

Abstract
Prevailing literature suggests diversified cellular functions for the adenomatous polyposis coli (APC) gene. Among them a recently discovered unique role of APC is in DNA repair. The APC gene can modulate the base excision repair (BER) pathway through an interaction with DNA polymerase β (Pol-β) and flap endonuclease 1 (Fen-1). Taken together with the transcriptional activation of APC gene by alkylating agents and modulation of BER activity, APC may play an important role in carcinogenesis and chemotherapy by determining whether cells with DNA damage survive or undergo apoptosis. In this review, we summarize the evidence supporting this novel concept and suggest that these results will have implications for the development of more effective strategies for chemoprevention, prognosis, and chemotherapy of certain types of tumors.
Keywords: Adenomatous polyposis coli, Base excision repair, DNA polymerase β, Carcinogenesis, Chemotherapy
1. Introduction
APC is expressed constitutively within the normal colonic epithelium. The APC gene product is a 310-kDa-homodimeric protein, which is localized in the cytoplasm and the nucleus [1], [2], [3], [4], [5], [6] and [7]. Appropriate levels of wild-type APC are critical to cytoskeletal integrity [1] and [8], cellular adhesion [9] , and Wingless/Wnt signaling [7], [10] and [11]. Wild-type APC binds to EB1, which regulates microtubule polymerization [12], and a tumor suppressor protein, DLG, which regulates cell cycle progression from the Go/G1 to the S phase of the cell cycle [13]. Truncation in APC dominantly interferes with mitotic spindle function by regulating microtubule dynamics during mitosis [14] and [15]. In addition, APC may act as a negative regulator of β-catenin signaling in the transformation of colonic epithelial cells [16] and [17] and in melanoma progression [18]. The β-catenin/Tcf4 complex regulates the proto-oncogene and cell cycle regulator c-myc [19], the G1/S-regulating cyclin D1 [20], the gene encoding the matrix-degrading metalloproteinase, matrilysin [21], the AP-1 transcription factors c-jun and fra-1 and the urokinase-type plasminogen activator receptor gene [22].
There is a considerable body of evidence that indicates that mutations of APC are associated with colorectal cancer. As described by Muto et al. [23], colorectal cancer develops through a series of histologically distinct stages from “adenoma to carcinoma” and it has been suggested that this progression through the histologic stages is associated with the temporal order in which mutations occur in different genes [24] and [25]. Mutations in APC, as well as Ki-ras, deleted in colorectal cancer (DCC), and p53 genes all play important roles in the development of colorectal cancer and appear to act at different stages of tumorigenesis [7] and [11]. Mutation of the APC gene is an early event in familial adenomatous polyposis (FAP), a syndrome in which there is an inherited predisposition to colon cancer [26] and [27]. Mutations in the APC gene are also found in 60 to 80% of sporadic colorectal cancers and adenomas [26], [28] and [29].
The site of the mutation of the APC gene appears to be of importance in the development of the colorectal cancer. An association has been shown between the severe polyposis phenotype and germline mutations in the mutation cluster region (MCR) of the APC gene [30], [31] and [32]. Recently, the biological significance of nonsense and frame-shift mutations in MCR has been correlated with truncated and nonfunctional APC protein. Allelic loss of APC gene results in the complete loss of its function. The truncating mutation at nucleotide 3920, identified by in vitro synthesized protein method, converts isoleucine to lysine at codon 1307(I1307K) [33]. This germ-line mutation causes structural abnormalities that impair its biological activity. Selective pressure for an MCR mutant has been proposed based on the germline mutation in familial adenomatous polyposis [34]. Patients with mutations outside of the MCR region exhibit milder phenotypes. A recent study has shown that animals with homozygous truncating mutations at codon 1638 of APC do not develop tumors and survive through adulthood [35]. These data suggest that a truncation mutation in MCR is necessary for the loss of β-catenin binding and nuclear localization signals, resulting in loss of function and tumor progression [36]. It is now established that mutations in APC may be necessary for the early onset of polyposis.
2. Regulation of the APC gene in response to DNA alkylating agents
Earlier it has been reported that the APC gene is inducible, and its transcription can be enhanced in response to DNA-alkylating agents such as N-methyl-N’-nitro-N-nitrosoguanidine (MNNG), methylmethane sulfonate (MMS) and dimethylhydrazine (DMH) [37]. The promoter region of the APC gene which is fully characterized for various cis-regulatory elements, has a TATA-less promoter and two p53-binding elements, as well as consensus binding sites for Octamer, AP2, Sp1, a CAAT-box, and three nucleotide sequences for E-Box A, B and M [38]. Subsequent studies to understand the mechanism of APC gene regulation suggest that its regulation in response to DNA-alkylating agents can occur through several different mechanisms. Notably, DNA alkylation-induced transcriptional regulation of the APC gene expression in colon cancer cells is mediated through p53-binding elements [39]. This finding was the first evidence of a direct link between p53 and APC. However, overexpression of the upstream stimulating factors 1 and 2 (USF-1 and USF-2) in colon cancer cells that bind to the E-box B site can also upregulate the transcription of the APC gene [38]. The DNA damage-induced APC gene regulation was further supported by another study in which a cigarette smoke constituent, 7,12-dimethyl benzanthracene (DMBA), was shown to upregulate the expression of this gene in spontaneously immortalized normal human breast epithelial cells. In these cells, the DMBA-response on the APC gene expression was mediated through the GC-box binding protein Sp3 [40]. These studies provide clear evidence that DNA alkylation damage can result in enhanced transcription of APC and that this effect can be mediated by various mechanisms. Thus, p53, E Box B and GC-box-binding proteins can upregulate the APC gene expression. However, the involvement of additional mechanisms cannot be ruled out.
3. Role of APC in base-excision repair
Exogenous and endogenous mutagenic agents attack the genomes of all living cells resulting in the generation of damaged DNA bases. These damaged DNA bases may be cytotoxic and/or miscoding and are thought to be a major source of intermediates in carcinogenesis and tumorigenesis [41]. DNA repair systems efficiently remove damaged DNA via several different pathways, which reverse the vast majority of genetic lesions formed during the life span of a cell [42]. Base excision repair (BER) is the main pathway for the repair of endogenous abasic DNA damage. Most DNA repair mechanisms, including the base excision repair (BER) pathway, involve the participation of enzymes and other proteins that recognize structural alterations in DNA [43], [44] and [45]. It has been estimated that approximately one million abasic sites are generated per mammalian cell per day [46]. Abasic sites are unstable and degrade spontaneously into DNA-strand breaks by β-elimination that retards DNA polymerases. The abasic sites are highly mutagenic because they result in non-template DNA and RNA synthesis. Despite the large number of abasic sites generated per cell per day, the number of mutations is extremely low. This discordance underscores the importance of the elaborate mechanisms that the cell has devised to repair abasic sites.
In mammalian cells, BER can proceed through at least two pathways designated as the “single nucleotide (SN)-BER” and “multinucleotide or long-patch (LP)-BER” pathways (Fig. 1). These two pathways are differentiated by the repair patch size, as well as by the contribution of different proteins to the pathway [47] and [48]. In both pathways, repair is initiated by the recognition and removal of the modified base by a DNA glycosylase generating an abasic site (AP-site). Subsequently, APE-1 cleaves the DNA backbone generating 3′-OH and 5’-deoxyribose phosphate (5′-dRP) ends [49], [50] and [51]. Subsequently, the remaining 5′-dRP residue is cleaved by a 5’-deoxyribose phosphate lyase (dRP-lyase) activity of Pol-β to yield a 5’-phosphorylated gapped-DNA strand [52]. Pol-β then incorporates the correct base at the site of the damaged base with its polymerizing activity and DNA ligase-I or III sealing the nick [53] and [54]. This repair process becomes more complicated once the AP-site becomes oxidized or reduced. Under these circumstances, the dRP-lyase activity of Pol-β is interrupted, and the repair of DNA is accomplished through LP-BER. The Pol-β-dependent strand-displacement synthesis generates a longer repair patch and a 5’-overhang of a single-stranded DNA-flap with a modified sugar at its 5’-end. Pol-β can also be replaced by Pol-δ/ε for strand-displacement synthesis [55]. The 5’-overhang DNA-flap is cleaved by flap endonuclease 1 (Fen-1), and finally the nick is sealed by DNA ligase I or III [42], [47], [56], [57], [58], [59] and [60]. The reduced activity of any one of the BER proteins either due to reduced expression or interaction with other proteins can lead to decrease in BER. In many colon and lung tumors and cell lines, a defective BER is associated with the development of these cancers [61], [62], [63] and [64].
Fig. 1. Model of BER pathways.

DNA repair of abasic sites diverge after the generation of the 3’-hydroxyl required for replacement synthesis. The SN- or LP-BER pathways and their known protein components are summarized. Panel A shows SN-BER, in which APC-mediated blockage site of dRP-lyase activity is indicated. Panel B shows LP-BER, in which APC-mediated blockage sites of strand-displacement synthesis and Fen-1 activity are indicated. APE1, apurinic/apyrimidinic endonuclease 1; Fen-1, Flap endonuclease 1; Pol-β, DNA polymerase β; UDG, uridine DNA glycosylase.
3.1 APC interacts with Pol-β and Fen-1
The role of APC in DNA repair, particularly in base excision repair (BER) pathway is a novel finding, which was initially discovered by determining its interaction with proliferating cell nuclear antigen (PCNA). Since PCNA is known to participate in BER as a cofactor and to stimulate its activity [65], it was presumed a possibility that the interaction of APC with PCNA may block BER; however, the later studies concluded that the interaction of APC with PCNA did not affect BER. Concomitantly, the possible interaction of APC with other BER proteins such as Pol-β and flap endonuclease 1 (Fen-1), the two key enzymes of the BER pathway, were identified (Fig. 1) [66], [67], [68] and [69].
3.2 APC blocks Pol-β-directed SN- and LP-BER
The dRP-lyase activity of Pol-β is a rate-limiting step in SN-BER [70] and [71]. Biochemical [72], [73] and [74] and crystallographic studies [75] indicate that Lys72 plays a critical role in the dRP-lyase reaction mechanism. This reaction proceeds via a Schiff-base intermediate between Pol-β and the 5’-dRP residue of the substrate, whereby the side chain of Lys72 provides the nucleophile for the completion of the reaction [76]. By modeling analysis it has indicated that the interaction of APC with Pol-β is mediated by a stretch of residues near Lys72 (Fig. 3) [71]. The crystal structure of Pol-β indicates that Lys35, Lys68, Lys72 and Lys84 coordinate the 5’-phosphate of a gapped-DNA and may modulate its dRP-lyase activity [77]. Since the interaction of APC with Thr79, Lys81 and Arg83 falls near the dRP-lyase active site pocket, the influence of APC binding on dRP-lyase activity has been examined. The interaction of APC's DRI-domain with amino acid residues Thr79, Lys81 and Arg83 of Pol-β blocked Pol-β-directed dRP-lyase activity [68]. The mechanism of blockage of LP-BER by APC is more complex than the blockage of SN-BER [68]. In LP-BER, APC interacts with Pol-β and blocks Pol-β-directed strand-displacement synthesis. It also interacts with Fen-1 and blocks its 5’-flap endonuclease and 3’-5’ exonuclease activities [67]. Thus, by blocking both activities of Pol-β as well as Fen-1, APC blocks LP-BER [68]. Currently, the precise consequence of the blocked SN- and LP-BER on cellular fate is not clear.
Fig. 3. Ribbon representation of Pol-β highlighting the position of key interaction site of APC.

Based on yeast two-hybrid analysis with APCwt and APC(I-A,Y-A) mutant expression plasmids (amino acid 1190−1324, in which the 1259I and 1269Y of DRI-domain are substituted with A in the mutant), we found the interaction domain of Pol-β with APC located within the stretch of amino acid residues 80−120. To further identify critical residues of Pol-β that might be involved in the interaction with APC, the solvent surface accessibility of residues suspected from the yeast two-hybrid analysis to interact with APC was examined. Since the crystal structure of APC has not been solved, it is not feasible to identify probable interactions through possible docking modes. The crystal structure of a substrate complex of Pol-β indicates that it is composed of two-domains with distinct enzymatic activities necessary for SN-BER: an amino-terminal lyase domain and a carboxyl-terminal polymerase domain. Residues suspected of interacting with APC are in a stretch of amino acids 80−120 that connect these domains. From the structure of the ternary substrate complex, two regions - Set-1 (amino acid Thr79, Lys81 and Arg83) and Set-2 (amino acid Arg89, Gln90 and Asp92) - are identified that exhibit high solvent accessibility. The protein backbone of Set-1 region can be seen in the structure. Alteration of the backbone dynamics of this region is expected to affect Pol-β-dependent substrate binding and/or catalysis. Set-1 residues are displayed on a ribbon representation of a ternary substrate complex of Pol-β (pdb accession code 2FMS). The lyase and polymerase domains are colored yellowish green and blue, respectively, and the DNA backbone is red and pink. Site-directed mutagenesis studies suggested that Set-1 amino acid residues (Thr79, Lys81 and Arg83) are important for the interaction with APC.
4. Physiological significance of the APC-mediated blockage of SN- and LP-BER activities
Most mutations of the APC gene occur in the MCR region and result in the production of a truncated protein. This truncation of the protein compromises several functions of APC and contributes to chromosomal instability [78] and [79]. Whether APC-mediated blockage of SN- and LP-BER pathways has any physiological consequence in development of colorectal cancer is not yet examined. The DRI-domain of APC, which is the site of interaction with Pol-β and Fen-1, is located in the N-terminal region and is spared by mutations of MCR that result in truncation of the protein. Thus, most mutant APC proteins (those with an intact DRI-domain), as well as wild-type APC, are capable of modulating BER (Fig. 2).
Fig. 2. Schematic representation of the structure of APC.
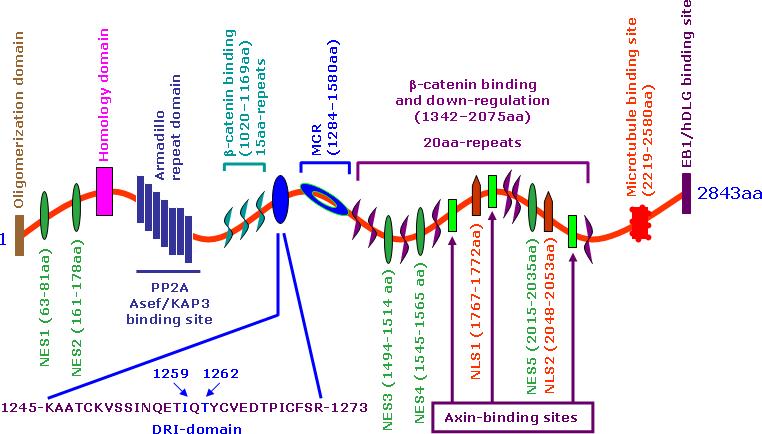
The 2843 amino acid sequence displays an armadillo domain near the N-terminus. There are two β-catenin binding domains. The first 15-amino acid repeat can bind β-catenin, but its functional significance is still obscure, while the 20-amino acid repeat can bind β-catenin with a high affinity upon phosphorylation. The DRI-domain, just upstream of MCR, is involved in the regulation of the BER pathway. Asef, APC-stimulated guanine nucleotide exchange factor; DLG, Drosophila discs large; EB1, end-binding protein 1, KAP3A, kinesin superfamily-associated protein 3A; NES, nuclear export signal; NLS, nuclear localization signal; PP2-B56α, protein phosphatase 2A B56α subunit.
Earlier studies have indicated that DNA-alkylating agents can enhance the level of APC in colorectal cancer cells. The induced level of APC thus can block SN- and LP-BER activities and affect cellular responses. Depending upon the cellular context and the extent of DNA damage, the consequence of APC/BER interaction could lead to either enhanced alkylation-induced carcinogenesis or apoptosis (Fig. 4). The former scenario is indicated by our studies of the effects of cigarette smoke condensate (CSC), a surrogate for cigarette smoke, which induces APC gene expression. We have found that the enhanced levels of APC block LP-BER and that this might contribute to the transformation of spontaneously immortalized normal breast epithelial cells [80]. On the other hand, treatment with methylmethane sulfonate (MMS) increases APC levels, blocks BER and induces apoptosis in human colon cancer cell lines and mouse embryonic fibroblast cells [66] and [81]. Collectively, the results of these studies suggest a previously unrecognized function of APC as a pivotal determinant of the fate of a cell with damaged DNA. The role of APC in carcinogenesis is paradoxical; however, its role in apoptosis supports tumor suppressor function which needs to be further ascertained in future studies.
Fig. 4.

A hypothetical model for the role of APC blockage of BER in carcinogenesis and chemotherapy.
5. Concluding remarks
The studies discussed in this review are critical for understanding the mechanisms that define whether a cell that has sustained DNA damage survives and enters into the carcinogenic cascade or whether it becomes apoptotic. The current understanding is that the type and level of DNA damage plays a critical role in defining the outcome. That is, in response to DNA damage, the cells can adequately repair the damage and survive with no apparent consequence to the organism as a whole. When a threshold limit of DNA damage is incurred, the cell tries to repair the damage; however, according to the currently accepted understanding, a few cells may escape the repair, acquire additional cell survival characteristics, and become tumorigenic. If the extent of DNA damage sustained is higher than the repair capacity, the cell becomes apoptotic. Thus, in those cells that has DNA damage above the threshold limit, the APC level increases and blocks Pol-β-directed BER leading to apoptosis (Fig. 4). In summary, since APC plays a central role in the development of colon cancer, a fuller understanding of its novel role in DNA repair mechanisms will provide valuable information for more effective prognosis and treatment of colon cancers without affecting normal colonic epithelial cells.
Acknowledgements
Our studies are an extension of the efforts of our colleagues who conduct research in the areas of APC and BER. We recognize that we have not fully acknowledged their outstanding contributions in this brief review. We extend our sincere thanks to Dr. William A. Beard (Laboratory of Structural Biology, NIEHS, National Institutes of Health, DHHS, Research Triangle Park, North California) for providing us the ribbon structure of DNA polymerase β presented in Figure 3. These studies were performed with support to Satya Narayan by NCI-NIH (CA-097031 and CA-100247) and Flight Attendants Medical Research Institute, Miami, FL. We also extend our sincere thanks to the reviewers for their suggestions which improved the quality of the manuscript.
Abbreviations
- APC
adenomatous polyposis coli
- APE 1
apurinic/apyrimidinic endonuclease 1
- BER
base excision repair
- DRI-domain
DNA repair inhibitory-domain
- dRP-lysae
5’-deoxyribose phosphate lyase
- Fen-1
flap endonuclease 1
- LP
long-patch
- MCR
mutation cluster region
- SN
single-nucleotide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement. We have no any financial and personal relationships with other people or organizations that could inapropriately influence (bias) our work.
References
- 1.Munemitsu S, Souza B, Muller O, Albert I, Rubinfeld B, Polakis P. The APC gene product associates with microtubules in vivo and promotes their assembly in vitro. Cancer Res. 1994;54:3676–3681. [PubMed] [Google Scholar]
- 2.Neufeld KL, White RL. Nuclear localizations of the adenomatous polyposis coli protein. Proc. Natl. Acad. Sci. USA. 1997;94:3034–3039. doi: 10.1073/pnas.94.7.3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henderson BR. Nuclear-cytoplasmic shuttling of APC regulates β-catenin subcellular localization and turnover. Nat. Cell Biol. 2000;2:653–660. doi: 10.1038/35023605. [DOI] [PubMed] [Google Scholar]
- 4.Neufeld KL, Nix DA, Bogerd H, Kang Y, Beckerle MC, Cullen BR, White RL. Adenomatous polyposis coli protein contains two nuclear export signals and shuttles between the nucleus and cytoplasm. Proc. Natl. Acad. Sci. USA. 2000;97:12085–12090. doi: 10.1073/pnas.220401797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosin-Arbesfeld R, Townsley F, Bienz M. The APC tumour suppressor has a nuclear export function. Nature. 2000;406:1009–1012. doi: 10.1038/35023016. [DOI] [PubMed] [Google Scholar]
- 6.Wong MH, Hermiston ML, Syder AJ, Gordon JI. Forced expression of the tumor suppressor adenomatous polyposis coli protein induces disordered cell migration in the intestinal epithelium. Proc. Natl. Acad. Sci. USA. 1996;93:9588–9593. doi: 10.1073/pnas.93.18.9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Narayan S, Roy D. Role of APC and DNA mismatch repair genes in the development of colorectal cancers. Mol. Cancer. 2003;2:41–56. doi: 10.1186/1476-4598-2-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith KJ, Levy DB, Maupin P, Pollard TD, Vogelstein B, Kinzler KW. Wild-type but not mutant APC associates with the microtubule cytoskeleton. Cancer Res. 1994;54:3672–3675. [PubMed] [Google Scholar]
- 9.Barth AIM, Pollack AL, Altschuler Y, Mostov KE, Nelson WJ. NH2-terminal deletion of β-catenin results in stable colocalization of mutant β-catenin with adenomatpous polyposis coli protein and altered MDCK cell adhesion. J. Cell. Biol. 1997;136:693–706. doi: 10.1083/jcb.136.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goss KH, Groden J. Biology of the adenomatous polyposis coli tumor suppressor. J. Clin. Oncol. 2000;18:1967–1979. doi: 10.1200/JCO.2000.18.9.1967. [DOI] [PubMed] [Google Scholar]
- 11.Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Hum. Mol. Genet. 2001;10:721–733. doi: 10.1093/hmg/10.7.721. [DOI] [PubMed] [Google Scholar]
- 12.Nakamura M, Zhou XZ, Lu KP. Critical role for the EB1 and APC interaction in the regulation of microtubule polymerization. Curr. Biol. 2000;11:1062–1067. doi: 10.1016/s0960-9822(01)00297-4. [DOI] [PubMed] [Google Scholar]
- 13.Ishidate T, Matsumine A, Toyoshima K, Akiyama T. The APC-hDLG complex negatively regulates cell cycle progression from the Go/G1 to S phase. Oncogene. 2000;19:365–372. doi: 10.1038/sj.onc.1203309. [DOI] [PubMed] [Google Scholar]
- 14.Kaplan KB, Burds AA, Swedlow JR, Bekir SSB, Sorger PK, Nathke IS. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat. Cell Biol. 2001;3:429–432. doi: 10.1038/35070123. [DOI] [PubMed] [Google Scholar]
- 15.Green RA, Wollman R, Kaplan KB. APC and EB1 function together in mitosis to regulate spindle dynamics and chromosome alignment. Mol. Biol. Cell. 2005;16:4609–4622. doi: 10.1091/mbc.E05-03-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 17.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 18.Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabilization of β-catenin by genetic defects in melanoma cell lines. Science. 1997;275:1790–1792. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- 19.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 20.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crawford HC, Fingleton BM, Rudolph-Owen LA, Goss KJ, Rubinfeld B, Polakis P, Matrisian LM. The metalloproteinase matrilysin is a target of β-catenin transactivation in intestinal tumors. Oncogene. 1999;1999;18:2883–2891. doi: 10.1038/sj.onc.1202627. [DOI] [PubMed] [Google Scholar]
- 22.Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ, Hanski C. Target genes of β-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc. Natl. Acad. Sci. USA. 1999;96:1603–1608. doi: 10.1073/pnas.96.4.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muto T, Bussey HJ, Morson BC. The evolution of cancer of the colon and rectum. Cancer. 1995;36:2251–2270. doi: 10.1002/cncr.2820360944. [DOI] [PubMed] [Google Scholar]
- 24.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 25.Bodmer WF. The somatic evolution of cancer. The Harveian Oration of 1996. J. Royal Coll. Physicians (London) 1996;31:82–89. [PMC free article] [PubMed] [Google Scholar]
- 26.Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum. Mol. Genet. 1992;1:229–233. doi: 10.1093/hmg/1.4.229. [DOI] [PubMed] [Google Scholar]
- 27.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 28.Fearnhead NS, Wilding JL, Bodmer WF. Genetics of colorectal cancer: hereditary aspects and overview of colorectal tumorigenesis. Br. Med. Bull. 2002;64:27–43. doi: 10.1093/bmb/64.1.27. [DOI] [PubMed] [Google Scholar]
- 29.Taso J, Shibata D. Further evidence that one of the earliest alterations in the colorectal carcinogenesis involves APC. Am. J. Pathol. 1994;145:531–534. [PMC free article] [PubMed] [Google Scholar]
- 30.Ficari F, Cama A, Valanzano R, Curia MC, Palmirotta R, Aceto G, Esposito DL, Crognale S, Lombardi A, Messerini L, Mariani-Costantini R, Tonelli P, Battista F. APC gene mutations and colorectal adenomatosis in familial adenomatous polyposis. Br. J. Cancer. 2000;82:348–353. doi: 10.1054/bjoc.1999.0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagase H, Miyoshi Y, Horii A, Aoki T, Ogawa M, Utsunomiya J, Baba S, Sasazuki T, Nakamura Y. Correlation between the location of germ-line mutations in the APC gene and the number of colorectal polyps in familial adenomatous polyposis patients. Cancer Res. 1992;52:4055–4057. [PubMed] [Google Scholar]
- 32.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 33.Frayling IM, Beck NE, Ilyas M, Dove-Edwin I, Goodman P, Pack K, Bell JA, Williams CB, Hodgson SV, Thomas HJ, Talbot IC, Bodmer WF, Tomlinson IP. The APC variants I1307K and E1317Q are associated with colorectal tumors, but not always with a family history. Proc. Natl. Acad. Sci. U S A. 1998;95:10722–10727. doi: 10.1073/pnas.95.18.10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamlum H, Ilyas M, Rowan A, Clark S, Johnson V, Bell J, Frayling I, Efstathiou J, Pack K, Payne S, Roylance R, Gorman P, Sheer D, Neale K, Phillips R, Talbot I, Bodmer WF, Tomlinson I. The type of somatic mutation at APC in familial adenomatous polyposis is determined by the site of the germline mutation: a new facet to Knudson's ‘two-hit’ hypothesis. Nat. Med. 1991;5:1071–1075. doi: 10.1038/12511. [DOI] [PubMed] [Google Scholar]
- 35.Smits S, Kielman MF, Breukel C, Zurcher C, Neufeld K, Jagmohan-Changur S, Hofland N, van Dijk J, White R, Edelmann W, Kucherlapati R, Khan PM, Fodde R. Apc1638T: a mouse model delineating critical domains of the adenomatous polyposis coli protein involved in tumorigenesis and development. Genes Dev. 1999;13:1309–1321. doi: 10.1101/gad.13.10.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fodde R, Brabletz T. Wnt/β-catenin signaling in cancer stemness and malignant behavior. Curr. Opin. Cell Biol. 2007;19:150–158. doi: 10.1016/j.ceb.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 37.Narayan S, Jaiswal AS. Activation of adenomatous polyposis coli (APC) gene expression by the DNA-alkylating agent N-methyl-N'-nitro-N-nitrosoguanidine requires p53. J. Biol. Chem. 1997;272:30619–30622. doi: 10.1074/jbc.272.49.30619. [DOI] [PubMed] [Google Scholar]
- 38.Jaiswal AS, Narayan S. Upstream stimulating factor-1 (USF1) and USF2 bind to and activate the promoter of the adenomatous polyposis coli (APC) tumor suppressor gene. J. Cell. Biochem. 2001;81:262–277. doi: 10.1002/1097-4644(20010501)81:2<262::aid-jcb1041>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 39.Jaiswal AS, Narayan S. p53-dependent transcriptional regulation of the APC promoter in colon cancer cells treated with DNA alkylating agents. J. Biol. Chem. 2001;276:18193–18199. doi: 10.1074/jbc.M101298200. [DOI] [PubMed] [Google Scholar]
- 40.Jaiswal AS, Balusu R, Narayan S. 7,12-Dimethylbenzanthracene-dependent transcriptional regulation of adenomatous polyposis coli (APC) gene expression in normal breast epithelial cells is mediated by GC-box binding protein Sp3. Carcinogenesis. 2006;27:252–261. doi: 10.1093/carcin/bgi225. [DOI] [PubMed] [Google Scholar]
- 41.Krokan HE, Standal R, Slupphaug G. DNA glycosylases in the base excision repair of DNA. Biochem. J. 1997;325:1–16. doi: 10.1042/bj3250001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 43.Sancar A, Sancar GB. DNA repair enzymes. Annu. Rev. Biochem. 1988;57:29–67. doi: 10.1146/annurev.bi.57.070188.000333. [DOI] [PubMed] [Google Scholar]
- 44.Sarasin AR, Hanawalt PC. Carcinogens enhance survival of UV-irradiated simian virus 40 in treated monkey kidney cells: induction of a recovery pathway? Proc. Natl. Acad. Sci. USA. 1978;75:346–350. doi: 10.1073/pnas.75.1.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Friedberg EC, Walker GC, Siede W. DNA repair and Mutagenesis. ASM Press; Washington, DC: 1995. [Google Scholar]
- 46.Holmquist GP. Endogenous lesions, S-phase-independent spontaneous mutations, and evolutionary strategies for base excision repair. Mutat. Res. 1998;400:59–68. doi: 10.1016/s0027-5107(98)00051-7. [DOI] [PubMed] [Google Scholar]
- 47.Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) EMBO J. 1997;16:3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biade S, Sobol RW, Wilson SH, Matsumoto Y. Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J. Biol. Chem. 1998;273:898–902. doi: 10.1074/jbc.273.2.898. [DOI] [PubMed] [Google Scholar]
- 49.Kane CM, Linn S. Purification and characterization of an apurinic/apyrimidinic endonuclease from HeLa cells. J. Biol. Chem. 1981;256:3405–3414. [PubMed] [Google Scholar]
- 50.Demple B, Herman T, Chen DS. Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes. Proc. Natl. Acad. Sci. USA. 1991;88:11450–11454. doi: 10.1073/pnas.88.24.11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strauss PR, Beard WA, Patterson TA, Wilson SH. Substrate binding by human apurinic/apyrimidinic endonuclease indicates a Briggs-Haldane mechanism. J. Biol. Chem. 1997;272:1302–1307. doi: 10.1074/jbc.272.2.1302. [DOI] [PubMed] [Google Scholar]
- 52.Matsumoto Y, Kim K. Excision of deoxyribose phosphate residues by DNA polymerase β during DNA repair. Science. 1995;269:699–702. doi: 10.1126/science.7624801. [DOI] [PubMed] [Google Scholar]
- 53.Kim K, Biade S, Matsumoto Y. Involvement of Flap Endonuclease 1 in Base Excision DNA Repair. J. Biol. Chem. 1998;273:8842–8848. doi: 10.1074/jbc.273.15.8842. [DOI] [PubMed] [Google Scholar]
- 54.Podlutsky AJ, Dianova II, Podust VN, Bohr VA, Dianov DL. Human DNA polymerase β initiates DNA synthesis during long-patch repair of reduced AP sites in DNA. EMBO J. 2001;20:1477–1482. doi: 10.1093/emboj/20.6.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pascucci B, Stucki M, Jónsson ZO, Dogliotti E, Hübscher U. Long patch base excision repair with purified human proteins. DNA ligase I as patch size mediator for DNA polymerases delta and epsilon. J. Biol. Chem. 1999;274:33696–33702. doi: 10.1074/jbc.274.47.33696. [DOI] [PubMed] [Google Scholar]
- 56.Bambara RA, Murante RS, Henricksen LA. Enzymes and reactions at the eukaryotic DNA replication fork. J. Biol. Chem. 1997;272:4647–4650. doi: 10.1074/jbc.272.8.4647. [DOI] [PubMed] [Google Scholar]
- 57.Lieber MR. The FEN-1 family of structure-specific nucleases in eukaryotic DNA replication, recombination and repair. Bioessays. 1997;19:233–240. doi: 10.1002/bies.950190309. [DOI] [PubMed] [Google Scholar]
- 58.Memisoglu A, Samson L. Base excision repair in yeast and mammals. Mutat. Res. 2000;451:39–51. doi: 10.1016/s0027-5107(00)00039-7. [DOI] [PubMed] [Google Scholar]
- 59.Norbury CJ, Hickson ID. Cellular responses to DNA damage. Annu. Rev. Pharmacol. Toxicol. 2001;41:367–401. doi: 10.1146/annurev.pharmtox.41.1.367. [DOI] [PubMed] [Google Scholar]
- 60.Shen B, Singh P, Liu R, Qiu J, Zheng L, Finger LD, Alas S. Multiple but dissectible functions of FEN-1 nucleases in nucleic acid processing, genome stability and diseases. Bioessays. 2005;27:717–729. doi: 10.1002/bies.20255. [DOI] [PubMed] [Google Scholar]
- 61.Bhattacharyya N, Chen HC, Comhair S, Erzurum SC, Banerjee S. Variant forms of DNA polymerase β in primary lung carcinomas. DNA Cell Biol. 1999;18:549–554. doi: 10.1089/104454999315097. [DOI] [PubMed] [Google Scholar]
- 62.Wang L, Patel U, Ghosh L, Banerjee S. DNA polymerase β mutations in human colorectal cancer. Cancer Res. 1992;52:4824–4827. [PubMed] [Google Scholar]
- 63.Srivastava DK, Husain I, Arteaga CL, Wilson SH. DNA polymerase β expression differences in selected human tumors and cell lines. Carcinogenesis. 1999;20:1049–1054. doi: 10.1093/carcin/20.6.1049. [DOI] [PubMed] [Google Scholar]
- 64.Canitrot Y, Hoffmann JS, Calsou P, Hayakawa H, Salles B, Cazaux C. Nucleotide excision repair DNA synthesis by excess DNA polymerase β: a potential source of genetic instability in cancer cells. FASEB J. 2000;14:1765–1774. doi: 10.1096/fj.99-1063com. [DOI] [PubMed] [Google Scholar]
- 65.Chapados BR, Hosfield DJ, Han S, Qiu J, Yelent B, Shen B, Tainer JA. Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell. 2004;116:39–50. doi: 10.1016/s0092-8674(03)01036-5. [DOI] [PubMed] [Google Scholar]
- 66.Narayan S, Jaiswal AS, Balusu R. Tumor suppressor APC blocks DNA polymerase β-dependent strand displacement synthesis during long patch but not short patch base excision repair and increases sensitivity to methylmethane sulfonate. J. Biol. Chem. 2005;280:6942–6949. doi: 10.1074/jbc.M409200200. [DOI] [PubMed] [Google Scholar]
- 67.Jaiswal AS, Balusu R, Armas ML, Kundu CN, Narayan S. Mechanism of adenomatous polyposis coli (APC)-mediated blockage of long-patch base excision repair. Biochemistry. 2006a;2006;45:15903–15914. doi: 10.1021/bi0607958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Balusu R, Jaiswal AS, Armas ML, Kundu CN, Bloom LB, Narayan S. Structure/function analysis of adenomatous polyposis coli (APC) interaction with DNA polymerase β and its implication in base excision repair. Biochemistry. 2007;46:13961–13974. doi: 10.1021/bi701632e. [DOI] [PubMed] [Google Scholar]
- 69.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 70.Maitra M, Gudzelak A, Li SX, Matsumoto Y, Eckert KA, Jager J, Sweasy JB. Threonine 79 is a hinge residue that governs the fidelity of DNA polymerase β by helping to position the DNA within the active site. J. Biol. Chem. 2002;277:35550–35560. doi: 10.1074/jbc.M204953200. [DOI] [PubMed] [Google Scholar]
- 71.Beard WA, Wilson SH. Structure and mechanism of DNA polymerase β. Chem. Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 72.Piersen CE, Prasad R, Wilson SH, Lloyd RS. Evidence for an imino intermediate in the DNA polymerase β deoxyribose phosphate excision reaction. J. Biol. Chem. 1996;271:17811–17815. doi: 10.1074/jbc.271.30.17811. [DOI] [PubMed] [Google Scholar]
- 73.Prasad R, Beard WA, Chyan JY, Maciejewski MW, Mullen GP, Wilson SH. Functional analysis of the amino-terminal 8-kDa domain of DNA polymerase β as revealed by site-directed mutagenesis. DNA binding and 5'-deoxyribose phosphate lyase activities. J. Biol. Chem. 1998;273:11121–11126. doi: 10.1074/jbc.273.18.11121. [DOI] [PubMed] [Google Scholar]
- 74.Deterding LJ, Prasad R, Mullen GP, Wilson SH, Tomer KB. Mapping of the 5'-2-deoxyribose-5-phosphate lyase active site in DNA polymerase β by mass spectrometry. J. Biol. Chem. 2000;275:10463–10471. doi: 10.1074/jbc.275.14.10463. [DOI] [PubMed] [Google Scholar]
- 75.Prasad R, Batra VK, Yang XP, Krahn JM, Pedersen LC, Beard WA, Wilson SH. Structural insight into the DNA polymerase β deoxyribose phosphate lyase mechanism. DNA Repair. 2005;4:1347–1357. doi: 10.1016/j.dnarep.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 76.Mizushina Y, Kamisuki S, Kasai N, Shimazaki N, Takemura M, Asahara H, Linn S, Yoshida S, Matsukage A, Koiwai O, Sugawara F, Yoshida H, Sakaguchi K. A plant phytotoxin, solanapyrone A, is an inhibitor of DNA polymerase β and lambda. J. Biol. Chem. 2002;277:630–638. doi: 10.1074/jbc.M105144200. [DOI] [PubMed] [Google Scholar]
- 77.Pelletier H, Sawaya MR. Characterization of the metal ion binding helix-hairpin-helix motifs in human DNA polymerase β by X-ray structural analysis. Biochemistry. 1996;35:12778–12787. doi: 10.1021/bi960790i. [DOI] [PubMed] [Google Scholar]
- 78.Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993;260:816–819. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- 79.Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, van Es JH, Breukel C, Wiegant J, Giles RH, Clevers H. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat. Cell Biol. 2001;3:433–438. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- 80.Kundu CN, Balusu R, Jaiswal AS, Gairola CG, Narayan S. Cigarette smoke condensate-induced level of adenomatous polyposis coli blocks long-patch base excision repair in breast epithelial cells. Oncogene. 2007;26:1428–1438. doi: 10.1038/sj.onc.1209925. [DOI] [PubMed] [Google Scholar]
- 81.Kundu CN, Balusu R, Jaiswal AS, Narayan S. Adenomatous polyposis coli-mediated hypersensitivity of mouse embryonic fibroblast cell lines to methylmethane sulfonate treatment: implication of base excision repair pathways. Carcinogenesis. 2007;28:2089–2095. doi: 10.1093/carcin/bgm125. [DOI] [PubMed] [Google Scholar]