

Abstract
Stromal interaction molecule 1 (STIM1) and Orai1 have been identified as crucial elements of the store-operated Ca2+ entry (SOCE) pathway, but the mechanism of their functional interaction remains controversial. It is now well established that, upon depletion of the stores, both molecules can accumulate and colocalize in specific areas (puncta) where the endoplasmic reticulum comes in close proximity to the plasma membrane. Some models propose a direct interaction between STIM1 and Orai1 as the most straightforward mechanism for signal transduction from the stores to the plasma membrane. To test some of the predictions of a conformational coupling model, we assessed how tight the relationships are between STIM1 and Orai1 expression, puncta formation, and SOCE activation. Here we present evidence that STIM1 accumulates in puncta equally well in the presence or absence of Orai1 expression, that STIM1 accumulation is not sufficient for Orai1 accumulation in the same areas, and that normal Ca2+ release-activated Ca2+ current (ICRAC) can be activated in STIM1-deficient cells. These data challenge the idea of direct conformational coupling between STIM1 and Orai1 as a viable mechanism of puncta formation and SOCE activation and uncover greater complexity in their relationship, which may require additional intermediate elements.
Keywords: store-operated Ca2+ entry
store-operated ca2+ entry (SOCE) is activated upon depletion of endoplasmic reticulum (ER) Ca2+ stores (for review see Ref. 25). Recently, two important components of SOCE were identified, stromal interaction molecule 1 (STIM1) as a Ca2+ sensor in the ER, which is capable of triggering a cascade of reactions leading to SOCE activation, and Orai1 (also called CRACM1) as a plasma membrane (PM) channel that is activated by the store-operated pathway (for recent review see Ref. 13). STIM1 is a protein predominantly located in the ER membrane; it has one transmembrane domain and an EF hand motif in its NH2 terminus, which allows STIM1 to bind Ca2+ in the ER lumen and to function as a low-affinity Ca2+ sensor in the stores. Upon Ca2+ depletion, STIM1 can lose Ca2+ from its EF hand, oligomerize, and accumulate into punctate structures in the ER membrane located in close proximity (10–25 nm) to the PM (1, 14–16, 20, 22, 23, 29, 36, 40). Orai1 is a PM protein that is thought to be a pore-forming subunit of Ca2+ release-activated Ca2+ channel (CRAC) (9, 19, 20, 39, 41). In resting cells, Orai1 is homogenously distributed in the PM, and, after store depletion, it was shown to accumulate in puncta and colocalize with STIM1. Molecular knockdown of either STIM1 or Orai1 has been shown to impair SOCE in a wide variety of cell types (see Ref. 13 for complete review), but their relationship is far from being understood.
Several groups have proposed direct physical interaction between STIM1 and Orai1 to be a mechanism for SOCE activation (10, 12, 18, 34, 35, 41). This idea was strongly supported by several important findings. First, coimmunoprecipitation of overexpressed STIM1 and Orai1 was shown in S2 cells (41) and human embryonic kidney 293 (HEK293) cells (39). Second, overexpression of both proteins resulted in remarkable amplification of ICRAC (28, 34, 44). Third, Förster resonance energy transfer (FRET) between fluorescently tagged STIM1 and Orai1 was reported (23), indicating their very close spatial proximity. Fourth, overexpression of the COOH terminus part of STIM1 along with full-length Orai1 was reported to be sufficient for SOCE activation (12). All these data strongly supported the idea of direct conformational coupling of STIM1 to Orai1 as a straightforward mechanism of signal transduction and SOCE activation. However, these studies did not consider the possibility that intermediate steps and additional molecular components may be required for STIM1/Orai1 colocalization, mutual interaction, and signal transduction from the ER to the PM. In an alternative model (31), SOCE activation was shown to require a diffusible messenger(s) and specific variant of phospholipase A2 (PLA2) as additional components for signal transduction from the ER to the PM (for all the details, see reviews in Refs. 3 and 4). The need for an additional intermediate(s) between STIM1 and Orai1 was also strongly supported by recent findings from Dr. Balla's group (38), which demonstrated that colocalization of STIM1 and Orai1 does not occur in narrow (4–6 nm) junctions but happens only in the areas where a larger (12–14 nm) gap exists between the ER and the PM. Thus the question remains unanswered as to whether direct physical interaction between STIM1 and Orai1 may be required and sufficient for their accumulation in puncta and SOCE activation or whether additional elements could participate in signal transduction from depleted stores to the PM channel.
This study was designed to test several important predictions of the conformational coupling model and to obtain new data that will either support or challenge the physical coupling of STIM1 and Orai1 as the sole mechanism for puncta formation and SOCE activation. If the mutual presence and direct interaction of STIM1 with Orai1 are indeed required and sufficient, one could expect that there should be a very strict correlation between expression of both these proteins, their accumulation in puncta, and activation of Orai1 and ICRAC. In contrast, the results of our experiments yielded numerous examples of STIM1 accumulation in puncta independently of Orai1 and Orai1 and ICRAC activation independently of STIM1, which challenge the predictions of the conformational coupling model. New evidence suggests that the SOCE mechanism may be a much more complex phenomenon and may require the presence of an additional intermediate element(s) that can mediate puncta formation and signal transduction from STIM1 in the ER to Orai1 in the PM.
MATERIALS AND METHODS
Cells and transfections.
HEK293 cells were obtained from American Type Culture Center (ATCC) and grown in DMEM containing 4.5 mg/ml glucose supplemented with 10% heat-inactivated fetal bovine serum (GIBCO), 10 U/ml penicillin, and 10 mg/ml streptomycin at 37°C in the atmosphere of 5% CO2. Transient transfections of HEK293 cells were performed using jetPEI transfection factor (PolyPlus) according to the protocol suggested by the manufacturer. Standard transfection rate was ∼70–90%.
Rat basophilic leukemia (RBL-2H3) cells were obtained from ATCC, cultured, and transfected as described previously (6, 7). Briefly, cells were maintained in minimum essential medium supplemented with 10% heat-inactivated fetal bovine serum (GIBCO), 1% l-glutamine, 10 U/ml penicillin, and 10 mg/ml streptomycin at 37°C. RBL cells were transiently transfected using Nucleofector (Amaxa Biosystems) according to the protocol provided by the manufacturer. Standard transfection rate was ∼50–70%. For visual identification of transfected cells for patch clamp experiments, green fluorescent protein (GFP) constructs (2 μg) were added along with all constructs to mark transfected cells.
DNA constructs.
Sequences encoding Orai1 (GenBank acc. no. NM032790) and STIM1 (GenBank acc. no. NM003156) were amplified by PCR and cloned in frame into the pEGFP-N1 and/or pmCherry-N1 vectors (Clontech) with the use of BglII and EcoRI restriction sites. Recombinant DNA plasmids were purified with a QIAGEN Plasmid Maxi Kit (Qiagen).
Molecular downregulation of STIM1 and Orai1.
RBL-2H3 cells were transfected with either scrambled RNA (Ambion), or siRNA against rat STIM1 (5′-AAGGCTCTGGATACAGTGCTC-3′) or siRNA against rat Orai1 (5′-GUCCACAACCUCAACUCCTT-3′) as previously described(7).
HEK293 cells were cotransfected with plasmid DNA encoding STIMGFP (or STIM1Cherry), along with either scrambled RNA (Ambion) or siRNA against human Orai1 (5′-CCCUUCGGCCUGAUCUUUAUCGUCU-3′) (Invitrogen). Lipofectamine 2000 (Invitrogen) was used for transfection of HEK293 cells, and Nucleofector (Amaxa Biosystems) was used for RBL cells. Cells were investigated 48–72 h after transfection.
Imaging experiments.
A Nikon TE2000 wide-field system with a 60× oil immersion objective (1.4 NA) was used to image individual cells. Before imaging experiments, cells were passed onto glass-bottom dishes (MatTek) and studied 24–36 h after transfection. During imaging experiments, cells were maintained in extracellular buffer (125 mM NaCl, 5 mM KCl, 1.5 mM MgCl2, 20 mM HEPES, 10 mM glucose, and 1.5 mM CaCl2, pH 7.4) at room temperature. Fluorescence was monitored using the following filter sets (Chroma): 1) ET470/40 (excitation), ET525/50 (emission), and T495LP (dichroic) for GFP and 2) ET560/40 (excitation), ET630/75 (emission), and T585LP (dichroic) for mCherry. Stack of images was taken at Z intervals of 0.3 μm and then deconvolved with the AutoQuant Module in the NIS-elements software (Nikon).
Ca2± influx studies.
Cells were loaded with Fura-2 AM, and changes in intracellular Ca2+ (measured as F340/F380 ratio) were monitored as previously described (6, 7). Briefly, a dual-excitation fluorescence imaging system (Intracellular Imaging) was used for studies of individual cells. The changes in intracellular Ca2+ were expressed as ΔRatio, which was calculated as the difference between the peak F340/F380 ratio after and before extracellular Ca2+ was added. Data were summarized from the large number of individual cells (20–40 cells in a single run, repeated in 3–5 identical independent experiments using at least 3 different cell transfections).
Electrophysiology.
Whole-cell currents were recorded in RBL cells using standard whole-cell (dialysis) patch-clamp technique as previously described (6). An Axopatch 200B amplifier was used, and data were filtered at 1 kHz and digitized at 5 kHz. Pipettes were used with tip resistance of 2–4 MΩ. After breaking into the cell, the holding potential was 0 mV, and ramp depolarizations (from −100 to +100 mV, 200 ms) were applied every 3 s. The amplitude of the current was expressed in pA/pF. The time course of current development was analyzed at −80 mV for each individual cell, and summary data for 5–10 cells are shown in the figures. Average current/voltage (I/V) relationships are shown during ramp depolarization after the current reached its maximum. Passive leakage current with zero reversal potential (at the moment of breaking into the cell or after CRAC inhibition with 10 μM diethylstilbestrol; Ref. 42) was subtracted. Intracellular (pipette) solution contained (in mM): 145 CsGlutamate, 3 MgCl2, 10 HEPES, (pH 7.2), and 0.1 BAPTA, which by itself does not cause store depletion (6). In experiments with Orai1 overexpression, 20 mM BAPTA was used in the pipette. In experiments in which calcium influx factor (CIF) presence was indicated, HPLC-purified CIF extract from activated platelets (prepared as specified in previously, Ref. 6) was added to the pipette at 1:20 dilution. Standard extracellular solution was (in mM): 20 CaCl2, 130 NaCl, 5 HEPES, 3 CsCl, 1 MgCl2 (pH 7.4). In experiments with Orai1 overexpression, extracellular Ca2+ was reduced to 1 mM. Experiments were performed at 20–22°C.
Puncta analysis.
Number, size, and area of puncta of fluorescently tagged STIM1 within HEK293 cells treated with thapsigargin (TG) were analyzed using ImageJ software (National Institutes of Health). Images were processed with the use of a convolve filter followed by the particle analysis function. The same setting [size: 3–14 (pixel2), circularity: 0–0.3] was used for all images. Summary data were normalized to cell surface and presented as average ± SE from five representative cells in each group.
Statistical analysis.
Summary data are presented as means ± SE. Student's t-test was used to determine the statistical significance of the obtained data. Data were considered significant at P < 0.01 (see significance levels in the individual figure legends).
RESULTS
In this study, HEK293 and RBL-2H3 cell lines were used as complementary models suitable for a wide range of experimental approaches that allowed us to either overexpress fluorescently labeled Orai1 and/or STIM1 or to knock down endogenous molecules to study correlation between STIM1 and Orai1 expression, puncta formation, and activation of ICRAC and SOCE.
Store depletion and puncta formation.
To verify that depletion of the stores by itself [without accompanying Ca2+ rise in cytosol that happens upon cell stimulation with agonists or sarco(endo)plasmic reticulum Ca2+-ATPase inhibitors] can cause puncta formation, we compared puncta formation during cell treatment with TG, which causes a physical loss of Ca2+ from the stores due to its passive release into cytosol, and N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN), a membrane-permeable low-affinity Ca2+ buffer, which has been shown to buffer Ca2+ in ER without its physical release and without affecting Ca2+ in the cytosol. HEK293 cells were transfected with fluorescently tagged STIM1 and Orai1 (STIM1Cherry and Orai1GFP, respectively), and their accumulation and colocalization was monitored at the bottom plane of the cells. Fig. 1A shows that TG (5 μM) application to HEK cells caused dramatic changes in distribution patterns of STIM1Cherry and Orai1GFP, both of which migrated to the punctuate structures that are thought to represent regions where the ER is in close proximity to the PM (see review by Lewis, Ref. 13). The same STIM1Cherry and Orai1GFP accumulation and colocalization in puncta were observed when the cells were treated with TPEN (Fig. 1B), which has been shown to mimic TG effects and trigger SOCE in different cell types (6, 11).
Fig. 1.

Stromal interaction molecule 1 (STIM1)-dependent puncta formation and colocalization of STIM1 and Orai1. Fluorescent images of representative human embryonic kidney 293 (HEK293) cells coexpressing STIM1Cherry (red) and Orai1GFP (green) showing translocation and colocalization of both proteins in characteristic puncta after treatment with thapsigargin (TG) (5 μM for 5 min) (A) and N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN) (1 mM for 5 min) (B). The scale bar is 10 μm. GFP, green fluorescent protein.
Thus not only store depletion (with TG) but also simple reduction in ER intraluminal free Ca2+ concentration (by TPEN) without Ca2+ changes in cytosol can trigger STIM1 and Orai1 accumulation in the same puncta.
Orai1 is not required for STIM1 accumulation in puncta.
To test how strict the correlation is between expression of STIM1 and Orai1, puncta formation, and SOCE activation, we examined the process of Orai1 and STIM1 accumulation in puncta. For this purpose, TG-induced translocation of fluorescently tagged STIM1 (STIM1Cherry or STIM1GFP) into puncta and activation of SOCE were investigated in HEK293 cells, in which Orai1 was up- or downregulated. First, we confirmed that, even when STIM1 was expressed alone in HEK293 (Fig. 2A), TG caused its accumulation in puncta, which was consistent with findings by others (27, 33). In these STIM1-overexpressing HEK cells, TG also triggered SOCE activation, which suggested a significant amount of endogenous Orai1 being present and involved in HEK cell responses. Overexpression of exogenous Orai1GFP along with STIM1Cherry (like in Fig. 1A) resulted in about 50% increase of SOCE (Fig. 2B), consistent with creation of a monster ICRAC when these two molecules are coexpressed (28, 34, 44); Ca2+ influx increased from ΔR = 3.24 ± 0.14, n = 53 in STIM1-expressing cells to ΔR = 6.37 ± 0.84, n = 41 in cells expressing both, Orai1 and STIM1 (Fig. 3A). However, overexpression of Orai1 and dramatic increase in SOCE was not accompanied by any visible changes in the pattern, number, or size of STIM1GFP puncta (Fig. 2B and Fig. 3, B and C). Surprisingly, when, instead of overexpression, Orai1 was knocked down (by siRNA) and SOCE could not be detected (Fig. 2C and Fig. 3A), we found that STIM1GFP still formed puncta (Fig. 2C) that was identical to that in cells overexpressing Orai1 (Fig. 2B). Thus STIM1 was able to accumulate and form identical puncta independently on the presence or absence of Orai1 and SOCE activation (Fig. 3). The fact that normal STIM1 puncta are formed in the cells in which endogenous Orai1 was knocked down strongly suggests that Orai1 is not needed for STIM1 accumulation in puncta.
Fig. 2.
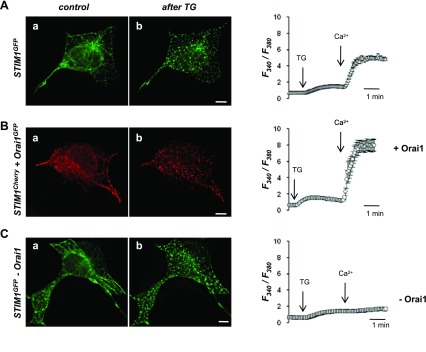
Puncta formation of STIM1 is independent of Orai1 expression. Left: representative images of subcellular distribution of fluorescently tagged STIM1 in HEK293 cells before (a) and after TG (b) treatment (5 μM for 5 min). Cells were cotransfected with: plasmid DNA encoding STIM1GFP and scrambled RNA (A), STIM1Cherry and Orai1GFP (+Orai1) (B), and STIM1GFP and siRNA against Orai1 (−Orai1) (C). The scale bar is 10 μm. Right: traces show representative average changes in intracellular Ca2+ (F340/F380) measured simultaneously in 15–20 cells transfected with plasmid DNA encoding STIM1GFP and scrambled RNA (A), STIM1Cherry and Orai1GFP (+Orai1) (B), and STIM1GFP and siRNA against Orai1 (−Orai1) (C).
Fig. 3.

Orai1 plays a key role in store-operated Ca2+ entry (SOCE) but not in STIM1 puncta formation. A: summary data show the maximum TG-induced Ca2+ influx in HEK293 cells described in Fig. 2. Cells were transfected as follows: fluorescently tagged STIM1 and scrambled RNA (+STIM1), fluorescently tagged STIM1 and Orai1 (+STIM1, +Orai1), and fluorescently tagged STIM1 and siRNA against Orai1 (+STIM1, −Orai1). Control corresponds to Ca2+ entry in nontransfected cells. Data are shown as the ΔRatio (±SE) values, which were calculated as the difference between the peak F340/F380 ratio after extracellular Ca2+ was added and its level immediately before Ca2+ addition. B: analysis of TG-induced STIM1 puncta in HEK293 cells corresponding to cells presented in Fig. 2. Number of STIM1 puncta plotted vs. total puncta area per 1 cell show that puncta formation is neither dependent on up- nor downregulation of Orai1 (+Orai1 and −Orai1, respectively). Scrambled corresponds to cells cotransfected with STIM1GFP and scrambled RNA. C: average size of STIM1 puncta in cells analyzed in B.
STIM1 accumulation in puncta is not sufficient for Orai1 accumulation.
When HEK cells were cotransfected with STIM1Cherry and Orai1GFP (using 0.4 μg of cDNA for STIM1 and 0.2 μg for Orai1, respectively), the majority of the cells did not show any significant accumulations of STIM1 or Orai1 under resting conditions and formed puncta only upon their stimulation with TG or TPEN, as shown in Fig. 1. However, in a significant number (20–40%) of resting (unstimulated) HEK293 cells, STIM1Cherry could be found in puncta-like patches (Fig. 4A), whereas Orai1GFP remained evenly distributed throughout the PM (Fig. 4C) and did not show any significant preference for the structures already preformed by STIM1Cherry. In these cells, TG application did not produce any additional change in the distribution pattern of STIM1Cherry (Fig. 4B), but it caused Orai1GFP to translocate and accumulate exactly in STIM1Cherry patches (Fig. 4D). Thus accumulation of STIM1 was clearly not sufficient for Orai1 accumulation in the same areas. The possibility of temporal and functional separation of STIM1 and Orai1 accumulation in puncta suggests that they can be driven by two independent processes.
Fig. 4.

Disconnection between STIM1 and Orai1 accumulation in puncta. Representative image of HEK293 cell in which STIM1Cherry was coexpressed with Orai1GFP and in which a puncta-like pattern of Stim1Cherry (A), but not Orai1 (C), was clearly visible in control conditions. Addition of TG (5 μM for 5 min) did not change the distribution of STIM1Cherry (B) but caused accumulation of Orai1GFP into the punctuate areas (D) that were identical to those preformed by STIM1Cherry before TG treatment. The scale bar is 10 μm.
ICRAC can be activated in STIM1-deficient cells.
To further test the role of STIM1 in activation of Orai1 channels, we studied SOCE and ICRAC (a whole-cell current through Orai1 channels) in control RBL cells and RBL cells in which STIM1 or Orai1 were knocked down. In full agreement with the reports on the important role of STIM1 and Orai1 in Ca2+ entry activated by depletion of the stores, molecular downregulation of STIM1 or Orai1 (using transient cell transfection with corresponding siRNAs) resulted in dramatic inhibition of Ca2+ influx triggered by TG in RBL cells (Fig. 5). To test whether STIM1 is indeed essential for activation of Orai1-encoded CRAC channels in RBL cells, we tested the impact of STIM1 downregulation on the activation of ICRAC via multiple stimuli. In our earlier studies (6), we demonstrated that ICRAC in RBL cells can be activated equally well by store depletion (with TG or BAPTA) or by cell dialysis with CIF, a diffusible messenger produced by depleted stores (4).
Fig. 5.

Downregulation of STIM1 or Orai1 impairs TG-induced SOCE in rat basophilic leukemia (RBL) cells. A: representative traces show the average changes in intracellular Ca2+ (F340/F380) recorded simultaneously in a number of individual RBL cells. The cells were pretreated with TG (5 μM for 5 min) in Ca2+-free solution before 2 mM Ca2+ was added (at the time indicated in the figure). TG-induced Ca2+ influx (average ± SE) is shown in cells transfected with scrambled siRNA (control) or siRNA either against STIM1 (−STIM1) or Orai1 (−Orai1). B: summary data showing the maximum TG-induced Ca2+ influx in RBL cells in experiments described in A. Each bar summarizes the results from 103–215 individual cells from 3–5 independent experiments. Basal corresponds to Ca2+ entry in cells treated with 0.5% DMSO without TG. Please notice that, in contrast with traces in A, data here show the ΔRatio (±SE) values, which were calculated as the difference between the peak F340/F380 ratio after extracellular Ca2+ was added and its level immediately before Ca2+ addition. Asterisks denote significant differences between control and −STIM1 (P = 0.007).
When RBL cells were treated with TG to initiate the signal by depletion of the stores, ICRAC developed in control but not in cells transfected with STIM1 siRNA (Fig. 6A), which was totally consistent with the absence of TG-induced Ca2+ entry in STIM1-deficient cells. Surprisingly, we found that a totally normal ICRAC developed in the same STIM1-deficient cells when depletion of the stores was bypassed. Figure 6B shows that RBL cell dialyzed with CIF (extracted from platelets with depleted stores, as described in our earlier publications, Ref. 6) activated ICRAC not only in control but also in STIM1-deficient RBL cells, in which TG-induced ICRAC and SOCE were inhibited. It is important to mention that, in contrast to STIM1, knockdown of either Orai1 (with siRNA) or calcium-independent PLA2-β (iPLA2β) (with antisense) significantly inhibited CIF-induced ICRAC (Fig. 7A), confirming that CIF activates the same iPLA2β-dependent Orai1-encoded CRAC channels that can also be activated by simple store depletion (with BAPTA or TG). Moreover, when Orai1 but not STIM1 was overexpressed, RBL cell dialysis with CIF resulted in activation of a “monster” ICRAC (Fig. 7B). Thus ICRAC appeared to be present and fully functional in STIM1-deficient cells and could be easily activated when the store-dependent step in the SOCE pathway was bypassed by cell dialysis with CIF.
Fig. 6.

Downregulation of STIM1 impairs store-operated Ca2+-selective current (ICRAC) induced by store depletion with TG (A) but not ICRAC activated by calcium influx factor (CIF) dialysis (B) in RBL-2H3 cells. A: traces show an average (±SE) ICRAC (pA/pF) developing at −80 mV during RBL dialysis with 1 μM TG (0.1 mM BAPTA in the pipette). RBL cells were transfected with scrambled siRNA (control, n = 7), or siRNA against STIM1 (−STIM1, n = 5). Diethylstilbestrol (DES, 10 μM) was applied to the bath at the end of the experiment as indicated. Right: summary current/voltage (I/V) relationships of the maximum ICRAC developed in these experiments. B: traces show an average (±SE) ICRAC (pA/pF) developing at −80 mV during RBL dialysis with exogenous CIF (HPLC-purified extract from human platelets with depleted stores). 0.1 mM BAPTA was present in the pipette that does not by itself cause depletion of the stores. RBL cells were transfected with scrambled siRNA (control, n = 7) or siRNA against STIM1 (−STIM1, n = 7). DES (10 μM) was applied to the bath at the end of experiment as indicated. Right: summary I/V relationships of the maximum ICRAC developed in these experiments.
Fig. 7.

Orai1 and calcium-independent PLA2-β (iPLA2β), but not STIM1, are essential for normal and monster ICRAC activation. A: summary data showing the amplitude of ICRAC (pA/pF) developing at −80 mV (in the presence of 20 mM extracellular Ca2+) during RBL dialysis with CIF and 0.1 mM BAPTA in RBL cells transfected with scrambled siRNA (control, n = 7), siRNA against STIM1 (−STIM1, n = 5), siRNA against Orai1 (−Orai1, n = 3), or antisense for iPLA2β (−iPLA2β, n = 3). B: summary data (n = 6) showing the amplitude of a monster ICRAC (pA/pF) developing at −80 mV (in the presence of 1 mM extracellular Ca2+) in RBL cells overexpressing Orai1 (but not STIM1) during their dialysis with CIF and 20 mM BAPTA. Please notice the difference in the scales of the Y-axes in A and B.
DISCUSSION
This study tested some predictions of the conformational coupling model, and the results revealed a much more complex behavior of STIM1 and Orai1 than what one could expect on the basis of their direct physical interaction as the sole requirement for puncta formation and ICRAC activation. Although we observed STIM1 and Orai1 accumulation and colocalization in punctate areas, we found examples of the lack of correlation between expression of STIM1 and Orai1 and their accumulation in puncta. First, Orai1 appeared to be not needed for STIM1 accumulation in puncta; STIM1 was able to form identical punctuate structures in HEK293 cells in which Orai1 was either knocked down or overexpressed. Importantly, STIM1 accumulation in puncta showed no correlation with SOCE activation. Furthermore, accumulation of STIM1 in puncta was not necessarily followed by Orai1 accumulation. Our results demonstrated that, in the absence of cell stimulation, these two events could be separated in time (when STIM1 is already in puncta while Orai1 is not), and cell stimulation (leading to store depletion) may be required for their final colocalization. It suggests that accumulation of STIM1 is not sufficient for Orai1 accumulation into the same punctuate structures, which in turn indicates that, at least in some cells, STIM1 does not directly drive Orai1 translocation. These examples demonstrate that STIM1 and Orai1 accumulation in the same areas can show a much higher degree of independence than could be expected if their colocalization and signal transduction was dictated by their direct physical coupling.
Since molecular knockdown of either Orai1 or STIM1 could totally impair SOCE activation, it does not seem likely that other variants of these molecules play a vital role in this process, but they can certainly be involved in regulation and fine tuning of SOCE mechanism, as proposed by several recent studies (5, 8, 17, 24, 26).
Existence of an additional structural and functional linker(s) between STIM1 and Orai1 can explain accumulating data that do not fit into their direct coupling models and may provide an alternative mechanism that can explain their functional independence. It can easily explain not only the findings reported here but also the findings of Dr. Balla's group (38), who recently demonstrated that STIM1 and Orai1 colocalization occur in the areas in which the ER and PM are separated by a gap that can be too big for their direct interaction. It is important to emphasize that the apparent requirement for the molecular linker between STIM1 and Orai1 does not contradict any of the studies that demonstrated their close spatial and functional proximity, including reported FRET between STIM1 and Orai1 (15, 23). Indeed, colocalization of fluorescently tagged proteins within ∼10 nM of space between the ER and PM membranes may allow energy transfer even without their physical interaction. An indirect evidence for such possibility comes from the studies by Muik et al. (15, 23), who presented a strong proof that only COOH (but not NH2) terminus of Orai1 is important for its colocalization with STIM1, but at the same time similar FRET could be detected between fluorescently tagged STIM1 and Orai1 when it was tagged at either COOH or NH2 terminus. Thus FRET can be observed between STIM1 and the different parts of Orai1, even if they are not involved in their interaction but may simply come in close proximity to STIM1.
The most striking evidence for a functional gap that may exist between STIM1 and Orai1 came from the experiments that demonstrated activation of normal ICRAC in STIM1-deficient cells. Indeed, if physical interaction of STIM1 with Orai1 is the mechanism of SOCE, activation of ICRAC by any means should be impossible in STIM1-deficient cells. In contrast, the result showed that CRAC channels could be equally well activated in cells with or without STIM1 when ER-delimited STIM1-dependent steps following depletion of the stores were bypassed by cell dialysis with CIF (production of which we have recently shown to be an early STIM1-dependent event in the ER, Ref. 7). The fact that CIF dialysis can cause activation of a monster ICRAC in cells that overexpress only Orai1 (without STIM1) is also inconsistent with direct conformational coupling models and suggests a different explanation for monster ICRAC phenomenon. It further supports the role of STIM1 as a trigger for production of CIF (7), which can activate Orai1 via iPLA2β-dependent mechanism. All these results are totally consistent with our earlier findings (6) showing that, even in the absence of store depletion, activation of normal ICRAC can be achieved by direct activation of iPLA2β with either CIF or CaM inhibitory peptide, which are both able to displace inhibitory CaM from iPLA2β (for the details on the CIF-iPLA2β-dependent model of SOCE activation, please see Ref. 4). So far none of the conformational coupling models could account for the crucial role of iPLA2β in SOCE activation, which was confirmed by different investigators in numerous cell types (2, 6, 21, 29–32, 37, 43) as well as by genetic screening of Drosophila melanogaster published by Vig et al. (39). Thus iPLA2β may be one of the molecular and/or functional linkers between STIM1 and Orai1, which need to be further investigated.
Although direct coupling of ER-resident STIM1 to PM-resident Orai1 may be rightfully considered as the most straightforward mechanism for signal transduction, new evidence suggests that the SOCE mechanism may be a much more complex phenomenon. It may require the presence and functional activity of additional intermediate element(s) that can mediate STIM1 and Orai1 accumulation in puncta and can orchestrate signal transduction from the ER to the PM leading to the eventual activation of SOCE.
GRANTS
This study was supported by the research grants from the National Institutes of Health (HL54150 and HL71793 to V. Bolotina) and by training grant from the National Institutes of Health (HL007224 to V. Zarayskiy).
Acknowledgments
We thank Dr. Michael Kirber for help with imaging experiments and expert comments on the manuscript and Prof. Andrzej Ozyhar from Wroclaw University of Technology for a kind gift of vectors encoding fluorescent proteins.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Baba Y, Hayashi K, Fujii Y, Mizushima A, Watarai H, Wakamori M, Numaga T, Mori Y, Iino M, Hikida M, Kurosaki T. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc Natl Acad Sci USA 103: 16704–16709, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boittin FX, Petermann O, Hirn C, Mittaud P, Dorchies OM, Roulet E, Ruegg UT. Ca2+-independent phospholipase A2 enhances store-operated Ca2+ entry in dystrophic skeletal muscle fibers. J Cell Sci 119: 3733–3742, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Bolotina VM Orai1, STIM1 and iPLA2[beta]: a view from a different perspective. J Physiol 586: 3035–3042, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolotina VM, Csutora P. CIF and other mysteries of the store-operated Ca2+-entry pathway. Trends Biochem Sci 30: 378–387, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca(2+) levels. Cell 131: 1327–1339, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Csutora P, Zarayskiy V, Peter K, Monje F, Smani T, Zakharov S, Litvinov D, Bolotina VM. Activation mechanism for CRAC current and store-operated Ca2+ entry: calcium influx factor and Ca2+-independent phospholipase A2b- mediated pathway. J Biol Chem 281: 34926–34935, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Csutora P, Peter K, Kilic H, Park KM, Zarayskiy V, Gwozdz T, Bolotina VM. Novel role for STIM1 as a trigger for calcium influx factor production. J Biol Chem 283: 14524–14531, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dehaven WI, Smyth JT, Boyles RR, Putney JW Jr. Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J Biol Chem 282: 17548–17556, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441: 179–185, 2006. [DOI] [PubMed] [Google Scholar]
- 10.Hisatsune C, Mikoshiba K. Novel compartment implicated in calcium signaling—is it an “induced coupling domain”? Sci STKE 2005: e53, 2005. [DOI] [PubMed]
- 11.Hofer AM, Fasolato C, Pozzan T. Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ stores: a study using simultaneous measurements of Icrac and intraluminal [Ca2+]. J Cell Biol 140: 325–334, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang GN, Zeng W, Kim JY, Yuan JP, Han L, Muallem S, Worley PF. STIM1 carboxyl-terminus activates native SOC, Icrac and TRPC1 channels. Nat Cell Biol 8: 1003–1010, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Lewis RS The molecular choreography of a store-operated calcium channel. Nature 446: 284–287, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Li Z, Lu J, Xu P, Xie X, Chen L, Xu T. Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J Biol Chem 282: 29448–29456, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA 104: 9301–9306, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 15: 1235–1241, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lis A, Peinelt C, Beck A, Parvez S, Monteilh-Zoller M, Fleig A, Penner R. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr Biol 17: 794–800, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopez JJ, Salido GM, Pariente JA, Rosado JA. Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. J Biol Chem 281: 28254–28264, 2006. [DOI] [PubMed] [Google Scholar]
- 19.Lorin-Nebel C, Xing J, Yan X, Strange K. CRAC channel activity in C. elegans is mediated by Orai1 and STIM1 homologues and is essential for ovulation and fertility. J Physiol 580: 67–85, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol 174: 815–825, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez J, Moreno JJ. Role of Ca2+-independent phospholipase A2 and cytochrome P-450 in store-operated calcium entry in 3T6 fibroblasts. Biochem Pharmacol 70: 733–739, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW Jr. Large store-operated calcium-selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem 281: 24979–24990, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muik M, Frischauf I, Derler I, Fahrner M, Bergsmann J, Eder P, Schindl R, Hesch C, Polzinger B, Fritsch R, Kahr H, Madl J, Gruber H, Groschner K, Romanin C. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem 283: 8014–8022, 2008. [DOI] [PubMed] [Google Scholar]
- 24.Oh-Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S, Rao A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol 9: 432–443, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parekh AB, Putney JW Jr. Store-operated calcium channels. Physiol Rev 85: 757–810, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Parvez S, Beck A, Peinelt C, Soboloff J, Lis A, Monteilh-Zoller M, Gill DL, Fleig A, Penner R. STIM2 protein mediates distinct store-dependent and store-independent modes of CRAC channel activation. FASEB J 22: 752–761, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peinelt C, Lis A, Beck A, Fleig A, Penner R. 2-Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J Physiol 586: 3061–3073, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, Koblan-Huberson M, Lis A, Fleig A, Penner R, Kinet JP. Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat Cell Biol 8: 771–773, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ross K, Whitaker M, Reynolds NJ. Agonist-induced calcium entry correlates with STIM1 translocation. J Cell Physiol 211: 569–576, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singaravelu K, Lohr C, Deitmer JW. Regulation of store-operated calcium entry by calcium-independent phospholipase A2 in rat cerebellar astrocytes. J Neurosci 26: 9579–9592, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smani T, Zakharov S, Csutora P, Leno E, Trepakova ES, Bolotina VM. A novel mechanism for the store-operated calcium influx pathway. Nat Cell Biol 6: 113–120, 2004. [DOI] [PubMed] [Google Scholar]
- 32.Smani T, Zakharov SI, Leno E, Csutora P, Trepakova ES, Bolotina VM. Ca2+-independent phospholipase A2 is a novel determinant of store-operated Ca2+ entry. J Biol Chem 278: 11909–11915, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Smyth JT, Dehaven WI, Bird GS, Putney JW Jr. Ca2+-store-dependent and -independent reversal of Stim1 localization and function. J Cell Sci 121: 762–772, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem 281: 20661–20665, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Spassova MA, Soboloff J, He LP, Xu W, Dziadek MA, Gill DL. STIM1 has a plasma membrane role in the activation of store-operated Ca(2+) channels. Proc Natl Acad Sci USA 103: 4040–4045, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stathopulos PB, Li GY, Plevin MJ, Ames JB, Ikura M. Stored Ca2+ depletion-induced oligomerization of STIM1 via the EF-SAM region: an initiation mechanism for capacitive Ca2+ entry. J Biol Chem 281: 35855–35862, 2006. [DOI] [PubMed] [Google Scholar]
- 37.Vanden Abeele F, Lemonnier L, Thebault S, Lepage G, Parys J, Shuba Y, Skryma R, Prevarskaya N. Two types of store-operated Ca2+ channels with different activation modes and molecular origin in LNCaP human prostate cancer epithelial cells. J Biol Chem 279: 30326–30337, 2004. [DOI] [PubMed] [Google Scholar]
- 38.Varnai P, Toth B, Toth DJ, Hunyady L, Balla T. Visualization and manipulation of plasma membrane-endoplasmic reticulum contact sites indicates the presence of additional molecular components within the STIM1-Orai1 complex. J Biol Chem 282: 29678–29690, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312: 1220–1223, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol 174: 803–813, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 443: 226–229, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zakharov SI, Smani T, Dobrydneva Y, Monje F, Fichandler C, Blackmore PF, Bolotina VM. Diethylstilbestrol is a potent inhibitor of store-operated channels and capacitative Ca2+ influx. Mol Pharmacol 66: 702–707, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Zarayskiy V, Monje F, Peter K, Csutora P, Khodorov BI, Bolotina VM. Store-operated Orai1 and IP3R-operated TRPC1 channel: separation of the Siamese twins. Channels (Austin) 1: 246–252, 2007. [DOI] [PubMed] [Google Scholar]
- 44.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci USA 103: 9357–9362, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]