

Abstract
Inositol 1,4,5-trisphosphate receptors (IP3Rs) regulate diverse physiological functions, including contraction and proliferation. There are three IP3R isoforms, but their functional significance in arterial smooth muscle cells is unclear. Here, we investigated relative expression and physiological functions of IP3R isoforms in cerebral artery smooth muscle cells. We show that 2-aminoethoxydiphenyl borate and xestospongin C, membrane-permeant IP3R blockers, reduced Ca2+ wave activation and global intracellular Ca2+ ([Ca2+]i) elevation stimulated by UTP, a phospholipase C-coupled purinergic receptor agonist. Quantitative PCR, Western blotting, and immunofluorescence indicated that all three IP3R isoforms were expressed in acutely isolated cerebral artery smooth muscle cells, with IP3R1 being the most abundant isoform at 82% of total IP3R message. IP3R1 knockdown with short hairpin RNA (shRNA) did not alter baseline Ca2+ wave frequency and global [Ca2+]i but abolished UTP-induced Ca2+ wave activation and reduced the UTP-induced global [Ca2+]i elevation by ∼61%. Antibodies targeting IP3R1 and IP3R1 knockdown reduced UTP-induced nonselective cation current (Icat) activation. IP3R1 knockdown also reduced UTP-induced vasoconstriction in pressurized arteries with both intact and depleted sarcoplasmic reticulum (SR) Ca2+ by ∼45%. These data indicate that IP3R1 is the predominant IP3R isoform expressed in rat cerebral artery smooth muscle cells. IP3R1 stimulation contributes to UTP-induced Icat activation, Ca2+ wave generation, global [Ca2+]i elevation, and vasoconstriction. In addition, IP3R1 activation constricts cerebral arteries in the absence of SR Ca2+ release by stimulating plasma membrane Icat.
Keywords: cerebral artery smooth muscle cells, calcium wave, short hairpin RNA
inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs) are expressed in many cell types and regulate several physiological functions, including development, muscle contraction, cell proliferation, and differentiation (1, 45, 47, 56). In vascular smooth muscle cells, IP3Rs modulate vasoconstriction and proliferation by altering spatial and temporal properties of local and global Ca2+ signals (51, 53). Vasoconstrictors, including norepinephrine, phenylephrine, UTP, and vasopressin, stimulate Ca2+ waves and Ca2+ oscillations in smooth muscle cells of portal vein, rat tail, mesenteric and cerebral arteries, and cultured A7r5 smooth muscle cells (5, 7, 21, 23, 29). Vasoconstrictors also elevate arterial smooth muscle cell global intracellular Ca2+ concentration ([Ca2+]i) and induce vasoconstriction by stimulating IP3Rs (7, 43, 54). IP3 also activates cation channels in vascular smooth muscle cells through mechanisms that do not require the stimulation of sarcoplasmic reticulum (SR) Ca2+ release (27, 54). In cerebral artery smooth muscle cells, IP3R activation stimulates TRPC3 channels, leading to Na+ influx, membrane depolarization, voltage-dependent Ca2+ channel activation, and vasoconstriction (54). Thus IP3Rs regulate ion channel activity, Ca2+ signals, and physiological functions in the vasculature. However, despite the functional importance of IP3Rs in arterial smooth muscle cells, specific IP3R isoforms that are expressed in this cell type and their functional significance are poorly understood.
Three IP3R isoforms, designated IP3R1, IP3R2, and IP3R3, are encoded by distinct genes (6, 37). IP3R isoform expression varies widely between different cell types. Cerebellar Purkinje neurons express predominantly IP3R1, pancreatic β-cells primarily express IP3R3, and cardiac myocytes express IP3R2 (14, 48). All three IP3R isoforms are expressed in whole aorta, mesenteric and basilar arteries, and cultured aortic smooth muscle cells (17, 56). In primary cultured portal vein smooth muscle cells, IP3R1 and IP3R2 were detected by immunofluorescence, whereas IP3R3 was absent (32). In contrast, in primary cultured rat ureter smooth muscle cells and cultured A7r5 cells, IP3R1 and IP3R3 were expressed and IP3R2 was absent (32, 51).
IP3R isoforms may also regulate vascular smooth muscle Ca2+ signaling in a cell-specific manner. IP3R2 activation contributes to ACh-induced Ca2+ oscillations in cultured portal vein smooth muscle cells (32). In contrast, IP3R1 was required for ACh-induced Ca2+ elevations in cultured portal vein smooth muscle cells (32), ATP-induced Ca2+ transients in cultured aortic smooth muscle cells (56), and vasopressin-evoked Ca2+ release and capacitative Ca2+ entry in cultured A7r5 smooth muscle cells (51). These studies suggest that IP3R isoform expression may differ between smooth muscle cell types, leading to tissue-specific regulation of intracellular Ca2+ signaling.
The major goal of this study was to determine the functional significance of IP3R isoforms that are expressed in smooth muscle cells of resistance-size cerebral arteries. We demonstrate that all three IP3R isoforms are expressed in rat cerebral artery smooth muscle cells and that IP3R1 is, by far, the most abundant. We also show that IP3R1 suppression using antibodies and short hairpin RNA (shRNA) reduces UTP-induced elevations in Ca2+ wave frequency and global [Ca2+]i, cation current (Icat) activation, and vasoconstriction. These data identify IP3R1 as the principal molecular and functional IP3R isoform in cerebral artery smooth muscle cells.
MATERIALS AND METHODS
Tissue preparation.
Animal protocols were reviewed and approved by the Animal Care and Use Committee of the University of Tennessee Health Science Center. Sprague-Dawley rats (200–250 g body wt) were euthanized with pentobarbital sodium (150 mg/kg). The brain was removed and placed into ice-cold (4°C) oxygenated (21% O2-5% CO2-74% N2) physiological saline solution (PSS) containing (in mM) 119 NaCl, 4.7 KCl, 24 NaHCO3, 1.2 KH2PO4, 1.6 CaCl2, 1.2 MgSO4, and 11 glucose (with pH adjusted to 7.4 with NaOH). Posterior cerebral, middle cerebral, and cerebellar arteries (∼50–200 μm diameter) were removed and cleaned of connective tissue. When appropriate, endothelium was removed by introducing an air bubble into the arterial lumen for 2 min, followed by a 30-s wash with PSS. Cerebral artery smooth muscle cells were isolated as previously described (10).
RT-PCR and real-time PCR.
IP3R fragments were amplified by nested PCR using the Taq DNA polymerase kit (Invitrogen). Reactions were run at 94°C for 30 s, 56°C for 30 s, and 72°C for 1 min 30 s for 35 cycles each, with the following primers: CACCCAAAGAAGAGCTGCTCC (forward, F1), CTGTCATCTGGTCCTTTAGTTCTGAC (reverse, R1), CTCTGTTTGCTGCAAGGGTGATC (F2), and GTCCTTCACTTTCACCAGCACG (R2) for IP3R1; CCTGTTCTCCATCATCGGCTTC (F1), TGTCATCTGCTCCTTCAGCTCTG (R1), ATTGTCACCGTGCTGAACCAG (F2), and GCCACAGATGAAGCAGGTTGTC (R2) for IP3R2; and ATCCTGCTCTTTGACCTCATCTACC (F1), GTTCTTGTTCTTGATCATCTGAGCCAC (R1), CCTGAGATCCTAGAAGAGGACGAG (F2), and TCTCCAGACCACAGATGAAGCAC (R2) for IP3R3. PCR products were separated on 2% agarose gels, and bands were visualized by staining with ethidium bromide.
For real-time PCR, IP3R isoform-specific primers were as follows: GAGATGAGCCTGGCTGAGGTTCAA (forward) and TGTTGCCTCCTTCCAGAAGTGCGA (reverse) for IP3R1, CAACCAGACCCTGGAGAGCTTGAC (forward) and GTCAGGAACTGGCAGATGGCAGGT (reverse) for IP3R2, AGACCCGCTGGCCTACTATGAGAA (forward), GTCAGGAACTGGCAGATGGCAGGT (reverse) for IP3R3, and ATCCTGTGGCATTCCATGAAACTAC (forward) and AGGAGCCAGGGCAGTAATCTC (reverse) for β-actin. Each PCR mixture (25 μl) contained 12.5 μl of SYBR Green PCR Master Mix (Applied Biosystems), 400 nM primer, and an equal volume of cDNA template. Reactions were performed in triplicate on a sequence detection system (Prism 7700, Applied Biosystems) at 95°C for 10 min and 40 cycles of 20 s at 95°C, 20 s at 65°C, and 45 s at 72°C. A negative control, with smooth muscle cell RNA, instead of cDNA, was carried out in each experiment. The integrity of each PCR was verified by using dissociation curve analysis. Relative IP3R isoform mRNA expression was calculated from the difference between fluorescence threshold (Ct) values (ΔCt) of each IP3R isoform sample using the ΔCt method.
Immunoblotting.
Rat whole brain or cerebral arteries were homogenized in Laemmli sample buffer (2.5% SDS, 10% glycerol, 0.01% bromphenol blue, and 5% β-mercaptoethanol in 100 mM Tris·HCl, pH 6.8) and centrifuged at 10,000 g for 10 min. Proteins (30 μg/lane) were separated by 4–15% gradient SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride membranes using a Mini Trans Blot Cell (Bio-Rad, Hercules, CA). Membranes were then incubated with polyclonal anti-IP3R1 (1:1,000 dilution; Alomone Labs), IP3R2 (1:500 dilution; Santa Cruz Biotechnology), or monoclonal anti-IP3R3 (1:500 dilution, BD Transduction Laboratories) antibodies overnight at 4°C in Tris-buffered solution (TBS) with 0.1% Tween 20 (TBS-T) and 5% nonfat dry milk. After three washes with TBS-T, membranes were incubated for 1 h with horseradish peroxidase-conjugated secondary antibody (1:10,000 dilution; Pierce Biotechnology) and then washed with TBS-T. Membranes were developed using enhanced chemiluminescence (Amersham, Arlington Heights, IL). Membranes were reprobed with monoclonal anti-actin antibody (1:10,000 dilution; Chemicon International) and then with horseradish peroxidase-conjugated anti-mouse IgG. Band intensities were quantified by digital densitometry using Quantity One version 4.4.1 software. IP3R isoform band intensity was normalized to actin.
Immunofluorescence.
Freshly isolated smooth muscle cells were fixed with 3.7% paraformaldehyde in PBS (Invitrogen, Carlsbad, CA) for 15 min and permeabilized with 0.1% Triton X-100 for 1 min at room temperature. After 1 h of incubation in PBS containing 5% BSA, smooth muscle cells were treated overnight at 4°C with antibodies to IP3R1 (UC Davis/NINDS/NIMH NeuroMab Facility). IP3R2 (Affinity Bio), or IP3R3 (BD Transduction Laboratories), each at a dilution of 1:100 in PBS containing 5% BSA. After they were washed and blocked with 10% goat serum, smooth muscle cells were incubated with secondary antibodies (1:100 dilution; Invitrogen-Molecular Probes) as follows: goat anti-mouse IgG with Alexa Fluor 546 for IP3R1, goat anti-rabbit IgG with Alexa Fluor 488 for IP3R2, or anti-mouse IgG2a with Alexa Fluor 555 for IP3R3. The cells were washed and mounted, and fluorescence images were obtained using a Zeiss LSM Pascal scanning confocal microscope. Negative controls were prepared by omission of primary antibodies.
IP3R1 knockdown.
Two different IP3R1 silencing vectors were constructed by GenScript (Piscataway, NJ) using pRNA-U6.1/Neo as a backbone plasmid to encode shRNA targeting IP3R1. The vectors encode CGGTCGAAATGTCCAGTATA (IP3R1shV1) or GCAGCAACGTGATGAGATCTA (IP3R1shV2). A pRNA-U6.1/Neo vector that encodes a scrambled sequence (TGAACATCAGTGCTAGGTTAC) was used as a control (IP3R1scrm). To deliver plasmids into arterial wall cells, reverse permeabilization was used (54). IP3R1shV1 (10 μg/ml) was used for Western blotting, patch clamp, and diameter measurements, and IP3R1shV1 (5 μg/ml) + IP3R1shV2 (5 μg/ml) was used for Western blotting and Ca2+ imaging experiments, with IP3R1scrm (10 μg/ml) as control for each.
Patch-clamp electrophysiology.
Icat was measured in the presence or absence of UTP (30 μM) using the conventional whole cell patch-clamp configuration (Axopatch 200B, Clampex 8.2). The pipette solution contained (in mM) 140 CsCl, 10 HEPES, 10 glucose, 5 MgATP, and 5 EGTA (with pH adjusted to 7.2 with CsOH), with free Ca2+ adjusted to 100 nM using Ca2+-sensitive (catalog no. 476041, Corning) and reference (catalog no. 476370, Corning) electrodes. Bath solution contained (in mM) 140 NaCl, 1.8 CaCl2, 1.2 MgCl2, 10 HEPES, and 10 glucose (with pH adjusted to 7.4 with NaOH). For experiments studying Icat regulation in myocytes treated with IP3R1 scrambled and suppression vectors, thapsigargin (100 nM) was included in the pipette solution to induce SR Ca2+ depletion. Where appropriate, IP3R1 antibody (Alomone Labs) or a denatured IP3R1 antibody control (95°C, 10 min) was included in the pipette solution (1:100 dilution). Whole cell currents were measured by applying 940-ms voltage ramps between −120 and +20 mV with a holding potential of −40 mV. Current amplitude was analyzed offline using Clampfit 9.2.
Laser-scanning confocal Ca2+ imaging.
Cerebral artery segments were cannulated and incubated in the dark with fluo-4 AM (10 μM) and 0.05% Pluronic F-127 for 1 h. Intracellular Ca2+ signals in cerebral artery smooth muscle cells were imaged using a Noran Oz laser-scanning confocal microscope, as described previously (11). Fluo-4 AM was excited with 488-nm light, and emitted light >510 nm was collected. Images (112.6 × 105.6 μm) were acquired at a rate of 30 s−1. Under each condition, at least two different representative areas of the same arterial segment were each scanned for 10 s. The same area of artery was scanned only once to avoid any laser-induced changes in Ca2+ signaling, and the effects of pharmacological agents were measured in paired experiments. Ca2+ wave frequency and global [Ca2+]i were analyzed offline using custom software kindly provided by Dr. M. T. Nelson (University of Vermont), as previously described (11). Global [Ca2+]i was calculated using the following equation: [Ca2+]i = KR/{K/([Ca2+]rest + 1 − R)}, where K is the apparent affinity of fluo-4 AM for Ca2+ (770 nM) (10), R is the fractional fluorescence increase (F/F0), and [Ca2+]rest is [Ca2+]i at F0 (193 nM) (10).
Fura-2 imaging.
IP3R1scrm- or IP3R1shV-treated cerebral artery segments were incubated in HEPES-buffered solution (in mM: 134 NaCl, 6 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, with pH adjusted to 7.4 with NaOH) containing fura-2 AM (5 μM) and 0.05% Pluronic F-127 for 30 min and then washed for 15 min. Fura-2 was alternately excited at 340 or 380 nm using a personal computer-driven hyperswitch (Ionoptix, Milton, MA). Background-corrected ratios were collected every 1 s at 510 nm using a photomultiplier tube. SR Ca2+ load was estimated by measuring the amplitude of caffeine (10 mM)-induced [Ca2+]i transients, as we described previously (11).
Pressurized artery diameter measurement.
Endothelium-denuded cerebral artery segments were cannulated at each end in a perfusion chamber. The chamber was continuously perfused with PSS and maintained at 37°C. Intravascular pressure was altered using an attached reservoir and monitored using a pressure transducer. Wall diameter was measured at 1 Hz using a charge-coupled device camera and the edge-detection function of IonWizard (Ionoptix). Pharmacological agents were applied via chamber perfusion. Passive diameter was determined by applying Ca2+-free PSS supplemented with 5 mM EGTA. The magnitude of myogenic tone was calculated using the following equation: myogenic tone (%) = (1 − active diameter/passive diameter) × 100.
Reagents.
Unless otherwise specified, all reagents were purchased from Sigma-Aldrich (St. Louis, MO). Fluo-4 AM, fura-2 AM, and Pluronic F-127 were purchased from Molecular Probes (Eugene, OR), and papain was obtained from Worthington Biochemical (Lakewood, NJ).
Statistical analysis.
Values are means ± SE. Student's t-test and Student-Newman-Keuls test were used for comparison of paired or unpaired data and multiple data sets, respectively. P < 0.05 was considered significant.
RESULTS
IP3R stimulation contributes to UTP-induced Ca2+ wave activation and global [Ca2+]i elevation in rat cerebral artery smooth muscle cells.
Ca2+ signals in smooth muscle cells of intact cerebral arteries were acquired by imaging Fluo-4 AM fluorescence at a rate of 30 images per second. UTP (30 μM) elevated mean Ca2+ wave frequency in smooth muscle cells from 0.07 to 0.70 Hz, or ∼10-fold (Fig. 1, A and B). UTP also elevated mean global [Ca2+]i from 193 nM (10) to ∼318 nM (Fig. 1C). 2-Aminoethoxydiphenyl borate (2-APB) and xestospongin C (XeC), IP3R blockers both prevented UTP-induced Ca2+ wave activation (Fig. 1, A and B). The mean UTP-induced global [Ca2+]i elevation was reduced by ∼54% by XeC and blocked by 2-APB (Fig. 1C). XeC did not significantly change spontaneous Ca2+ wave frequency (0.07 ± 0.05 Hz, n = 4, P > 0.05), whereas 2-APB abolished these events (0 ± 0 Hz, n = 4, P < 0.05). These data indicate that IP3R activation contributes to UTP-induced Ca2+ waves and global [Ca2+]i elevation in cerebral artery smooth muscle cells.
Fig. 1.
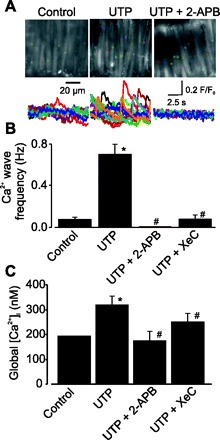
UTP activates Ca2+ waves and elevates global intracellular Ca2+ concentration ([Ca2+]i) as a result of inositol 1,4,5-trisphosphate (IP3) receptor (IP3R) activation in cerebral artery smooth muscle cells. A: Ca2+ signals recorded in smooth muscle cells of an intact cerebral artery under control conditions and in the presence of UTP (30 μM) and UTP + 2-aminoethoxydiphenyl borate (2-APB, 100 μM). Top: colored boxes (2.2 × 2.2 μm, 10 × 10 pixels) indicate locations of changes in fluorescence ratio (F/F0) measured over 10 s in arterial smooth muscle cells. Bottom: colored traces showing changes in F/F0 for respective colored boxes over 10 s. Images were acquired at 30 Hz. B: mean data illustrating inhibition of UTP-induced Ca2+ waves in smooth muscle cells of cerebral artery segments by 2-APB (100 μM) and xestospongin C (XeC, 20 μM). C: UTP-induced global [Ca2+]i elevations are abolished by 2-APB and attenuated by XeC. Changes in global [Ca2+]i were calculated from a control value previously determined using fura-2 (10). Values are means ± SE; n = 27, 13, 4, and 9 for control, UTP, UTP + 2-APB, and UTP + XeC, respectively. *P < 0.05 vs. control. #P < 0.05 vs. UTP.
IP3R isoform expression in rat cerebral artery smooth muscle cells.
We sought to examine the physiological function of individual IP3R isoforms in mediating UTP regulation of arterial smooth muscle cell Ca2+ signals. RT-PCR was performed on lysate isolated from small groups (∼100) of individually collected cerebral artery smooth muscle cells, as we have done previously (9). Transcripts for all three IP3R isoforms were amplified using conventional RT-PCR (Fig. 2A). Quantitative real-time PCR indicated that IP3R1 mRNA was the most abundant, at ∼82% of total IP3R message (Fig. 2B). Western blotting was performed using IP3R isoform-specific antibodies to detect proteins in rat cerebral artery lysate. Consistent with data obtained from RT-PCR experiments, protein bands for IP3R1, IP3R2, and IP3R3 were detected in whole brain and rat cerebral artery lysates, with the IP3R1 band the most prominent and IP3R2 the least intense (Fig. 2C). Immunofluorescence also identified IP3R1, IP3R2, and IP3R3 protein in freshly isolated cerebral artery smooth muscle cells (Fig. 2D). Collectively, these data indicate that IP3R1 is the most abundant IP3R isoform expressed in rat cerebral artery smooth muscle cells.
Fig. 2.
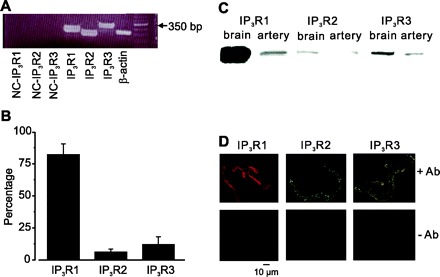
PCR, Western analysis, and immunofluorescence identify IP3R1, IP3R2, and IP3R3 expression in cerebral artery smooth muscle cells. A: RT-PCR detection of IP3R1 (285 bp), IP3R2 (241 bp), IP3R3 (319 bp), and β-actin (257 bp) in isolated smooth muscle cells. NC, negative control using smooth muscle cell RNA, instead of cDNA. Right: 50-bp-increment ladders are shown. B: real-time PCR data indicating percent message for IP3R1, IP3R2, and IP3R3 in isolated smooth muscle cells. Values are means ± SE; n = 3 for each. C: Western blot detection of IP3R1, IP3R2, and IP3R3 in cerebral arteries and brain. D: immunofluorescence results showing IP3R1, IP3R2, and IP3R3 expression and localization in isolated smooth muscle cells.
IP3R1 knockdown using shRNA in intact cerebral arteries.
Two different silencing vectors (IP3R1shV1 and IP3R1shV2) were constructed to express IP3R1-specific shRNA. IP3R1shV1 and IP3R1shV2 target exons 32 and 24, respectively, in IP3R1. A vector that encodes scrambled shRNA (IP3R1scrm) was used as a control. Reversible permeabilization was used to introduce IP3R1scrm, IP3R1shV1, IP3R1shV2, and IP3R1shV1 + IP3R1shV2 into intact cerebral arteries. Western blot analysis indicated that IP3R1shV1 and IP3R1shV2 reduced mean IP3R1 protein to ∼67 and 53%, respectively, of that in arteries treated with IP3R1scrm (Fig. 3). Insertion of both IP3R1shV1 + IP3R1shV2 decreased mean IP3R1 expression to ∼48% of that in arteries treated with IP3R1scrm (Fig. 3). IP3R1 knockdown did not alter IP3R3 protein, as determined by reprobing membranes with an IP3R3-selective antibody (Fig. 3B).
Fig. 3.
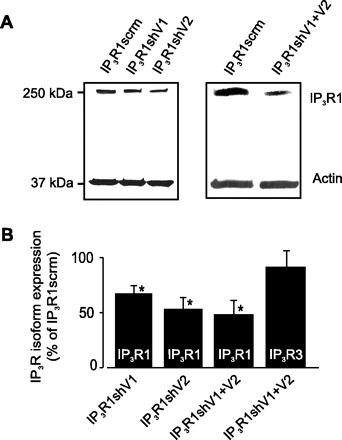
IP3R1 short hairpin RNA (shRNA) suppression vectors attenuate IP3R1 protein in cerebral arteries. A: Western blot illustrating that IP3R1shV1, IP3R1shV2, and IP3R1shV1 + IP3R1shV2 reduce IP3R1 protein in cerebral arteries. B: mean data illustrating that shRNAs targeting IP3R1 reduce IP3R1 expression, but do not alter IP3R3 expression, in cerebral arteries. Values are means ± SE; n = 5, 4, 3, and 3 for IP3R1shV1, IP3R1shV2, and IP3R1shV1 + IP3R1shV2 (IP3R1) and IP3R1shV1 + IP3R1shV2 (IP3R3), respectively. *P < 0.05 vs. IP3R1scrm.
IP3R1 knockdown attenuates UTP-induced Ca2+ wave activation and global [Ca2+]i elevation.
To study the function of IP3R1 in mediating UTP-induced Ca2+ wave and global [Ca2+]i elevations, we studied arteries in which IP3R1 expression was reduced with IP3R1shV1 + IP3R1shV2. IP3R1 knockdown did not alter spontaneous Ca2+ wave frequency but prevented UTP-induced Ca2+ wave activation and reduced the UTP-induced global [Ca2+]i elevation by ∼61% (Fig. 4, A and B). IP3R1 knockdown did not alter resting [Ca2+]i [fura-2 ratio at 340-nm emission to 380-nm emission = 1.03 ± 0.05 (n = 16) for IP3R1scrm and 1.08 ± 0.08 (n = 14, P > 0.05) for IP3R1shV] or SR Ca2+ load, as determined by measuring caffeine (10 mM)-induced [Ca2+]i transients (Fig. 4, C and D). These data indicate that IP3R1 activation is essential for UTP-induced Ca2+ signal modification in arterial smooth muscle cells.
Fig. 4.
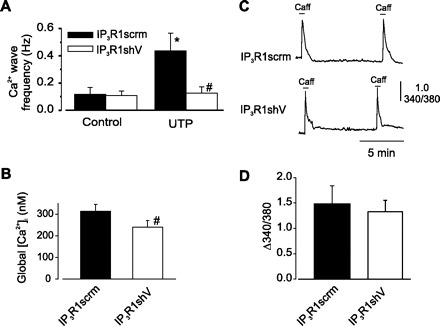
IP3R1 knockdown attenuates UTP- induced Ca2+ wave generation and global [Ca2+]i elevation in smooth muscle cells of intact cerebral artery segments. A: IP3R1 knockdown did not alter resting Ca2+ wave frequency but altered UTP-induced Ca2+ wave generation (n = 6). B: IP3R1 knockdown reduced UTP-induced global [Ca2+]i elevation (n = 6). Values show mean change in calibrated global [Ca2+]i from a control value of 193 nM (10). C: amplitude of caffeine (10 mM)-induced [Ca2+]i transients were similar in IP3R1scrm- and IP3R1shV-treated cerebral arteries. D: mean change in fura-2 ratio (Δ340/380) in cerebral arteries treated with IP3R1scrm (n = 16) and IP3R1shV (n = 14). Values are means ± SE. *P < 0.05 vs. control. #P < 0.05 vs. IP3R1scrm.
IP3R1 knockdown and an antibody targeting IP3R1 attenuate UTP-induced Icat activation.
We have previously shown that IP3-induced IP3R activation stimulates an Icat in cerebral artery smooth muscle cells (54). Therefore, we studied the importance of IP3R1 for this response. Inclusion of an anti-IP3R1 antibody in the pipette solution reduced mean baseline Icat density by ∼70% and attenuated the UTP-induced Icat elevation by ∼85% compared with a denatured antibody control (Fig. 5). Icat regulation was also studied in smooth muscle cells isolated from arteries in which IP3R1 expression was reduced using shRNA. Baseline current density in IP3R1scrm and IP3R1shV were similar [−3.8 ± 0.7 pA/pF (n = 11) and −3.9 ± 1.1 pA/pF (n = 4), respectively, P > 0.05; Fig. 6]. However, UTP-induced Icat activation was reduced by ∼87% in IP3R1shV-treated smooth muscle cells compared with cells treated with IP3R1scrm (Fig. 6). These data indicate that IP3R1 is required for UTP-induced Icat activation in cerebral artery smooth muscle cells.
Fig. 5.
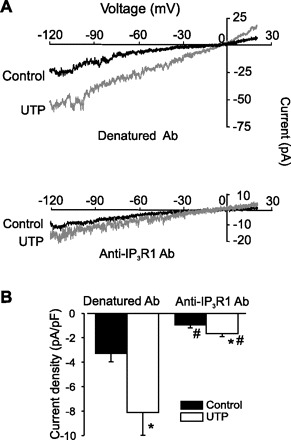
Anti-IP3R1 antibody inhibits UTP-induced cation current (Icat) in rat cerebral artery smooth muscle cells. A: exemplar traces obtained using whole cell patch-clamp configuration with anti-IP3R1 (1:100 dilution) or its heat-denatured control in the pipette before or after UTP (30 μM) application. B: mean data showing the effect of anti-IP3R1 antibody on Icat. Values are means ± SE; n = 11 (denatured control Ab) and 5 (all others). *P < 0.05 vs. control. #P < 0.05 vs. denatured Ab.
Fig. 6.
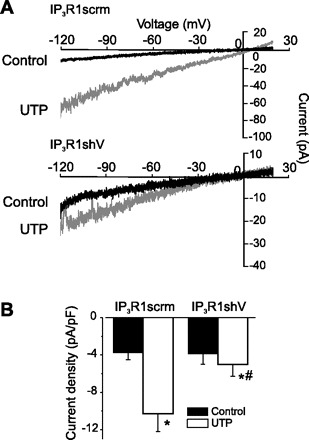
IP3R1 knockdown inhibits Icat activation by UTP in cerebral artery smooth muscle cells. A: exemplar traces illustrating UTP-induced Icat activation in IP3R1scrm- and IP3R1shV-treated smooth muscle cells. B: mean current density data for IP3R1scrm- and IP3R1shV-treated cells. *P < 0.05 vs. control. #P < 0.05 vs. IP3R1scrm.
IP3R1 knockdown attenuates UTP-induced vasoconstriction in arteries with intact and depleted SR Ca2+.
To investigate physiological functions of IP3R1, diameter regulation of pressurized (20 mmHg) endothelium-denuded arteries was studied. IP3R1 knockdown did not alter baseline myogenic tone [23 ± 1.8% (n = 6) vs. 19 ± 0.7% for scrambled control (n = 6), P > 0.05] but reduced mean UTP (1 μM)-induced constriction by ∼52% compared with scrambled control (Fig. 7). Thapsigargin (100 nM–1 μM), at concentrations that deplete SR Ca2+ in cerebral artery smooth muscle cells (54), did not alter arterial diameter (Fig. 7). In SR Ca2+-depleted arteries, IP3R1 knockdown also reduced mean UTP-induced vasoconstriction by ∼57% (100 nM thapsigargin) and 62% (1 μM thapsigargin). These data indicate that IP3R1 activation is essential for UTP-induced vasoconstriction in cerebral arteries.
Fig. 7.
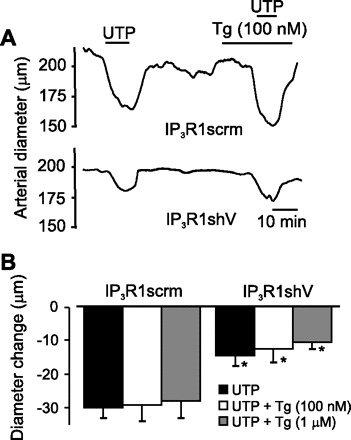
IP3R1 knockdown attenuates UTP-induced constriction in pressurized (20 mmHg) cerebral arteries. A: exemplar diameter measurements illustrating UTP-induced vasoconstriction in cerebral arteries treated with IP3R1scrm or IP3R1shV in the presence or absence of thapsigargin (Tg, 100 nM). B: mean diameter change data. Values are means ± SE; n = 6 (UTP and UTP + 100 nM Tg) and 4 (UPT + 1 μM Tg). *P < 0.05 vs. IP3R1scrm.
DISCUSSION
We have investigated the relative expression and physiological functions of IP3R isoforms in cerebral artery smooth muscle cells. We provide novel evidence that 1) IP3R1 is the predominant IP3R isoform, 2) IP3R1 activation is required for UTP-induced Icat activation, Ca2+ wave stimulation, and global [Ca2+]i elevation, and 3) IP3R1 activation contributes to UTP-induced vasoconstriction in arteries with intact and depleted SR Ca2+.
Previous studies detected IP3R isoforms that are expressed in the vasculature by performing RT-PCR and immunofluorescence on whole vessels or cultured vascular smooth muscle cells (17, 32, 51, 56). These studies suggested that all three IP3R isoforms are expressed in cultured aortic smooth muscle cells, whole aorta, and mesenteric and basilar arteries, whereas IP3R3 was absent in cultured portal vein smooth muscle cells (17, 32, 51, 56). Using quantitative RT-PCR, we show that all three IP3R isoforms are expressed in freshly isolated, selected cerebral artery smooth muscle cells, with IP3R1 message being the most abundant. Immunofluorescence also identified all three IP3R isoforms in isolated smooth muscle cells, and Western blotting of cerebral artery lysate supported the quantitative PCR data suggesting that IP3R1 was the most abundant protein. These data indicate that IP3R1 is by far the most prevalent IP3R isoform expressed in cerebral artery smooth muscle cells.
UTP-induced phospholipase C activation elevates diacylglycerol (DAG), leading to PKC activation, and increases IP3, leading to IP3R activation (35, 44). Vasoconstrictors, including UTP, block Ca2+ sparks, activate Ca2+ waves and oscillations, and elevate global [Ca2+]i in arterial smooth muscle cells (23, 31, 52). UTP-induced Ca2+ spark inhibition occurs as a result of PKC activation (23). Mechanisms that generate Ca2+ waves and the physiological functions of these Ca2+ signals in vascular smooth muscle cells are unclear. Vasoconstrictors elevate Ca2+ wave frequency, and depletion of SR Ca2+ abolishes these events (23, 30). Different proposals have been developed to explain mechanisms that generate and propagate Ca2+ waves (7, 15, 16, 18). One model suggests that ryanodine receptor (RyR) channel and IP3R activation is required for Ca2+ wave generation and propagation (7, 15, 16, 26). In this scenario, IP3 acts as a primer to trigger Ca2+ release through SR IP3Rs. Released Ca2+ then stimulates further Ca2+ release by activating RyR channels, and waves proceed independently of IP3Rs. Another proposal suggests that IP3R activation alone is sufficient to initiate and propagate waves through Ca2+-induced Ca2+ release (18, 26). Both concepts indicate that IP3Rs are required for Ca2+ wave generation. In cultured human coronary artery smooth muscle cells, IP3 generation and subsequent IP3R activation are required for UTP-induced SR Ca2+ release (44). Our data obtained using pharmacological blockers, antibodies, and protein knockdown indicate that IP3R1 is required for UTP-induced Ca2+ wave generation and propagation. IP3R1 knockdown did not change SR Ca2+ load, which would have indirectly affected Ca2+ wave frequency (23, 30). Thus data suggest that IP3R1 knockdown inhibits Ca2+ waves by reducing the number of IP3R1 channels that contribute to these signaling events. Intracellular IP3 concentration should be low in the absence of exogenous receptor agonists, although UTP may be released from endothelial cells under resting conditions, which may generate additional IP3 (24, 25, 38). Since IP3R1 knockdown and XeC did not alter baseline Ca2+ wave frequency in the absence of UTP, our data suggest that the contribution of IP3 to Ca2+ waves in the absence of agonists is small. These data support previous evidence that spontaneous Ca2+ waves occur due to RyR channel activation and that RyR channel activation alone can produce Ca2+ waves (8, 19). In contrast to effects of XeC and IP3R knockdown, 2-APB abolished spontaneous Ca2+ waves. This effect may occur as a result of nonspecific actions of 2-APB on mitochondria and nonselective cation channels, in addition to IP3R inhibition (33). In cultured portal vein smooth muscle cells, IP3R2 activation was required for ACh-induced global Ca2+ oscillations, whereas targeting of IP3R1 had no effect on these Ca2+ signals (32). These different findings raise the possibility that IP3Rs exhibit variable expression profiles and physiological functions in smooth muscle cells of anatomically different vessels and in cultured versus noncultured smooth muscle cells.
Collectively, data presented here and in our previous study indicate that IP3R1 activation contributes to agonist-induced Icat activation in cerebral artery smooth muscle cells (54). TRPC channels, including TRPC1, TRPC3, TRPC5, TRPC6, and TRPC7, are expressed in vascular smooth muscle cells and may contribute to Icat (2, 12, 28, 36, 39, 40, 54). In cerebral artery smooth muscle cells, TRPC3 channel knockdown attenuated endothelin-1- and IP3-induced [Ca2+]i elevation and constriction, indicating that this isoform is a major contributor to the IP3R-induced Icat in this cell type (54). The mechanism by which IP3R activation stimulates Icat in arterial smooth muscle cells is unclear. Studies using protein overexpression have demonstrated that a consensus sequence present on the COOH terminus of all TRPC channel isoforms recognizes an NH2-terminal binding domain found in all IP3R isoforms (46). It has been proposed that physical coupling of these two domains removes inhibitory calmodulin from the TRPC channel COOH terminus, leading to channel activation (46, 55). Whether a similar coupling mechanism underlies IP3R regulation of Icat and TRPC3 channels in arterial smooth muscle cells remains to be determined but is one possibility.
Our data indicate that IP3R1 suppression with antibodies or shRNA attenuated UTP-induced Icat activation by ∼85%. This level of inhibition is large, given that UTP-induced phospholipase C activation also elevates DAG, and DAG and 1-oleoyl-2-acetyl-sn-glycerol (OAG), a DAG analog, activated Icat in rat cerebral and rabbit coronary artery smooth muscle cells, portal vein smooth muscle cells, and cells overexpressing TRPC channels (3, 20, 34, 42). In cerebral artery smooth muscle cells, PKC activation stimulates an Icat by increasing the apparent micromolar Ca2+ sensitivity of TRPM4 channels (13, 42). In our patch-clamp experiments, EGTA was used to clamp free Ca2+ at a physiological concentration of 100 nM. Therefore, UTP-induced PKC activation would not be expected to activate TRPM4 channels, and the strong inhibitory effect of IP3R1 inhibition on UTP-induced Icat may be due to an effect on TRPC channels alone. However, PKC regulation of TRPC channels is complex, with studies reporting activation, inhibition, or no effect on TRP channels, including those using smooth muscle cells from a variety of blood vessels (4, 34, 41, 49, 50). In rabbit coronary artery smooth muscle cells, IP3 potentiated DAG-induced Icat activation, and heparin blocked this activation, indicating that IP3Rs mediated this response (34). In cerebral artery smooth muscle cell inside-out patches, IP3 and OAG did not activate cation channels, and OAG did not facilitate IP3-induced cation channel activation (54). Whether interactions occur between IP3Rs and PKC in Icat activation and what the mechanisms are in cerebral artery smooth muscle cells remain to be determined. Nevertheless, our data indicate that IP3R activation is a key mechanism mediating agonist-induced Icat activation in these cells.
An elevation in global [Ca2+]i stimulates vasoconstriction, whereas a reduction in global [Ca2+]i leads to vasodilation. UTP elevated global [Ca2+]i, and this effect was blocked by 2-APB and attenuated by XeC and IP3R1 knockdown. To study physiological functions of IP3R1 that occur through SR Ca2+ release-dependent and -independent mechanisms, diameter measurements were obtained at an intravascular pressure of 20 mmHg to reduce the influence of SR Ca2+ depletion regulating diameter through Ca2+ spark inhibition, as we have done previously (54). UTP-induced vasoconstriction was inhibited by IP3R1 knockdown, consistent with effects on Ca2+ signals. More importantly, UTP-induced vasoconstriction was inhibited by IP3R1 knockdown, even though SR Ca2+ depletion did not alter UTP-induced vasoconstriction. In addition, SR Ca2+ depletion did not alter arterial diameter at 20 mmHg, consistent with previous data (54). Taken together, data indicate that 1) IP3R1 activation contributes to UTP-induced vasoconstriction, 2) at 20 mmHg, SR Ca2+ release does not contribute to UTP-induced vasoconstriction, 3) SR Ca2+ load does not regulate arterial diameter at low pressure, and 4) UTP-induced IP3R1 activation constricts cerebral arteries through an SR Ca2+ release-independent mechanism. We propose that, in cerebral artery smooth muscle cells, UTP stimulates IP3R1, leading to Icat activation, membrane depolarization, voltage-dependent Ca2+ channel activation, a global [Ca2+]i elevation, and vasoconstriction. We also show that UTP-induced Ca2+ waves require IP3R1 activation and SR Ca2+ release (22). Since SR Ca2+ depletion did not alter UTP-induced vasoconstriction, our data suggest that SR Ca2+ release and, thus, Ca2+ waves do not contribute to UTP-induced vasoconstriction. These data support our previous observation that SR Ca2+ release does not contribute to IP3-induced vasoconstriction at low pressure (54). We have also shown that elevating pressure to 60 mmHg increases the contribution of SR Ca2+ release to IP3-induced vasoconstriction (54). Conceivably, vasoconstrictor-induced Ca2+ waves may directly contribute to contraction at higher pressures.
Physiological functions of IP3R2 and IP3R3 were not directly investigated in the present study. By comparing effects of XeC, a blocker of all IP3R isoforms, and those of IP3R1 knockdown and antibodies, data suggest that IP3R1 is the principal molecular isoform mediating UTP-induced Icat activation, Ca2+ wave stimulation, and contraction in cerebral artery smooth muscle cells. Partial IP3R1 knockdown almost completely blocked UTP-induced Icat and Ca2+ wave activation and partially attenuated vasoconstriction. IP3R2 and IP3R3 are expressed and therefore, these proteins are highly likely to perform physiological functions in arterial myocytes. We do not rule out physiological functions for IP3R2 and IP3R3. However, our data indicate a principal function for IP3R1 in mediating these responses.
In summary, the present study provides evidence that all three IP3R isoforms are expressed in rat cerebral artery smooth muscle cells and that IP3R1 is the most abundant subtype. We also show that IP3R1 activation contributes to UTP-induced Icat activation, Ca2+ wave stimulation, global [Ca2+]i elevation, and vasoconstriction. These data indicate that IP3R1 activation is necessary for UTP-induced vasoconstriction.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants HL-67061 and HL-077678 to J. H. Jaggar and American Heart Association Southeast Affiliate Postdoctoral Fellowship 0625672B to A. Adebiyi.
Acknowledgments
We thank Dr. John Bannister for critical reading of the manuscript.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Acharya JK, Jalink K, Hardy RW, Hartenstein V, Zuker CS. InsP3 receptor is essential for growth and differentiation but not for vision in Drosophila Neuron 18: 881–887, 1997 [DOI] [PubMed] [Google Scholar]
- 2.Albert AP, Large WA. Store-operated Ca2+-permeable non-selective cation channels in smooth muscle cells. Cell Calcium 33: 345–356, 2003 [DOI] [PubMed] [Google Scholar]
- 3.Albert AP, Large WA. Synergism between inositol phosphates and diacylglycerol on native TRPC6-like channels in rabbit portal vein myocytes. J Physiol 552: 789–795, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albert AP, Large WA. Inhibitory regulation of constitutive transient receptor potential-like cation channels in rabbit ear artery myocytes. J Physiol 560: 169–180, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blatter LA, Wier WG. Agonist-induced [Ca2+]i waves and Ca2+-induced Ca2+ release in mammalian vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 263: H576–H586, 1992 [DOI] [PubMed] [Google Scholar]
- 6.Blondel O, Takeda J, Janssen H, Seino S, Bell GI. Sequence and functional characterization of a third inositol trisphosphate receptor subtype, IP3R-3, expressed in pancreatic islets, kidney, gastrointestinal tract, and other tissues. J Biol Chem 268: 11356–11363, 1993 [PubMed] [Google Scholar]
- 7.Boittin FX, Macrez N, Halet G, Mironneau J. Norepinephrine-induced Ca2+ waves depend on InsP3 and ryanodine receptor activation in vascular myocytes. Am J Physiol Cell Physiol 277: C139–C151, 1999 [DOI] [PubMed] [Google Scholar]
- 8.Cheng H, Lederer MR, Lederer WJ, Cannell MB. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am J Physiol Cell Physiol 270: C148–C159, 1996 [DOI] [PubMed] [Google Scholar]
- 9.Cheng X, Liu J, Asuncion-Chin M, Blaskova E, Bannister JP, Dopico AM, Jaggar JH. A novel CaV1.2 N terminus expressed in smooth muscle cells of resistance size arteries modifies channel regulation by auxiliary subunits. J Biol Chem 282: 29211–29221, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheranov SY, Jaggar JH. Mitochondrial modulation of Ca2+ sparks and transient KCa currents in smooth muscle cells of rat cerebral arteries. J Physiol 556: 755–771, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheranov SY, Jaggar JH. Sarcoplasmic reticulum calcium load regulates rat arterial smooth muscle calcium sparks and transient KCa currents. J Physiol 544: 71–84, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dietrich A, Kalwa H, Rost BR, Gudermann T. The diacylgylcerol-sensitive TRPC3/6/7 subfamily of cation channels: functional characterization and physiological relevance. Pflügers Arch 451: 72–80, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Earley S, Straub SV, Brayden JE. Protein kinase C regulates vascular myogenic tone through activation of TRPM4. Am J Physiol Heart Circ Physiol 292: H2613–H2622, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87: 593–658, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldbeter A, Dupont G, Berridge MJ. Minimal model for signal-induced Ca2+ oscillations and for their frequency encoding through protein phosphorylation. Proc Natl Acad Sci USA 87: 1461–1465, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gordienko DV, Bolton TB. Crosstalk between ryanodine receptors and IP3 receptors as a factor shaping spontaneous Ca2+-release events in rabbit portal vein myocytes. J Physiol 542: 743–762, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grayson TH, Haddock RE, Murray TP, Wojcikiewicz RJ, Hill CE. Inositol 1,4,5-trisphosphate receptor subtypes are differentially distributed between smooth muscle and endothelial layers of rat arteries. Cell Calcium 36: 447–458, 2004 [DOI] [PubMed] [Google Scholar]
- 18.Hajnoczky G, Thomas AP. Minimal requirements for calcium oscillations driven by the IP3 receptor. EMBO J 16: 3533–3543, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heppner TJ, Bonev AD, Santana LF, Nelson MT. Alkaline pH shifts Ca2+ sparks to Ca2+ waves in smooth muscle cells of pressurized cerebral arteries. Am J Physiol Heart Circ Physiol 283: H2169–H2176, 2002 [DOI] [PubMed] [Google Scholar]
- 20.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397: 259–263, 1999 [DOI] [PubMed] [Google Scholar]
- 21.Iino M, Kasai H, Yamazawa T. Visualization of neural control of intracellular Ca2+ concentration in single vascular smooth muscle cells in situ. EMBO J 13: 5026–5031, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaggar JH. Intravascular pressure regulates local and global Ca2+ signaling in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol 281: C439–C448, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Jaggar JH, Nelson MT. Differential regulation of Ca2+ sparks and Ca2+ waves by UTP in rat cerebral artery smooth muscle cells. Am J Physiol Cell Physiol 279: C1528–C1539, 2000 [DOI] [PubMed] [Google Scholar]
- 24.Lazarowski ER, Boucher RC. UTP as an extracellular signaling molecule. News Physiol Sci 16: 1–5, 2001 [DOI] [PubMed] [Google Scholar]
- 25.Lazarowski ER, Homolya L, Boucher RC, Harden TK. Direct demonstration of mechanically induced release of cellular UTP and its implication for uridine nucleotide receptor activation. J Biol Chem 272: 24348–24354, 1997 [DOI] [PubMed] [Google Scholar]
- 26.Lechleiter JD, Clapham DE. Molecular mechanisms of intracellular calcium excitability in X. laevis oocytes. Cell 69: 283–294, 1992 [DOI] [PubMed] [Google Scholar]
- 27.Liu M, Albert AP, Large WA. Facilitatory effect of Ins(1,4,5)P3 on store-operated Ca2+-permeable cation channels in rabbit portal vein myocytes. J Physiol 566: 161–171, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maruyama Y, Nakanishi Y, Walsh EJ, Wilson DP, Welsh DG, Cole WC. Heteromultimeric TRPC6-TRPC7 channels contribute to arginine vasopressin-induced cation current of A7r5 vascular smooth muscle cells. Circ Res 98: 1520–1527, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Mauban JR, Lamont C, Balke CW, Wier WG. Adrenergic stimulation of rat resistance arteries affects Ca2+ sparks, Ca2+ waves, and Ca2+ oscillations. Am J Physiol Heart Circ Physiol 280: H2399–H2405, 2001 [DOI] [PubMed] [Google Scholar]
- 30.McCarron JG, Bradley KN, MacMillan D, Chalmers S, Muir TC. The sarcoplasmic reticulum, Ca2+ trapping, and wave mechanisms in smooth muscle. News Physiol Sci 19: 138–147, 2004 [DOI] [PubMed] [Google Scholar]
- 31.Miriel VA, Mauban JR, Blaustein MP, Wier WG. Local and cellular Ca2+ transients in smooth muscle of pressurized rat resistance arteries during myogenic and agonist stimulation. J Physiol 518: 815–824, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morel JL, Fritz N, Lavie JL, Mironneau J. Crucial role of type 2 inositol 1,4,5-trisphosphate receptors for acetylcholine-induced Ca2+ oscillations in vascular myocytes. Arterioscler Thromb Vasc Biol 23: 1567–1575, 2003 [DOI] [PubMed] [Google Scholar]
- 33.Peppiatt CM, Collins TJ, Mackenzie L, Conway SJ, Holmes AB, Bootman MD, Berridge MJ, Seo JT, Roderick HL. 2-Aminoethoxydiphenyl borate (2-APB) antagonises inositol 1,4,5-trisphosphate-induced calcium release, inhibits calcium pumps and has a use-dependent and slowly reversible action on store-operated calcium entry channels. Cell Calcium 34: 97–108, 2003 [DOI] [PubMed] [Google Scholar]
- 34.Peppiatt-Wildman CM, Albert AP, Saleh SN, Large WA. Endothelin-1 activates a Ca2+-permeable cation channel with TRPC3 and TRPC7 properties in rabbit coronary artery myocytes. J Physiol 580: 755–764, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev 50: 413–492, 1998 [PubMed] [Google Scholar]
- 36.Reading SA, Earley S, Waldron BJ, Welsh DG, Brayden JE. TRPC3 mediates pyrimidine receptor-induced depolarization of cerebral arteries. Am J Physiol Heart Circ Physiol 288: H2055–H2061, 2005 [DOI] [PubMed] [Google Scholar]
- 37.Ross CA, Danoff SK, Schell MJ, Snyder SH, Ullrich A. Three additional inositol 1,4,5-trisphosphate receptors: molecular cloning and differential localization in brain and peripheral tissues. Proc Natl Acad Sci USA 89: 4265–4269, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saiag B, Shacoori V, Bodin P, Catheline M, Burnstock G. Lack of uptake, release and action of UTP at sympathetic perivascular nerve terminals in rabbit ear artery. Eur J Pharmacol 358: 139–145, 1998 [DOI] [PubMed] [Google Scholar]
- 39.Saleh SN, Albert AP, Peppiatt CM, Large WA. Angiotensin II activates two cation conductances with distinct TRPC1 and TRPC6 channel properties in rabbit mesenteric artery myocytes. J Physiol 577: 479–495, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saleh SN, Albert AP, Peppiatt-Wildman CM, Large WA. Diverse properties of store-operated TRPC channels activated by protein kinase C in vascular myocytes. J Physiol 586: 2463–2476, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi J, Mori E, Mori Y, Mori M, Li J, Ito Y, Inoue R. Multiple regulation by calcium of murine homologues of transient receptor potential proteins TRPC6 and TRPC7 expressed in HEK293 cells. J Physiol 561: 415–432, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Slish DF, Welsh DG, Brayden JE. Diacylglycerol and protein kinase C activate cation channels involved in myogenic tone. Am J Physiol Heart Circ Physiol 283: H2196–H2201, 2002 [DOI] [PubMed] [Google Scholar]
- 43.Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature 372: 231–236, 1994 [DOI] [PubMed] [Google Scholar]
- 44.Strobaek D, Olesen SP, Christophersen P, Dissing S. P2-purinoceptor-mediated formation of inositol phosphates and intracellular Ca2+ transients in human coronary artery smooth muscle cells. Br J Pharmacol 118: 1645–1652, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takei K, Shin RM, Inoue T, Kato K, Mikoshiba K. Regulation of nerve growth mediated by inositol 1,4,5-trisphosphate receptors in growth cones. Science 282: 1705–1708, 1998 [DOI] [PubMed] [Google Scholar]
- 46.Tang J, Lin Y, Zhang Z, Tikunova S, Birnbaumer L, Zhu MX. Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of trp channels. J Biol Chem 276: 21303–21310, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tasker PN, Taylor CW, Nixon GF. Expression and distribution of InsP3 receptor subtypes in proliferating vascular smooth muscle cells. Biochem Biophys Res Commun 273: 907–912, 2000 [DOI] [PubMed] [Google Scholar]
- 48.Taylor CW, Genazzani AA, Morris SA. Expression of inositol trisphosphate receptors. Cell Calcium 26: 237–251, 1999 [DOI] [PubMed] [Google Scholar]
- 49.Trebak M, Hempel N, Wedel BJ, Smyth JT, Bird GS, Putney JW Jr. Negative regulation of TRPC3 channels by protein kinase C-mediated phosphorylation of serine 712. Mol Pharmacol 67: 558–563, 2005 [DOI] [PubMed] [Google Scholar]
- 50.Venkatachalam K, Zheng F, Gill DL. Regulation of canonical transient receptor potential (TRPC) channel function by diacylglycerol and protein kinase C. J Biol Chem 278: 29031–29040, 2003 [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, Chen J, Wang Y, Taylor CW, Hirata Y, Hagiwara H, Mikoshiba K, Toyo-oka T, Omata M, Sakaki Y. Crucial role of type 1, but not type 3, inositol 1,4,5-trisphosphate (IP3) receptors in IP3-induced Ca2+ release, capacitative Ca2+ entry, and proliferation of A7r5 vascular smooth muscle cells. Circ Res 88: 202–209, 2001 [DOI] [PubMed] [Google Scholar]
- 52.Welsh DG, Brayden JE. Mechanisms of coronary artery depolarization by uridine triphosphate. Am J Physiol Heart Circ Physiol 280: H2545–H2553, 2001 [DOI] [PubMed] [Google Scholar]
- 53.Wilkerson MK, Heppner TJ, Bonev AD, Nelson MT. Inositol trisphosphate receptor calcium release is required for cerebral artery smooth muscle cell proliferation. Am J Physiol Heart Circ Physiol 290: H240–H247, 2006 [DOI] [PubMed] [Google Scholar]
- 54.Xi Q, Adebiyi A, Zhao G, Chapman KE, Waters CM, Hassid A, Jaggar JH. IP3 constricts cerebral arteries via IP3 receptor-mediated TRPC3 channel activation and independently of sarcoplasmic reticulum Ca2+ release. Circ Res 102: 1118–1126, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Z, Tang J, Tikunova S, Johnson JD, Chen Z, Qin N, Dietrich A, Stefani E, Birnbaumer L, Zhu MX. Activation of Trp3 by inositol 1,4,5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain. Proc Natl Acad Sci USA 98: 3168–3173, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou H, Nakamura T, Matsumoto N, Hisatsune C, Mizutani A, Iesaki T, Daida H, Mikoshiba K. Predominant role of type 1 IP3 receptor in aortic vascular muscle contraction. Biochem Biophys Res Commun 369: 213–219, 2008 [DOI] [PubMed] [Google Scholar]