noninduced cells (Fig. 1A). Punctate labeling in apparent appositional membrane was sometimes observed (inset, Fig. 1C). Cx37 was readily detected in immunoblots of total protein (whole cell) as well as in the Triton X-100-insoluble fraction (Fig. 1C) isolated from induced cells, but it was generally absent from noninduced cells (4 of 21 distinct cell isolates showed a faint Cx37 band with ≥25 μg total protein loaded). The presence of a Triton-insoluble component and occasional punctate labeling of appositional areas suggested that Cx37 may be assembled by Rin cells into functional gap junctions (43).
Fig. 1.

Connexin 37 (Cx37) is significantly induced, forms occasional plaques, and appears in the Triton X-100-insoluble fraction of cell homogenates. A–C: cells were stained with Hoechst dye (nuclear stain, blue) and immunostained with Cx37-18264 antibody (Cy3-conjugated secondary, pink); overlays of the two images are presented. Cx37 labeling is evident in nearly all cells induced with either 0.25 μg/ml (B) or 2 μg/ml (C) doxycycline, but it is not detected in noninduced (A) cells. Obvious gap junction plaques were infrequent at both doxycyline concentrations; nevertheless, punctae at regions of cell-cell contact were occasionally observed (inset in C; magnification is 3.5-fold higher than in A, where scale bar represents 20 μm). D: Western blot of total [whole cell (WC)] and Triton-insoluble (TX) protein isolated from induced (Dox+) and noninduced (Dox−) cells. Doxycycline induced a significant increase in Cx37 expression (compare WC Dox+ and WC Dox− lanes). Cx37 was also readily detected in the Triton X-100-insoluble fraction. (WC−, WC+, and TX+ lanes were loaded with 33, 21, and 33 μg protein, respectively.)
Cx37 forms functional gap junctions.
To confirm that the iRin37-F and -H clones formed functional gap junctions, junctional conductance was measured using dual whole cell voltage-clamp procedures. Despite significantly different levels of protein expression by these clones (supplemental data Fig. 1S-B), neither the incidence of coupling (50% for iRin-F; 64% for iRin-H) nor its extent differed between clones (Fig. 2A). As expected (33, 47, 57), the conductance of the fully open channel was ∼350 pS, and multiple open states were observed both in poorly coupled pairs (Fig. 2B) and in better coupled pairs following halothane application (to reduce open probability; data not shown). Parental Rin and iRin cells were not coupled (data not shown).
Fig. 2.
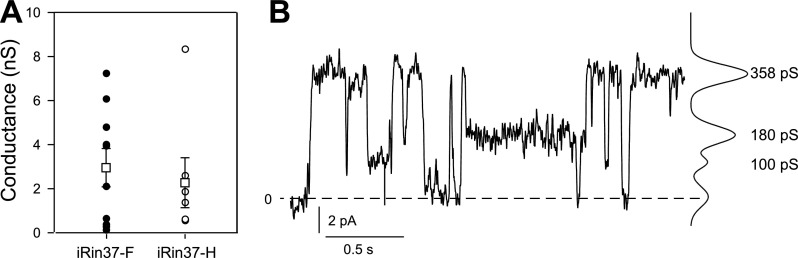
Functional gap junctions and channels are formed by rat insulinoma (Rin) cells induced to express Cx37 (iRin37). A: mean conductance and individual data points for junctions formed by pairs of iRin37-F and -H cells. Means were not different despite different expression levels (see supplemental data Fig. 1S). Data for iRin37-F clone were derived from 7 different cultures on 7 distinct days, and data for iRin37-H were from 2 different cultures and days. Multiple coverslips of cells were used each day. B: single-channel record and all points histogram from a pair of iRin 37-F cells. Conductance of the fully open channel was ∼350 pS, and at least two substates (100 and 180 pS) were observed.
Proliferation is suppressed in Cx37-expressing cells.
To determine whether expression of Cx37 slowed or prevented proliferation of Rin cells, we compared the proliferative properties of induced iRin37 cells to Rin, iRin, Rin40, Rin43, and noninduced iRin37 cells over a 21-day period. As shown in Fig. 3, iRin, Rin40, Rin43, Rin (all with or without doxycycline treatment), and noninduced iRin37 cells grew rapidly (between days 3 and 15) until density-dependent slowing of proliferation occurred. In contrast, proliferation of Cx37-expressing cells (induced iRin37-F and iRin37-H) was significantly depressed over this time period. Induction of Cx37 expression in cells growing at approximately 10-fold higher density (Fig. 4) also profoundly slowed proliferation, suggesting that growth suppression by Cx37 was density independent. Localization of Cx40 in the Rin40 cells and of Cx43 in the Rin43 cells was comparable to that of Cx37 in the induced iRin37-F cells (Fig. 5), and as observed for the iRin37-F cells, obvious plaques were infrequent. That Cx40- and Cx43-expressing Rin cells, which form functional gap junctions (7, 8, 12, 23), proliferate comparably to connexin-deficient Rin cells (Rin, iRin, and noninduced iRin37-F) suggests that formation of functional gap junctions, per se, between Rin cells is not sufficient to explain growth suppression by Cx37.
Fig. 3.

Proliferation of Rin cells was suppressed by Cx37 but not by Cx40 or Cx43 expression. In the absence of Cx37 expression, iRin37-F and -H cells proliferated comparably to the parental iRin cells (A). However, when Cx37 expression was induced (B), proliferation of the 37-F and -H clones was suppressed. Constitutive expression of rat Cx40 (Rin40) or rat Cx43 (Rin43) did not affect cell proliferation, despite the absence (C) or presence (D) of doxycycline. Each point represents the average of 3 wells of cells; where error bars (SE) are not evident they are obscured by the symbols.
Fig. 4.

Cx37-induced suppression of proliferation is density independent and has a rapid onset. iRin37-F cells were plated at 3 × 104 cells per well. Doxycycline was added to one third of wells 24 h later (Dox+ data) and to another third 12 days later (Dox: 12d−, 9d+ data); the remaining wells were untreated (no Dox). Cell number was monitored at 3-day intervals for 21 days for all treatment groups. Proliferation of cells exposed to doxycycline was suppressed irrespective of cell density at the time of induction.
Fig. 5.

Cx37, Cx40, and Cx43 are comparably localized and obvious gap junction plaques are largely absent in iRin37-F, Rin40, and Rin43 cells. Shown are iRin37-F (A–C), Rin40 (D–F), and Rin43 (G–I) cells. Cx-specific labeling is shown in A, D, and G and as overlays (pink) with Hoechst nuclear stain (blue) in B, E, and H; phase contrast images of immunostained fields are shown in C, F, and I. Scale bar represents 20 μm and applies to all panels.
Cx37 does not induce apoptosis.
A previous study demonstrated that adenoviral delivery of human Cx37 to human umbilical vein endothelial cells induced apoptosis (51). To determine whether failed proliferation of induced iRin37-F cells might reflect induction of apoptosis, we grew cells for 7 days in the absence of doxycycline, added (or not) doxycycline for 2 or 5 days, and used FACS on propidium iodide-stained cells (Fig. 6) to determine whether an apoptotic population was present. Apoptotic cells were generally absent in both induced (for either 2 or 5 days) and noninduced cells [%apoptotic cells at 2 days: no doxycycline (Dox) 0.042 ± 0.031 (n = 5), with Dox 0.022 ± 0.016 (n = 6), P > 0.5; at 5 days: no Dox 0.028 ± 0.020 (n = 5), with Dox 0.216 ± 0.19 (n = 7), P > 0.3]. To verify that apoptotic cells were not preferentially lost from the surface of the dish, a similar comparison was made for unattached cells, and again apoptotic cells were generally absent [%apoptotic cells in unattached cells at 2 days: no Dox 0.105 ± 0.069 (n = 6), with Dox 0.148 ± 0.133 (n = 6), P > 0.7; at 5 days: no Dox 0.043 ± 0.018 (n = 7), with Dox 0.70 ± 0.36 (n = 7), P > 0.097]. Thus, although apoptotic cells can be detected as a sub-G0/G1 peak in this cell type (Fig. 6, inset) such peaks were rare in iRin37-F cells (Fig. 6), and their presence did not correlate with Cx37 expression.
Fig. 6.

Cell cycle position of iRin37-F cells induced or not to express Cx37 for 48 h. Although a sub-G1 peak, indicative of apoptosis, was sometimes observed in all types of Rin cells (inset shows iRin cells), such a peak was not predictably observed in iRin37-F cells (induced or not). In these samples, the percentage of cells in G0/G1, S, and G2 for the noninduced iRin37-F cells was 71.8%, 17.4%, and 10.8%, respectively, and for the induced iRin37 cells was 76.18%, 13.65%, and 10.17%, respectively.
Cell cycle position of Cx37-expressing Rin cells.
Since Cx37-mediated suppression of Rin cell proliferation did not involve apoptosis, we hypothesized that Cx37 arrested or profoundly slowed cell cycle progression. We used FACS of propidium iodide-stained iRin37-F cells to determine cell cycle distribution (50). Following 48 h of doxycyline induction, we observed no significant difference in cell cycle distribution between induced and noninduced cells (Fig. 6 and Table 1). However, when cells were induced to express Cx37 for 5 days, we observed a small, but significant, accumulation of induced cells in G0/G1, with a concomitant decrease in S-phase cells.
Table 1.
Cx37 expression accumulates iRin37-F cells in G0/G1
| iRin37-F | n | G0/G1 | S | G2 |
|---|---|---|---|---|
| 48 h | ||||
| Noninduced | 8 | 75±2 | 14±2 | 10±1.8 |
| Induced | 9 | 78±2 | 15±2 | 8±1 |
| 5 days | ||||
| Noninduced | 5 | 73±2 | 20±2 | 7±1 |
| Induced | 7 | 79±0.4* | 14±0.3* | 7±0.3 |
Values are means ± SE expressed as the percentage of total cell population in each phase. iRin37-F, connexin 37 (Cx37)-expressing rat insulinoma (Rin) clone.
Significantly different from noninduced cells.
The propidium iodide-FACS strategy for cell cycle analysis cannot distinguish between cells in G0 versus G1 of the cell cycle. To determine whether Cx37 expression caused the Rin cells to leave the cell cycle (and enter G0), induced and noninduced iRin37-F cells were stained for PCNA, a cell cycle protein that is expressed at peak levels in late G1 and S and absent from G0 cells. Intense PCNA staining was observed in both induced and noninduced iRin37-F cells (Fig. 7), and virtually all cells were stained. These data indicate that Cx37 expression did not cause cells to exit the cell cycle into G0, but rather promoted arrest at the G1/S boundary.
Fig. 7.

Cx37-expressing cells do not exit the cell cycle into G0. iRin37-F cells were plated on glass coverslips, induced (C and D) or not (A and B) to express Cx37 for 48 h, and stained to reveal PCNA expression (brown), which is absent in G0 cells. Cells were counterstained with hematoxylin (blue in B and D) and observed at ×10 and ×40 (insets). PCNA expression was clearly evident in noninduced cells (A) and induced cells (C). No nonspecific staining was observed in cells exposed only to secondary antibody (B and D). Data are representative of 4 experiments. Scale bar (in D) corresponds to 50 μm for ×10 images and to 9 μm for insets.
Given the profound suppression of proliferation mediated by Cx37, the cell cycle and proliferation data above suggested that cell cycle time must be significantly prolonged in Cx37-expressing Rin cells. To evaluate this possibility, we determined the “typical” profile for proliferation of Rin and iRin37-F (induced and noninduced) cells by pooling and plotting the data from multiple proliferation assays. These plots show that iRin and noninduced iRin37-F cells proliferated comparably (Fig. 8A), whereas induced iRin37-F cells proliferated, but very slowly (Fig. 8B). The large standard errors evident at the later time points in the iRin and noninduced iRin37-F plots reflect the occurrence of density-dependent inhibition of proliferation in some but not all experiments. The large standard errors evident at the later time points for the induced iRin37-F cells reflected limited proliferation of induced iRin37-F cells toward the end of the assay period in some but not all experiments. This proliferation occurred despite continued expression of Cx37 in virtually all cells (Fig. 9). To identify the period during which exponential growth for each cell type occurred (a prerequisite for calculation of doubling time), the data are also presented in semilogarithmic plots (insets, Fig. 8, A and B). For both the iRin and noninduced iRin37-F cells, exponential growth was evident between days 3 and 15. For the induced iRin37-F cells, growth was exponential between days 6 and 15, although very slow, as well as between days 15 and 21 at a faster rate. Doubling time, calculated as (t2 − t1) [log2/log(q2/q1)] (where t is time and q is number of cells), was determined for each cell type using a 3-, 6-, or 9-day time differential for the periods of logarithmic growth (days 3–15 for Rin and noninduced iRin37 cells and days 6–21 for induced iRin37 cells). For the iRin and noninduced iRin37-F cells, all three time differentials revealed comparable doubling times of ∼2 days (6-day time differential is illustrated in Fig. 8C). For the induced iRin37-F cells, only the 6-day and 9-day time differentials gave a consistent pattern of doubling time (6-day time differential is illustrated in Fig. 8C). The data show that Cx37 increased doubling time to ∼9 days for the early period of exponential growth (between days 6 and 15) and to 3–4 days for the later period of growth (between days 15 and 21).
Fig. 8.

Doubling time is significantly increased in Cx37-expressing cells. A and B: pooled data from iRin and noninduced iRin37-F cells (A; iRin37−) and induced iRin37-F cells (B; iRin37+). Proliferation of Cx37-expressing cells is suppressed by nearly 23-fold; note the difference in y-axis scaling between A and B. iRin and iRin37+ data derive from 16 experiments, iRin37− from 14; time points with reduced sample sizes are noted in the figure. Logarithmic plots (insets) reveal exponential growth between days 3 and 15 for iRin and iRin37− cells; the iRin37+ cells transition between two periods (6–15 and 18–21 days) of exponential growth. C: calculated doubling time for each cell type for their logarithmic growth periods; a 6-day window of analysis was used, in which the 6th day is noted on the abscissa.
Fig. 9.

Cx37 expression continues in virtually all iRin37-F cells throughout a 21-day proliferation assay. A and C: cells stained with Hoechst dye. B and D: Cx37 expression for the same cells. Images in A and B were taken 24 h after induction, and in C and D, 21 days after induction. Note the increase in cell number between days 1 and 21. Scale bar represents 9 μm.
The durations of the phases of the cell cycle were calculated from the estimated doubling time and cell cycle distributions (determined at 5-day time point for induced iRin37-F; Table 1). For the noninduced iRin37-F cells with a doubling time of ∼2 days, the durations of G1, S, and G2 were 36, 10, and 3.5 h, respectively. Induction of Cx37 expression in these cells increased their doubling time by nearly 4-fold to ∼9 days. The distribution of cells in the cell cycle and these doubling time data indicate that the durations of all phases of the cell cycle were substantially increased by Cx37 expression to 166, 30, and 15 h for G1, S, and G2 phases, respectively. These data strongly suggest that Cx37 profoundly prolongs the cell cycle by prolonging all phases of the cell cycle and promoting arrest at the G1/S checkpoint.
Serum deprivation augments Cx37-mediated cell cycle arrest.
Progression through the G1/S checkpoint in mammalian cells requires activation of the cell cycle machinery through growth factor-activated signaling (42). Since connexins are targeted by and influence the activity of some components of the cell cycle machinery (5, 31, 35, 64–66), we hypothesized that Cx37 expression may render the Rin cells susceptible to regulation by growth factor signaling in a manner different from Cx43. To test this hypothesis, we examined the effects of serum deprivation on cell cycle position of induced iRin37-F, parental Rin, and Rin43 cells. Focusing initially on the Cx-specific differences, in 10% serum, expression of either Cx37 or Cx43 altered the distribution of cells in the cell cycle compared with the Cx-deficient Rin cells (Table 2). Cx43 expression decreased G0/G1 cells and increased S-phase cells, whereas Cx37 increased G0/G1 cells and decreased S-phase cells. These trends were greatly augmented by serum deprivation, such that each cell type in 0% serum differed from itself in 10% serum for G0/G1 and S-phase and from Rin cells in all phases (Table 2). Thus, both Cx43 and Cx37 confer on the Rin cells sensitivity to growth factors, but they do so with very different results. Cx43 did not change overall cell cycle time, so the decrease in G1 and increase in S-phase cells suggests that cells progress more quickly through G1 and more slowly through S-phase, an effect that is enhanced by serum deprivation. In contrast, Cx37 significantly prolonged doubling time (cell cycle duration), so the increase in G1 and decrease in S-phase cells suggests very slow progression through G1 and possible arrest therein when cells were serum deprived.
Table 2.
Cell cycle position of Rin, Rin43, and iRin37-F cells in 10% versus 0% serum
| n | G0/G1 | S | G2 | |
|---|---|---|---|---|
| Rin | ||||
| 10% Serum | 3 | 71.8±0.8 | 19.5±1.2 | 8.7±0.6 |
| 0% Serum | 9 | 69.8±1.0 | 21.5±1.2 | 8.5±0.5 |
| Rin43 | ||||
| 10% Serum | 3 | 64.9±2.1* | 25.3±1.8* | 9.9±0.8 |
| 0% Serum | 9 | 55.8±3.3†* | 33.1±3.0†* | 11.2±0.9†* |
| iRin37-F‡ | ||||
| 10% Serum | 10 | 75.8±1.4* | 15.7±0.7* | 8.4±1.1 |
| 0% Serum | 10 | 82.6±0.91†* | 11.0±0.9†* | 6.4±0.7* |
Values are means ± SE expressed as the percentage of total cell population in each phase. Rin43, Rin cells with constitutive expression of rat Cx43.
Significantly different from Rin cells at same serum concentration.
Significantly different from 10% serum of same cell type.
72-h Serum deprivation with 24-h induction; Rin43 and Rin experiments were 48-h serum deprivation.
To estimate where in G1 Cx37-expressing cells were arrested by serum deprivation, serum deprivation-release experiments were performed (Fig. 10). Although the distribution of Rin cells in the cell cycle was unaffected by serum deprivation, restoration of 10% serum to these cells resulted in a significant decrease (compared with the 4-h time point) in G1 and increase in S-phase cells at 24 h. In contrast, progression from G1 to S-phase following serum deprivation in Cx37-expressing cells was significantly delayed, to 48 h. In view of the estimated duration of G1, 166 h, these data strongly suggest that Cx37 expression results in accumulation of Rin cells near the G1/S boundary.
Fig. 10.

Cell cycle progression and response to serum starvation differ in Cx37-expressing (iRin37-F) vs. Cx37-deficient (Rin) cells. Shown are the percentages of the cell population in G1 (A), S (B), and G2 (C) following 72 h in 10% serum (open symbols on the left in each graph) or 0% serum (0-h time points) and as a function time following restoration of 10% serum to the serum-deprived cells. All cells were treated with doxycycline for the 24 h preceding the 0-h time point and throughout the remainder of the experiment. *Significant difference from non-serum-deprived cells (G1 and S, in iRin37 only). #Significant difference from the 0-h time point. †Significant difference between cell types at the indicated time point.
DISCUSSION
The growth-suppressive potential of gap junctions and their comprising connexin proteins has long been recognized (29, 30). The tissue specificity of such growth suppression indicates that its mechanistic basis reflects the regulatory profile of the tissue (e.g., activity of specific signaling pathways), whereas the connexin specificity suggests primary sequence-dependent differences in critical properties of these channel-forming proteins. It is likely that their ability to support intercellular exchange of metabolites and signaling molecules features prominently in some settings of connexin-mediated growth suppression; certainly, connexin-specific differences in channel permselectivity (12) are consistent with such a possibility. However, in recent years, several studies have concluded that connexin-mediated growth suppression can occur in a manner independent of the formation of functional gap junctions; sequence-dependent differences in protein-protein interactions and/or phosphorylation-dependent regulation are consistent with connexin specificity of growth suppression. Armed with the foreknowledge that in some tissues expression of Cx37 and Cx43 is oppositely regulated following exposure to a growth stimulus [Cx37 down- and Cx43 upregulated (15)], in the present study we assessed the potential for and mechanism of growth suppression by Cx37.
We show here that Cx37 expression profoundly slowed the proliferation of Rin cells, whereas expression of Cx40 or Cx43 had no effect on their proliferation. Cx37 exerted this growth-suppressive effect irrespective of cell density (from ∼750 to ∼38,000 cells/cm2) at the time of induced Cx37 expression and without inducing apoptosis. The molecular mechanisms underlying Cx37's growth-suppressive effect in Rin cells remain unknown. Although Cx37-mediated suppression of cell proliferation has not previously been reported, numerous studies have demonstrated that Cx43 suppresses proliferation of a variety of cell types. That Cx37 suppressed proliferation of Rin cells when Cx43 did not indicates that the mechanisms underlying growth suppression by these connexins differ in critical ways.
Communication-dependent and -independent mechanisms have been proposed to underlie Cx43-mediated growth suppression (29). Our data do not directly address whether formation of functional gap junctions by Cx37 is necessary for the growth-suppressive effect of Cx37. Since communication-competent (7) Rin43 and Rin40 cells proliferate comparably to communication-deficient Rin cells, our data suggest that gap junction formation is not sufficient for growth suppression. However, it is possible that by virtue of unique permselective or channel regulatory properties (3, 12, 13, 23, 58, 59), formation of gap junction channels by Cx37 could prove necessary for its growth-suppressive effect. Our data also do not address the possible contribution of functional hemichannels (rather than or in addition to gap junction channels) to growth suppression by Cx37. Resolution of whether the function of Cx37 gap junction channels or hemichannels may augment (or even limit) the growth-suppressive effects of Cx37 awaits studies with a correctly targeted channel-dead mutant. Regardless of whether Cx37 gap junction or hemichannels are required for growth suppression, it remains to be determined how the cytoplasmic regions of Cx37 interact with the cell cycle machinery to limit growth.
Our data indicate that Cx37 expression slows cell cycle progression and arrests the cell cycle at the G1/S boundary, especially when the cells are also deprived of growth factors. From the doubling time and (5-day) cell cycle data, Cx37 appears to extend all phases of the cell cycle: G1 duration increased by 454%, S-phase by 300%, and G2 duration by 420%. The effect of Cx37 expression on the distribution of cells in the cell cycle was further augmented by serum deprivation. In serum-deprived cells, Cx37 expression for only 24 h was sufficient to substantially increase the percentage of cells in G1. Transition of these accumulated G1 cells into S-phase after serum restoration was delayed by 24 h compared with Cx37-deficient cells. Given the calculated duration of the G1 phase of the cell cycle, in excess of 150 h, these data strongly suggest that Cx37 expression slows progression of Rin cells through all phases of the cell cycle and arrests progression at the G1/S checkpoint.
Transition through G1 and the G1/S checkpoint involves cyclins D and E and cyclin-dependent kinase (CDK)4/6, with cyclin A and CDK2 critical for S-phase progression. Stable introduction of Cx43 into some tumorigenic cell types slows or arrests their proliferation, a phenomenon linked to reduced levels of the D and A cyclins and CDK6 (5); increased expression of the CDK inhibitor p27 (31, 65); increased degradation of Skp2, a protein that regulates ubiquitination and therefore degradation of p27 (66); and decreased DNA synthesis (9). The mechanisms linking Cx43 to these changes in proliferation and expression of cyclins, CDKs, and CDK regulatory proteins have not been identified. However, in the study by Dang et al. (9), the carboxyl-terminal domain of Cx43 was shown to be sufficient to suppress DNA synthesis, suggesting that gap junction channel function was not necessary for growth suppression by Cx43. In subsequent studies, this group (10, 11) showed that preventing phosphorylation of serine 262 (by mutation to alanine) in the carboxyl-terminal domain of Cx43 augmented the inhibitory effect of Cx43 on DNA synthesis and cell proliferation, whereas mimicking phosphorylation at this site (by mutation to aspartate) alleviated the inhibition induced by Cx43. Serine 262 is a known target for protein kinase C-dependent phosphorylation (11); thus, these studies provide strong support for a link between growth factor-activated signaling cascades, connexin phosphorylation, and cell cycle progression.
Since Cx43 had no effect and Cx37 had a profound effect on Rin cell proliferation, our data suggest that Cx37 must lead to reduced activity of cell cycle machinery via mechanisms that differ in critical ways from those induced by Cx43. In this context (at least) two ideas merit further investigation: sequence-related differences in phosphorylation-dependent regulation of channel function and/or sequence-related differences in protein-protein interactions. Compared with Cx43, very few studies have addressed either property of Cx37. Cx37 is known to be a phosphoprotein (38, 57), but the sites targeted, the kinases involved, and the prompting stimuli (likely cell-specific) have not been explored. Sequence comparisons of mCx37 and mCx43 suggest interesting potential differences in regard to both phosphorylation and possible protein binding partners. Cx43 is targeted for phosphorylation by multiple growth-activated kinases (35), including protein kinase C (at residues 368 as well as 262), protein kinase A (at S364 and possibly S365, S369, and S373), p34cdc2/cyclin B kinase (at S262 and S255), casein kinase 1 (at S325, S328, and S330), mitogen-activated protein kinase (at S255, S279, and S282), and pp60src kinase (at Y247). These phosphorylation events (can) influence gap junction assembly, channel function, and gap junction/connexin degradation, but for most it is not clear that they influence cell cycle progression and cell proliferation. Even for S262, the molecular mechanisms triggered by phosphorylation at this site that ultimately lead to reduced DNA synthesis remain unexplored. Cx37 is predicted by consensus sequence prediction programs to have multiple serines with a high probability (>90%) of phosphorylation by some of these same kinases. In at least two cases, these predicted serines align with sites targeted in Cx43: specifically, S282 in Cx43 (targeted by MAPK) aligns with S275 in Cx37, and S368 in Cx43 (targeted by PKC) aligns with S328 in Cx37. The possible roles in growth suppression of these and several other serines, threonines, and tyrosines in Cx37 that are predicted as high-probability targets for phosphorylation but do not align with corresponding sites in Cx43 remain to be explored.
In addition to likely differences in phosphorylation of Cx37 versus Cx43 following activation of growth factor signaling cascades, it is highly probable that the binding partners of Cx43 differ from those bound by Cx37. Cx43 interacts directly or indirectly with cell adhesion plaque proteins [including tubulin, zonula occludens-1 (ZO-1) and ZO-2] to form complexes that potentially regulate the availability of transcription factors (including β−catenin, ZO-1 nucleic acid binding protein, and SMADs) (for review, see Refs. 17 and 29). The binding sites in Cx43 identified as critical to these interactions are not present in Cx37. The binding partners of Cx37 remain completely unexplored; whether one or more of these partners may be central to regulation of the activity of the cell-cycle machinery needs investigation.
That Cx43 and Cx37 suppress proliferation by at least some nonoverlapping mechanisms raises new interest in possible therapeutic use of these connexins to suppress tumor angiogenesis and tumor growth. Cx37 is prominently expressed in vascular endothelium, but its expression is turned off in settings of growth and injury (25). Gene-targeting strategies (2) designed to introduce Cx37 (for constitutive or inducible expression) into the endothelium of blood vessels that must grow to support tumor angiogenesis (24) could be used to profoundly slow or block this process, thereby restraining tumor growth. Clearly, introduction of this connexin into the tumor cells themselves could also be efficacious. It is worth noting that the degree to which Cx37 expression suppressed proliferation of Rin cells is far greater than previously reported for growth-suppressive effects of other connexins, including Cx43, irrespective of the cell type. Thus, Cx37 may, in at least some cell types, be a far more potent tumor suppressor than other connexins and consequently warrants further evaluation in this context. The Rin cell system is ideally suited to this pursuit by virtue of the Cx-specificity of growth suppression by Cx37 in Rin cells.
In summary, we show here that Cx37 suppresses proliferation of Rin cells by significantly extending all phases of the cell cycle and prolonging or arresting transition through the G1/S checkpoint, especially when the cells are also deprived of growth factor stimulation. That Cx37 suppresses Rin cell proliferation when Cx43 does not, despite its growth-suppressive effects in other tumorigenic cell types, suggests that the mechanisms underlying Cx37-mediated growth suppression must differ in critical ways from those of Cx43, differences revealed by the Rin cell system.
GRANTS
These studies were supported by a Grant-in-Aid from the American Heart Association (0550158Z, to J. M. Burt), a Predoctoral Fellowship from the American Heart Association (0715532Z, to J. S. Fang), and National Heart, Lung, and Blood Institute Grants HL-064232 (to A. M. Simon and J. M. Burt) and HL-058732 (to J. M. Burt).
Acknowledgments
We thank Drs. Ek-Vitorin and Kurjiaka for thoughtful suggestions and occasional assistance during the course of these experiments and Humberto Gherna for occasional technical assistance.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Alcolea S, Jarry-Guichard T, de Bakker J, Gonzalez D, Lamers W, Coppen S, Barrio L, Jongsma H, Gros D, Van Rijen H. Replacement of connexin40 by connexin45 in the mouse: impact on cardiac electrical conduction. Circ Res 94: 100–109, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Balestrieri ML, Napoli C. Novel challenges in exploring peptide ligands and corresponding tissue-specific endothelial receptors. Eur J Cancer 43: 1242–1250, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Beblo DA, Veenstra RD. Monovalent cation permeation through the connexin40 gap junction channel Cs, Rb, K, Na, Li, TEA, TMA, TBA, and effects of anions Br, Cl, F, acetate, aspartate, glutamate, and NO3. J Gen Physiol 109: 509–522, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brissette JL, Kumar NM, Gilula NB, Dotto GP. The tumor promoter 12-O-tetradecanoylphorbol-13-acetate and the ras oncogene modulate expression and phosphorylation of gap junction proteins. Mol Cell Biol 11: 5364–5371, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen SC, Pelletier DB, Peng A, Boynton AL. Connexin43 reverses the phenotype of transformed cells and alters their expression of cyclin/cyclin-dependent kinases. Cell Growth Differ 6: 681–690, 1995. [PubMed] [Google Scholar]
- 6.Clark SA, Burnham BL, Chick WL. Modulation of glucose-induced insulin secretion from a rat clonal beta-cell line. Endocrinology 127: 2779–2788, 1990. [DOI] [PubMed] [Google Scholar]
- 7.Cottrell GT, Burt JM. Heterotypic gap junction channel formation between heteromeric and homomeric Cx40 and Cx43 connexons. Am J Physiol Cell Physiol 281: C1559–C1567, 2001. [DOI] [PubMed] [Google Scholar]
- 8.Cottrell GT, Wu Y, Burt JM. Cx40 and Cx43 expression ratio influences heteromeric/heterotypic gap junction channel properties. Am J Physiol Cell Physiol 282: C1469–C1482, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Dang X, Doble BW, Kardami E. The carboxy-tail of connexin-43 localizes to the nucleus and inhibits cell growth. Mol Cell Biochem 242: 35–38, 2003. [PubMed] [Google Scholar]
- 10.Dang X, Jeyaraman M, Kardami E. Regulation of connexin-43-mediated growth inhibition by a phosphorylatable amino-acid is independent of gap junction-forming ability. Mol Cell Biochem 289: 201–207, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Doble BW, Dang X, Ping P, Fandrich RR, Nickel BE, Jin Y, Cattini PA, Kardami E. Phosphorylation of serine 262 in the gap junction protein connexin-43 regulates DNA synthesis in cell-cell contact forming cardiomyocytes. J Cell Sci 117: 507–514, 2004. [DOI] [PubMed] [Google Scholar]
- 12.Ek-Vitorin JF, Burt JM. Quantification of gap junction selectivity. Am J Physiol Cell Physiol 289: C1535–C1546, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Ek-Vitorin JF, King TJ, Heyman NS, Lampe PD, Burt JM. Selectivity of connexin 43 channels is regulated through protein kinase C-dependent phosphorylation. Circ Res 98: 1498–1505, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Figueroa XF, Isakson BE, Duling BR. Connexins: gaps in our knowledge of vascular function. Physiology (Bethesda) 19: 277–284, 2004. [DOI] [PubMed] [Google Scholar]
- 15.Gabriels JE, Paul DL. Connexin43 is highly localized to sites of disturbed flow in rat aortic endothelium but connexin37 and connexin40 are more uniformly distributed. Circ Res 83: 636–643, 1998. [DOI] [PubMed] [Google Scholar]
- 16.Gazdar AF, Chick WL, Oie HK, Sims HL, King DL, Weir GC, Lauris V. Continuous, clonal, insulin- and somatostatin-secreting cell lines established from a transplatable rat islet cell tumor. Proc Natl Acad Sci USA 77: 3519–3523, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giepmans BN Role of connexin43-interacting proteins at gap junctions. Adv Cardiol 42: 41–56, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Goodenough DA, Paul DL. Beyond the gap: functions of unpaired connexon channels. Nat Rev Mol Cell Biol 4: 285–294, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Gunning P, Leavitt J, Muscat G, Ng SY, Kedes L. A human beta-actin expression vector system directs high-level accumulation of antisense transcripts. Proc Natl Acad Sci USA 84: 4831–4835, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haefliger JA, Nicod P, Meda P. Contribution of connexins to the function of the vascular wall. Cardiovasc Res 62: 345–356, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Harris AL Emerging issues of connexin channels: biophysics fills the gap. Q Rev Biophys 34: 325–472, 2001. [DOI] [PubMed] [Google Scholar]
- 22.He DS, Jiang JX, Taffet S, Burt JM. Formation of heteromeric gap junction channels by connexins 40 and 43 in vascular smooth muscle cells. Proc Natl Acad Sci USA 96: 6495–6500, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heyman NS, Burt JM. Hindered diffusion through an aqueous pore describes invariant dye selectivity of Cx43 junctions. Biophys J 94: 840–854, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hillen F, Griffioen AW. Tumour vascularization: sprouting angiogenesis and beyond. Cancer Metastasis Rev 26: 489–502, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirschi KK, Xu CE, Tsukamoto T, Sager R. Gap junction genes Cx26 and Cx43 individually suppress the cancer phenotype of human mammary carcinoma cells and restore differentiation potential. Cell Growth Differ 7: 861–870, 1996. [PubMed] [Google Scholar]
- 26.Isakson BE, Kronke G, Kadl A, Leitinger N, Duling BR. Oxidized phospholipids alter vascular connexin expression, phosphorylation, and heterocellular communication. Arterioscler Thromb Vasc Biol 26: 2216–2221, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Jiang JX, Gu S. Gap junction- and hemichannel-independent actions of connexins. Biochim Biophys Acta 1711: 208–214, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.John SA, Kondo R, Wang SY, Goldhaber JI, Weiss JN. Connexin-43 hemichannels opened by metabolic inhibition. J Biol Chem 274: 236–240, 1999. [DOI] [PubMed] [Google Scholar]
- 29.Kardami E, Dang X, Iacobas DA, Nickel BE, Jeyaraman M, Srisakuldee W, Makazan J, Tanguy S, Spray DC. The role of connexins in controlling cell growth and gene expression. Prog Biophys Mol Biol 94: 245–264, 2007. [DOI] [PubMed] [Google Scholar]
- 30.King TJ, Bertram JS. Connexins as targets for cancer chemoprevention and chemotherapy. Biochim Biophys Acta 1719: 146–160, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Koffler L, Roshong S, Kyu Park I, Cesen-Cummings K, Thompson DC, Dwyer-Nield LD, Rice P, Mamay C, Malkinson AM, Ruch RJ. Growth inhibition in G(1) and altered expression of cyclin D1 and p27(kip-1) after forced connexin expression in lung and liver carcinoma cells. J Cell Biochem 79: 347–354, 2000. [DOI] [PubMed] [Google Scholar]
- 32.Kruger O, Plum A, Kim JS, Winterhager E, Maxeiner S, Hallas G, Kirchhoff S, Traub O, Lamers WH, Willecke K. Defective vascular development in connexin 45-deficient mice. Development 127: 4179–4193, 2000. [DOI] [PubMed] [Google Scholar]
- 33.Kumari SS, Varadaraj K, Valiunas V, Ramanan SV, Christensen EA, Beyer EC, Brink PR. Functional expression and biophysical properties of polymorphic variants of the human gap junction protein connexin37. Biochem Biophys Res Commun 274: 216–224, 2000. [DOI] [PubMed] [Google Scholar]
- 34.Kurjiaka DT, Steele TD, Olsen MV, Burt JM. Gap junction permeability is diminished in proliferating vascular smooth muscle cells. Am J Physiol Cell Physiol 275: C1674–C1682, 1998. [DOI] [PubMed] [Google Scholar]
- 35.Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol 36: 1171–1186, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lampe PD, Tenbroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol 149: 1503–1512, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langille BL, Reidy MA, Kline RL. Injury and repair of endothelium at sites of flow disturbances near abdominal aortic coarctations in rabbits. Arteriosclerosis 6: 146–154, 1986. [DOI] [PubMed] [Google Scholar]
- 38.Larson DM, Seul KH, Berthoud VM, Lau AF, Sagar GD, Beyer EC. Functional expression and biochemical characterization of an epitope-tagged connexin37. Mol Cell Biol Res Commun 3: 115–121, 2000. [DOI] [PubMed] [Google Scholar]
- 39.Lin SJ, Jan KM, Weinbaum S, Chien S. Transendothelial transport of low density lipoprotein in association with cell mitosis in rat aorta. Arteriosclerosis 9: 230–236, 1989. [DOI] [PubMed] [Google Scholar]
- 40.Loewenstein WR, Kanno Y. Intercellular communication and the control of tissue growth: lack of communication between cancer cells. Nature 209: 1248–1249, 1966. [DOI] [PubMed] [Google Scholar]
- 41.Martyn KD, Kurata WE, Warn-Cramer BJ, Burt JM, Tenbroek E, Lau AF. Immortalized connexin43 knockout cell lines display a subset of biological properties associated with the transformed phenotype. Cell Growth Differ 8: 1015–1027, 1997. [PubMed] [Google Scholar]
- 42.Murray A, Hunt T. The Cell Cycle: An Introduction. New York: Oxford University Press, 1993.
- 43.Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol 115: 1357–1374, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Plum A, Hallas G, Magin T, Dombrowski F, Hagendorff A, Schumacher B, Wolpert C, Kim J, Lamers WH, Evert M, Meda P, Traub O, Willecke K. Unique and shared functions of different connexins in mice. Curr Biol 10: 1083–1091, 2000. [DOI] [PubMed] [Google Scholar]
- 45.Qin H, Gunning P. The 3′-end of the human beta-actin gene enhances activity of the beta-actin expression vector system: construction of improved vectors. J Biochem Biophys Methods 36: 63–72, 1997. [DOI] [PubMed] [Google Scholar]
- 46.Reaume AG, De Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, Juneja SC, Kidder GM, Rossant J. Cardiac malformation in neonatal mice lacking connexin43. Science 267: 1831–1834, 1995. [DOI] [PubMed] [Google Scholar]
- 47.Reed KE, Westphale EM, Larson DM, Wang HZ, Veenstra RD, Beyer EC. Molecular cloning and functional expression of human connexin37, an endothelial cell gap junction protein. J Clin Invest 91: 997–1004, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rummery NM, Hill CE. Vascular gap junctions and implications for hypertension. Clin Exp Pharmacol Physiol 31: 659–667, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Scherer SS, Xu YT, Nelles E, Fischbeck K, Willecke K, Bone LJ. Connexin32-null mice develop demyelinating peripheral neuropathy. Glia 24: 8–20, 1998. [DOI] [PubMed] [Google Scholar]
- 50.Schorl C, Sedivy JM. Analysis of cell cycle phases and progression in cultured mammalian cells. Methods 41: 143–150, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seul KH, Kang KY, Lee KS, Kim SH, Beyer EC. Adenoviral delivery of human connexin37 induces endothelial cell death through apoptosis. Biochem Biophys Res Commun 319: 1144–1151, 2004. [DOI] [PubMed] [Google Scholar]
- 52.Shao Q, Wang H, McLachlan E, Veitch GI, Laird DW. Down-regulation of Cx43 by retroviral delivery of small interfering RNA promotes an aggressive breast cancer cell phenotype. Cancer Res 65: 2705–2711, 2005. [DOI] [PubMed] [Google Scholar]
- 53.Simon AM, Chen H, Jackson CL. Cx37 and Cx43 localize to zona pellucida in mouse ovarian follicles. Cell Commun Adhes 13: 61–77, 2006. [DOI] [PubMed] [Google Scholar]
- 54.Simon AM, Goodenough DA, Li E, Paul DL. Female infertility in mice lacking connexin 37. Nature 385: 525–529, 1997. [DOI] [PubMed] [Google Scholar]
- 55.Simon AM, McWhorter AR, Chen H, Jackson CL, Ouellette Y. Decreased intercellular communication and connexin expression in mouse aortic endothelium during lipopolysaccharide-induced inflammation. J Vasc Res 41: 323–333, 2004. [DOI] [PubMed] [Google Scholar]
- 56.Sohl G, Willecke K. Gap junctions and the connexin protein family. Cardiovasc Res 62: 228–232, 2004. [DOI] [PubMed] [Google Scholar]
- 57.Traub O, Hertlein B, Kasper M, Eckert R, Krisciukaitis A, Hulser D, Willecke K. Characterization of the gap junction protein connexin37 in murine endothelium, respiratory epithelium, and after transfection in human HeLa cells. Eur J Cell Biol 77: 313–322, 1998. [DOI] [PubMed] [Google Scholar]
- 58.Valiunas V, Beyer EC, Brink PR. Cardiac gap junction channels show quantitative differences in selectivity. Circ Res 91: 104–111, 2002. [DOI] [PubMed] [Google Scholar]
- 59.Wang HZ, Veenstra RD. Monovalent ion selectivity sequences of the rat connexin43 gap junction channel. J Gen Physiol 109: 491–507, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.White TW Unique and redundant connexin contributions to lens development. Science 295: 319–320, 2002. [DOI] [PubMed] [Google Scholar]
- 61.Willecke K, Heynkes R, Dahl E, Stutenkemper R, Hennemann H, Jungbluth S, Suchyna T, Nicholson BJ. Mouse connexin37: cloning and functional expression of a gap junction gene highly expressed in lung. J Cell Biol 114: 1049–1057, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wong CW, Christen T, Roth I, Chadjichristos CE, Derouette JP, Foglia BF, Chanson M, Goodenough DA, Kwak BR. Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat Med 12: 950–954, 2006. [DOI] [PubMed] [Google Scholar]
- 63.Zar JH Biostatistical Analysis. Englewood Cliffs, NJ: Prentice Hall, 1984.
- 64.Zhang YW, Kaneda M, Morita I. The gap junction-independent tumor-suppressing effect of connexin 43. J Biol Chem 278: 44852–44856, 2003. [DOI] [PubMed] [Google Scholar]
- 65.Zhang YW, Morita I, Ikeda M, Ma KW, Murota S. Connexin43 suppresses proliferation of osteosarcoma U2OS cells through post-transcriptional regulation of p27. Oncogene 20: 4138–4149, 2001. [DOI] [PubMed] [Google Scholar]
- 66.Zhang YW, Nakayama K, Nakayama K, Morita I. A novel route for connexin 43 to inhibit cell proliferation: negative regulation of S-phase kinase-associated protein (Skp 2). Cancer Res 63: 1623–1630, 2003. [PubMed] [Google Scholar]