

Abstract
The following review focuses on our current knowledge as to how the cell death regulatory machinery is activated to mediate irradiation-induced cell death. In particular, we will address recent developments related to the following questions: 1.) Which cell death regulatory genes mediate irradiation-induced cell death? 2.) What is the mechanism of irradiation–induced activation or suppression of cell death regulatory genes (proteins)? 3.) How does the condition of the cell death regulatory machinery affect the cell’s sensitivity or resistance to irradiation? Now more than ever, it seems clear that irradiation –induced apoptosis is a complex process involving all three major cell death regulatory pathways: the mitochondria pathway (Bcl-2/Apaf-1), the Iap pathway, and the death receptor pathway. Depending on the cellular context, one or multiple pathways may be activated to mediate irradiation-induced cell death. Therefore, a comprehensive understanding of these processes demands systematic strategies in contrast to traditional approaches that focused on one gene/protein. For this reason, we will also examine recent studies applying genomic (proteomic) methods in this area.
Keywords: Irradiation, Apoptosis, Bcl-2, Bax, Apaf-1, Death receptors, Fas, FasL, DNA microarray, DNA chip, Genomics, Systems biology, Transcription activation, Gene expression, Review
2. INTRODUCTION - CELL DEATH REGULATORY PATHWAYS
Cell death or apoptosis induced by irradiation can be viewed as a series of cascading events, beginning with direct cellular damage inflicted by irradiation, followed by sensing of cellular damage, signal transduction and checkpoint, activation of cell death regulatory genes and/or proteins, caspase activation and cellular destruction, and removal of apoptotic corpses. Topics related to cellular damage detection and signal transduction pathways have been meticulously covered in recent reviews (1,2). In this review, we will focus our attention on the activation of cell death regulatory genes/proteins that are directly responsible for cell death induced by irradiation.
2.1. Three pathways directly regulate caspase activation
Apoptosis is a fundamental biological process essential for normal development and tissue homeostasis (3–5). The execution of apoptosis is carried out by an unusual class of proteases, caspases (for cysteine aspartic acid-specific protease). Caspases are synthesized as inactive zymogens, which are widely expressed in both dying and living cells. Once activated, caspases will cleave structural proteins, enzyme inhibitors, etc. which in turn will lead to destruction, fragmentation and engulfing of the dying/dead cell (6). Crucial as it is, caspase activation is under the control of multiple cell death regulatory pathways, which are outlined in Figure 1. In the following paragraphs, we will refer to these pathways as the mitochondria pathway (the Bcl-2/Apaf-1 pathway), the Iap pathway, and the death receptor pathway.
Figure 1.

Simplified schematic presentation of the three major cell death regulatory pathways, which, when activated, all lead to the activation of caspases (in dashed square). Colors distinguish the organism to which the gene/protein belongs (eg. C. elegans, Drosophila, and Mammals). * Most mammalian genes have multiple names, only one is listed due to space limitation.
Cell death regulatory pathways are highly conserved across all metazoans. In fact, the crucial importance of caspases in mediating programmed cell death was first demonstrated in C. elegans. Animals mutated for Ced-3 were deficient in developmental cell death (7,8). Genetic analysis in C. elegans also delineated the regulatory pathway that controls the activation of Ced-3, namely the Ced-9/Ced-4 regulatory pathway (9). The mammalian orthologs of Ced-9 and Egl-1 are the Bcl-2 family proteins which regulate the integrity of mitochondria (10,11) (Figure 1). Pro-apoptotic Bcl-2 family members can cause cytochrome c release from mitochondria and subsequent activation of Apaf-1, the mammalian Ced-4 ortholog (12). Activated Apaf-1 will cause the cleavage and activation of Caspase-9 and in turn, the activation of effector caspases (Figure 1).
Iaps (Inhibitor of apoptosis protein) were first identified in insect viruses as potent inhibitors of apoptosis (13). Subsequently, other orthologs were identified in flies as well as mammals (14). Iaps inhibit the enzymatic activity of caspases through direct interaction, mediated by its N-terminal BIR (Baculovirus Iap Repeat) motifs (13). Iaps may also negatively regulate caspase activity by targeting them for proteosome-mediated degradation, a function carried out by the C-terminal RING (Really Interesting New Gene) domain present in some but not all Iaps (15). Iaps may be considered as a universal “Brake” to apoptosis. The inhibitory function of Iaps can be released by Iap antagonists. Antagonists of Iaps were first identified through genetic studies in Drosophila, namely reaper, hid, grim and more recently sickle (16–21). Interestingly, these four Drosophila IAP antagonist genes are located in proximity at the 75C1-2 chromosome region. The functional activity of these antagonist proteins is mediated by a tetra-peptide motif located in their N-terminal. Binding of this motif to IAP proteins relinquishes the inhibitory effect of IAP on caspases (20,22). This tetra-peptide motif, A-(V/T/I)-(P/A)-(F/Y/I/V), as well as the corresponding functional mechanism is conserved in mammalian systems (23). To date, at least two mammalian proteins have been identified that possess this tetra-peptide motif and function as Iap antagonists. However, unlike their fly counterparts, the tetra-peptide domain of Smac (Second mitochondria derived activator of caspase)/Diablo (24,25) and Omi1(HtrA2) (26–28) is not located on the N-terminal of the full length protein, it is instead exposed at the N-terminal only after post-translation processing.
The death receptors are a subclass of growth receptors that have an intracellular “death domain” signature motif (29–32). The binding of corresponding ligands will cause the oligomerization of the death receptors, which will bring together caspase 8 through adaptor proteins such as Fadd (Fas associated protein with death domain) and/or Tradd (TNF receptor associated protein with death domain) (33,34). The proximity of dormant (full length) caspase 8 molecules will cause auto-proteolytic activation and subsequent activation of downstream caspases (35).
Interestingly, it seems that all of the three major cell death regulatory pathways are conserved in Drosophila. A systematic search for cell death regulatory protein motifs (such as BH and death domains) in Drosophila genomic and EST sequences revealed that the fly genome has potential orthologs for all major cell death regulatory protein families (36). Most of these potential structural orthologs have been proven to be functionally conserved as well, including the Drosophila orthologs for Apaf-1/Ced-4 (37–39), Bcl-2 (40,41), Fadd/Tradd (42), RIP (43), Survivin (44), etc.
2.2. The interrelated cell death regulatory networks
The basic regulatory pathways are well conserved during evolution. However, the cell death regulatory machinery in mammals is conceivably much more complex than that in invertebrates. Structurally, the increased complexity can be largely attributed to the increased number of participants in each step of the pathway. For example, while only two Bcl-2 family members have been identified in C. elegans and Drosophila (genome projects for both organisms are finished), at least 19 Bcl-2 family members have been characterized in the human genome. Another indicator of increased complexity appears to be novel regulatory mechanisms of cell death regulatory genes/proteins. For example, in C. elegans, Ced-4 induces Ced-3 activation independent of cytochrome c. In contrast, cytochrome c interaction is very important for the function of its orthologs in humans (Apaf-1) and flies (Hac-1/Dark/Dapaf-1). This interaction is mediated through the WD repeat domain, which is present in both Apaf-1 and Hac-1, but not in Ced-4 (37–39).
Although assigning cell death regulatory genes/proteins to different pathways helps us conceptualize the global picture of cell death regulation, it should be stressed that the interrelations of these proteins are not linear in nature. Rather, complex interactions exist between regulatory pathways and the outcome of the life/death decision appears to be the result of an integrative process. For example, although caspase 8 is a regulatory caspase in the Fas/TNFR pathway, one of its substrates, Bid, is a pro-apoptotic component of the Bcl-2 pathway (45). The cleavage of Bid by caspase 8 releases the pro-apoptotic activity of Bid, which will counter-act the anti-apoptotic activity of Bcl-2 and induce mitochondrial damage and cytochrome c release (45,46). Similarly, in mammalian systems, the Iap antagonist Smac is released from mitochondria in response to apoptotic stimuli, a process that is regulated by the Bcl-2 family proteins (47). Thus Smac is part of the mitochondria pathway as well as an Iap antagonist.
Genetic model systems such as Drosophila offer considerable advantage for deciphering the interrelationship among pathways. For example, genetic analysis of the interrelationship between Iap antagonist Reaper and Hac-1 (Homologue of apaf-1 and ced-4; also known Dark and Dapaf-1) revealed that the function of these two pro-apoptotic genes converge at the same caspase (37). The integration of signals from their corresponding pathways is best explained by the analogy of “gas” and “brake” (Figure 2). Both Iaps and Hac-1 are expressed in almost all cells. While Hac-1 acts as “gas” for caspase activation, Iaps are the “brake” that inhibits precocious caspase activation. The stereotypic pattern of cell death observed during the development of Drosophila embryos can be regulated through either an increase of “gas” or removal of the “brake” by Iap antagonist, or both. Indeed, while Iap antagonists such as Reaper and Grim are specifically expressed in cells designated to die in the ventral nerve cord (16,18), Hac-1 expression in the pro-cephalic region is necessary for proper cell death in the brain (37). The functional interrelationship observed between Reaper and Hac-1 seems to be well conserved between mammalian Iap antagonists and Apaf-1 (48). Apaf-1 induced caspase activation is negatively regulated by mammalian Iaps, an inhibition that can be removed by antagonist Smac and HtrA2 (24,26–28,49,50).
Figure 2.
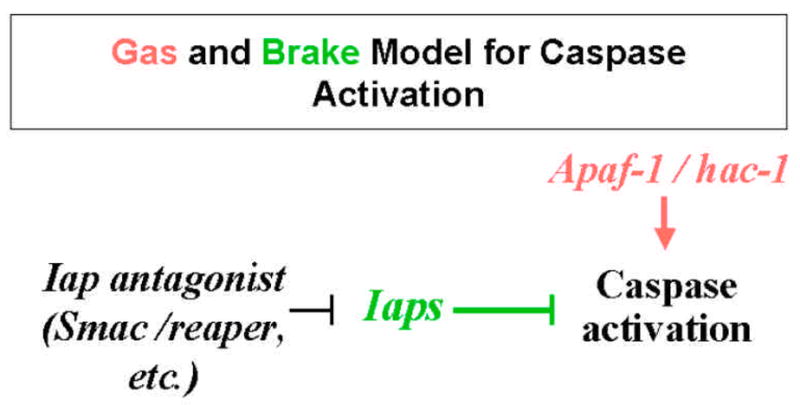
The integration of cell death regulatory control by Apaf-1/Hac-1 and Iap antagonists depicted in the “Gas” and “Brake” model. Apaf-1-like molecules function as “gas” for caspase activation, which is checked by the “brake” Iaps.
It should also be noted that while this review focuses on genes and pathways directly involved in controlling caspase activation (and thus apoptosis), components of these pathways are subject to regulation and/or modification by other cellular systems. These controlling mechanisms are essential for proper cell death regulation, as the decision to die or live is, and has to be, based on the input of multiple factors. For example, the sensitivity to irradiation-induced cell death is affected by the availability of growth factors. Because of that, there is a large (and seemingly ever increasing) number of cellular components that have been found capable of affecting the cell death process. However, these factors exert their influence via members in the core cell death regulatory pathways. In essentially all examined cases, tumor cell apoptosis induced by irradiation was accompanied by caspase activation (51). As we will discuss in the following section, activation of the core cell death machinery is the determinative step of irradiation induced cell death.
3. CELL DEATH REGULATORY GENES INVOLVED IN IRRADIATION -INDUCED APOPTOSIS
Irradiation may induce a variety of cellular damage and depending on specific cellular context, may elicit different signal transduction mechanisms that induce cell death (1,2). However, the final step of caspase activation and apoptosis in this process is always mediated through genes and proteins of the core cell death regulatory pathways that directly control the activation of caspases. In other words, the balance between the forces promoting and inhibiting caspase activation in a live cell has to be modified to shift the balance of life and death. This change in the activity of cell death regulatory proteins in response to irradiation can be mediated via transcriptional activation/suppression (52) (53), RNA stability (54), translational regulation (55), post-translational modification (56,57), protein stability (58) and protein subcellular localization (59).
Although it is true that, in experimental settings, cell death can eventually occur even when protein synthesis is blocked, this should not lead us to conclude that transcriptional activation is not a factor of utmost importance for irradiation-induced cell death. Many cell death regulatory genes are transcriptionally regulated in response to irradiation (52,60,61). The fact that P53 and other transcription factors play an important role in mediating apoptosis and determining tissue sensitivity to irradiation strongly argues that transcriptional regulation is a very important mechanism in vivo in mediating irradiation induced cell death. Indeed, in an in vivo system, cycloheximide treatment has been found to negate or significantly suppress irradiation–induced cell death in the testis and kidneys of neonate rats (62).
Another necessary caution that should be considered when identifying genes mediating irradiation induced cell death is to discern causal involvement v.s. consequential involvement. For example, Bid is truncated after irradiation in Jurkat cells. However, rather than causing the initial caspase activation, truncated Bid may be a result of caspase activation and subsequently acts as an augmentation agent in a reinforcing feedback loop (63,64).
3.1. The mitochondria pathway
3.1.1. Bcl-2 family
There are three subclasses within this family, which are distinct in protein motif composition and their corresponding role in regulating mitochondrial integrity and cell death (11). The first class are the “multi-domain” pro-apoptotic proteins, such as Bax and Bak, which all contain BH (Bcl-2 Homologous)3, BH2 and BH1 signature motifs. When activated, presumably through oligomerization, they cause cyctochrome c release and caspase activation. The second class are the anti-apoptotic family members, which all have a BH4 domain in addition to BH1-3 domains. Through forming heterodimers with the first class members, they inhibit the pro-apoptotic activity and stabilize the mitochondria membrane integrity. This inhibition, however, can be relinquished by the third class of the family, a group of pro-apoptotic proteins that only have the BH3 domain (65,66).
Bcl-2 family members have long been implicated in irradiation-induced cell death. Bcl-2 and Bax were among the first group of cell death regulatory genes identified as potential mediators of irradiation–induced cell death. Bax is transcriptionally induced by irradiation and is often accompanied by a suppression of Bcl-2 expression (52,53). The suppression of Bcl-2 and induction of Bax appears to be dependent on P53 function (67). Consensus P53 binding elements are present in the promoter region of the Bax gene, which are required for its P53 responsiveness (68).
In addition to Bcl-2 and Bax, BH3-only family members are also involved in irradiation induced cell death. For example, Bid is induced by irradiation in human T-lineage derived cells (69). Noxa, another member in this class, is induced by irradiation in a P53-dependent manner to cause cell death (70). It is worth noting that in C. elegans the expression of the BH3-only protein Egl-1, is also transcriptionally regulated (71).
The activity of the Bcl-2 family proteins is also subject to post-transcriptional regulation. The conserved A and U rich elements of the Bcl-2 mRNA regulate the stability of Bcl-2 RNA (54). More importantly, phosphorylation of Bcl-2 has significant impact on its anti-apoptotic activity, although the outcome of the impact may vary in different cellular contexts (56,57,72). Stress kinases such as JNK are activated in response to irradiation (73). The status of the JNK pathway has significant impact on irradiation-induced apoptosis (74). The finding that Bcl-2 is a direct substrate of JNK1 suggests that the stress-signaling pathway could, at least in part, exert its effect on cell death regulation through post-translational modification (75). The pro-apoptotic activity of Bax was reduced after phosphorylation by the P21-activated kinase (76). Addition of growth factor could induce phosphorylation of BAD and the dismissal of its pro-apoptotic activity (77). Conversely, the calcium-activated protein phosphatase calcineurin could undo this effect on Bad and induce cell death (78). These post-translational modifications may account for the influence of stress and growth factor signaling pathways on irradiation induced cell death (74,79). Finally, regulation of protein stability may also be involved. After UV irradiation, Bcl-2 protein is degraded by the proteasome (80). However, it is not clear whether the observed degradation is the cause of caspase activation or rather the result of the apoptotic destruction process.
The relative levels of Bcl-2 family members strongly affects the sensitivity of cells to irradiation induced cell death. Irradiation-induced cell death was significantly increased in mice mutated for Bcl-w, Bcl-2, and Bcl-XL (81,82). Mice mutated for Bax alone showed normal levels of cell death after irradiation (82). However, this is apparently due to the overlapping function of Bax and Bak, as MEFs derived from mice null for both Bax and Bak were highly resistant to irradiation induced cell death (65,66). Several studies have suggested that the ratio between Bcl-2 and Bax levels in cancer cells is associated with their sensitivity to irradiation (83). It is important to mention that the role of Bcl-2 family members in irradiation induced cell death appears to be conserved. Over-expression of the Drosophila ortholog of Bax/Bok, dBorg-1, in the eye disc also sensitizes the retinal cells to UV–induced cell death (41).
3.1.2. Ced-4/apaf-1/hac-1 family
Although the mammalian and Drosophila orthologs of Ced-4 were identified only recently (12,37–39), it did not take long to find that proteins in this family were also involved in irradiation induced cell death. Seemingly contradicting the fact that activation of Apaf-1 requires cytochrome c released from mitochondria (59), both Apaf-1 and its Drosophila counterpart, Hac-1 are regulated transcriptionally during embryonic development. Both are significantly up-regulated in the procephalic regions prior to the onset of massive cell death in the nervous system (37,84). Transcriptional regulation of Apaf-1 is apparently important for developmental cell death. Down-regulation of apaf-1 in the procephalic region leads to supernumerary neuronal cells in the fog (forebrain over-growth) mice (85). In a strikingly analogous manner, the procephalic expression of Hac-1 in Drosophila embryos is also required for neuronal cell death and brain development (37–39). In Drosophila embryos, Hac-1 is also up-regulated in response to UV irradiation in early stage embryos. This transcriptional activation is required for UV–induced cell death in embryos (86). Although currently there is no report of irradiation-induced transcriptional activation of Apaf-1 in mammalian systems, it certainly remains a possibility. Apaf-1 is a direct transcriptional target of E2F and P53 and is implicated in E2F induced cell death (87,88). It must be stressed that the induction of cell death regulatory genes by irradiation is highly dependent on tissue characteristics and cell differentiation status. In Drosophila, Hac-1 is only induced by UV irradiation during early embryogenesis. During middle stage embryogenesis, only Reaper is activated by UV to mediate the cell death response (86).
Sensitivity to irradiation -induced apoptosis is affected by the cellular levels of Apaf-1. Apaf-1 knock-out mice are resistant to UV–induced cell death (84), while over expression of Apaf-1 sensitized glioma cells to irradiation-induced cell death (89). Similarly, UV -induced cell death in early stage embryos was mostly abolished in hac-1 mutants (86).
3.2. The Iap pathway
3.2.1. the Iaps
Just as caspases are universally expressed, transcription of Iaps has been detected in essentially all tissues. Surprisingly, the expression of Ciap1 in several human cancer cell lines was significantly altered in response to ionizing irradiation (60). In addition, the expression of Xiap can be regulated at the translation step by irradiation. In the 5′UTR of the Xiap mRNA, there is an IRES (Internal Ribosome Entry Sequence), which could mediate mRNA cap-independent translation under stress conditions. Low doses of gamma irradiation increased Xiap expression through IRES-mediated translation mechanism, which conferred resistance to irradiation induced cytotoxicity (55).
The level of cellular Iaps appears to be a very important factor in determining the apoptotic response to irradiation. While gain-of-function Iaps inhibit irradiation-induced cell death (90), loss-of-function or down regulation of Iaps sensitizes the cell to irradiation induced cell death (91).
3.2.2. The Iap antagonists
Being the molecular “brake” for caspase activation, Iaps play an important inhibitory role in regulating apoptosis. In order for cell death to occur, this “brake” needs to be released by Iap antagonists. In Drosophila, the antagonists are regulated transcriptionally during development and are only expressed in cells that are designated to die (16,18–21). The only exception is Hid, which is also regulated by post-translational modification (92). Within 10–20 minutes of X-ray irradiation, mRNA of reaper, hid and sickle are up-regulated (16,21, unpublished observations). The H99 mutant embryos, which lack the reaper, hid and grim genes, are highly resistant to X-ray -induced cell death (16). Irradiation-induced activation of the reaper gene is mediated at least in part by direct transcriptional activation though Drosophila ortholog of P53 (93).
Mammalian functional orthologs of Drosophila Iap antagonists were identified recently. Two reported antagonists, Smac (24,94) and HtrA2/omi (26–28) both have the tetra-peptide motif shared by Drosophila Iap antagonists. However, unlike the Drosophila proteins, the tetra-peptide Iap-binding motif in Smac and HtrA2 is only revealed at the N-terminal after post-translational processing. The regulation of their activity in response to apoptosis-inducing stimuli seems to occur through translocation instead of transcription. In live cells, Smac is located in the mitochondria. HtrA2 is membrane bounded with a significant portion embedded in the mitochondria. Upon UV–irradiation both Smac and HtrA2 are released into the cytosol to act on the Iaps (24,26–28). The level of these Iap antagonists in mammalian cells apparently contributes to the sensitivity to irradiation, a high level of HtrA2 sensitizes the cell to irradiation induced cell death while lower levels convey resistance (26,27). It remains to be determined if there exists transcriptionally regulated mammalian Iap antagonists.
3.3. The death receptor pathway
The expression of both the death receptors and their ligands can be induced/increased by irradiation. Fas (CD95/Apo-1) was induced by UV irradiation in breast carcinoma cells (95) as well as in the spleen (61,96). Fas ligand (FasL) was induced by UV irradition in lymphoma cells (97). The induction of Fas may depend on P53, since it is absent in several cancer cell lines with mutated P53 (98) or cell lines derived from P53 −/− mice (99). However, it is clear that UV irradiation, especially UV-B could also activate a membrane-initiated pathway which leads to the activation of Map kinases, especially the so called stress-activated kinases, JNK and P38 (73,100,101). One transcription factor of this pathway, NF-kappa B, could also activate the transcription of Fas (102,103). Besides transcriptional activation, UV may directly activate the Fas pathway by inducing clustering of FAS independently of FasL (104).
In addition to Fas and FasL, TRAIL (TNF-related apoptosis inducing ligand) and DR5 (a death receptor that binds to TRAIL) were also involved in irradiation induced cell death. Transcription of Trail was activated after irradiation in several T lineage derived cells, including Jurkat and MOLT-4 (69). Treatment with TRAIL sensitized resistant lymphoma cells to irradiation –induced cell death (105). mRNA levels of Dr5 in the thymus, spleen and small intestines was increased almost 30 fold after gamma irradiation of wild type mice (61). However, the induction is absent in tissues from P53 null mouse, suggesting it is mediated through P53 transcriptional activation (61).
4. THE COMPLEXITY OF IRRADIATION-INDUCED CELL DEATH AND SYSTEMATIC APPROACHES
4.1. Overlapping protein functions and compensatory pathways
As reviewed above, most components of the three major cell death regulatory pathways have been implicated in irradiation-induced cell death (Table 1). Such a multigenic system poses a great challenge for traditional genetic and molecular biology approaches which address questions such as “Which gene is required for this process?”. For example, both Bax and Bak can promote cytochrome c release from mitochondria independently of each other (65,66,108). Thus only when both Bax and Bak are mutated, is UV-induced cell death in MEF cells blocked (65,66). Functional overlap also exists in other steps of cell death regulation, such as the death receptors and the Iap antagonists. Indeed, when the cell death machineries were compared among worm, fly, and human, it was apparent that the increase in protein family number and thus partial redundancy and increased system complexity is the favored trend during the evolution of complex organisms (109).
Table 1.
Cell death regulatory genes activated or suppressed after irradiation
| Mechanism/Pathway | Transcription | Post- transcription | Protein modification | Translocation |
|---|---|---|---|---|
| Mitochondria | Bcl-2, Bcl-XL, Bcl-W, Bax, Bak, Bid, Noxa, hac-1, etc. | Bid, Bcl-2, | ||
| Iap/antagonist | Ciap1, reaper, hid, sickle, etc. | Xiap | Smac, HtrA2 | |
| Death receptor | Fas, FasL, Trail, DR5, DcR2, DcR3, Tradd, etc. |
List of core cell death regulatory genes/proteins that have been reported as being activated or suppressed in response to irradiation. Drosophila genes are italicized.
Looking beyond individual steps, compensatory pathways may exist to mediate irradiation -induced cell death. First, different pathways in the signal transduction circuit could mediate irradiation-induced responses, perhaps depending on the nature of irradiation as well as the cellular context. For example, UV irradiation-induced cell death can be mediated via membrane –initiated or DNA damage–initiated signal transduction pathways (110). Reflecting the existence of alternative or compensatory pathways, there exists much conflicting evidence on whether P53 is required for irradiation-induced cell death (111–114).
Furthermore, even within the core cell death regulatory machinery that directly regulates caspase activation, alternative and compensatory pathways may exist under certain conditions. For instance, Fas activation is observed after irradiation in some cell lines in a P53 dependent manner, however Fas is not required for p53-mediated cell death (115,116). Evidently these results strongly indicate the existence of overlapping or alternative pathways. Compensatory pathways were also observed when irradiation induced cell death was studied in caspase knock-out mice. While caspase-3 is activated in wild type hepatocytes to mediate irradiation induced apoptosis, in caspase-3 −/− hepatocytes, caspase 6 and 7 were activated to induce cell death (117).
4.2. Tissue and cellular status specificity
The many conflicting evidence in the vast amount of literature on irradiation-induced cell death suggests that the mechanism of irradiation-induced apoptosis may vary according to tissue types and cellular status. Indeed it has long been observed in clinical radiation therapy that the sensitivity to irradiation is different among tissues (118). However, the molecular basis of tissue-specific sensitivity to irradiation is far from clear. Bouvard et al. reported that when whole-body irradiation was applied to 8 week old mice, Fas is induced in the spleen but not in other tissues such as the heart and liver (96). Burns et al. confirmed that while Fas transcription was up-regulated after irradiation in the spleen, it was not activated in the thymus and the small intestines, although apoptosis was induced in all three types of tissues (61). The evidence indicated that the molecular response is different not only between sensitive and resistant tissues, but also among sensitive tissues.
Using a DNA micro-array containing probes for approximately 1,200 human genes (~ 4% of the genome), Amundson et al. identified 48 genes whose expression was significantly changed after ionizing irradiation in the myeloid cell line ML-1 (60). Among the 48 genes identified were the cell death regulators Fas, Ciap1, Bak, and Bcl-XL. Based on the results obtained from the ML-1 cells, Amundson et al. further tested the expression of 12 “irradiation responsive” genes in a panel of 12 cell lines of different tissue origin and P53 status. They found that the response of these genes to ionizing irradiation or UV irradiation varied dramatically among different cell lines. For example, the same dosage of irradiation induced Ciap in some cell lines while suppressed it in others (60). There was also no obvious correlation between the P53 status in these cell lines and their varied response to irradiation.
Even for cell lines derived from the same tissue origin, the response to irradiation may differ. For example, Gong et al. reported that although radiation activated DR5 in MOLT-4 cells, it failed to do so in other Human T–lineage derived cells (69).
Differentiation status is another important factor in determining apoptotic response to irradiation. Differentiated keratinocytes are sensitive to UV-induced apoptosis, which is dependent on P53 function. In contrast, undifferentiated keratinocytes are relatively resistant to UV-induced cell death. The apoptosis induced by UV in the undifferentiated keratinocyte is independent of P53 function (119). This distinction is important for our understanding of carcinogenesis because non-melanoma skin cancer arises from the undifferentiated basal keratinocytes.
When measured using the Drosophila GeneChip (Affymetrix Inc.) with 13,000 probe sets (covering essentially the whole genome), the genomic responses to UV and x-ray irradiation are dramatically different among undifferentiated embryos, differentiating embryos and post-mitotic embryos (Zhou, et al., unpublished observation). While UVC irradiation could induce apoptosis in both undifferentiated and differentiating embryos, the mechanism is different. In early stage undifferentiated embryos, Hac-1 was induced by UV and this induction is required for UV-induced apoptosis. In middle stage differentiating embryos, however, Iap antagonist Reaper (but not Hac-1) was induced by UV to mediate cell death (86).
Overall, these findings suggest that the molecular mechanism activated by irradiation to induce cell death not only depends on the nature of the irradiation source, but also on the specific cellular context. While in some cell types, such as the MEF, one pathway is essential and required for irradiation induced cell death (65), in others alternative and compensatory pathways may be used to convey radiation sensitivity.
5. CONCLUSION AND PERSPECTIVES
Comprehensive understanding of the molecular mechanism mediating irradiation-induced apoptosis holds great promise for improved prognosis and treatment of many diseases. Although much information has been accumulated from research focusing on the role of a particular gene on irradiation-induced cell death, we are still far from a thorough understanding of this process. In addition to the complexity of cell death regulatory mechanisms, tissue type and cellular context differences also play a crucial role in determining the molecular mechanisms mediating irradiation–induced apoptosis. Reflecting the complexity of this process, single gene oriented approaches in predicting clinical radiosensitivity have been largely unsuccessful (51). Enlightened by accumulated experience and knowledge in this field and the availability of extensive genome information, we have just started to sketch and comprehend the complexity of this process. To make substantial progress in our understanding of radio-sensitivity and resistance, several basic questions remain to be addressed in depth: How different are tissue-specific responses to irradiation? What cellular factors determine the apoptosis regulatory mechanism activated by irradiation? While it is clear that some proteins play a crucial role in mediating irradiation-induced cell death under specific circumstances, the search for one gene or pathway that universally mediates irradiation induced cell death should be abandoned. Appreciating the intricate nature of this process, many problems should be approached from a systems biology point of view (120). That is, we should systematically monitor all involved components and decipher the interactions and interrelationships that account for the function as well as plasticity of this cellular process.
Acknowledgments
The authors are very grateful to comments and suggestions provided by Dr. John R. Nambu and Barbara A. Schreader. This work was partially supported by grants to L. Zhou from the College of Medicine, University of Florida.
References
- 1.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–9. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 2.Rich T, Allen RL, Wyllie AH. Defying death after DNA damage. Nature. 2000;407:777–83. doi: 10.1038/35037717. [DOI] [PubMed] [Google Scholar]
- 3.Vaux DL, Korsmeyer SJ. Cell Death in Development. Cell. 1999;96:245–254. doi: 10.1016/s0092-8674(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 4.Horvitz HR. Genetic control of programmed cell death in the nematode Caenorhabditis elegans. Cancer Res. 1999;59:1701s–1706s. [PubMed] [Google Scholar]
- 5.Bergmann A, Agapite J, Steller H. Mechanisms and control of programmed cell death in invertebrates. Oncogene. 1998;17:3215–3223. doi: 10.1038/sj.onc.1202586. [DOI] [PubMed] [Google Scholar]
- 6.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–90. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 7.Yuan J, Horvitz HR. The Caenorhabditis elegans Genes ced-3 and ced-4 Act Cell Autonomously to Cause Programmed Cell Death. Developmental Biol. 1990;138:33–41. doi: 10.1016/0012-1606(90)90174-h. [DOI] [PubMed] [Google Scholar]
- 8.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–52. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 9.Hengartner MO, Horvitz HR. Programmed cell death in Caenorhabditis elegans. Curr Opin Genet Dev. 1994;4:581–6. doi: 10.1016/0959-437x(94)90076-f. [DOI] [PubMed] [Google Scholar]
- 10.Chao DT, Korsmeyer SJ. BCL-2 family: regulators of cell death. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- 11.Adams JM, Cory S. Life-or-death decisions by the Bcl-2 protein family. Trends Biochem Sci. 2001;26:61–6. doi: 10.1016/s0968-0004(00)01740-0. [DOI] [PubMed] [Google Scholar]
- 12.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–13. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 13.Miller LK. An exegesis of IAPs: salvation and surprises from BIR motifs. Trends Cell Biol. 1999;9:323–8. doi: 10.1016/s0962-8924(99)01609-8. [DOI] [PubMed] [Google Scholar]
- 14.Hay BA. Understanding IAP function and regulation: a view from Drosophila. Cell Death Differ. 2000;7:1045–56. doi: 10.1038/sj.cdd.4400765. [DOI] [PubMed] [Google Scholar]
- 15.Huang H, Joazeiro CA, Bonfoco E, Kamada S, Leverson JD, Hunter T. The inhibitor of apoptosis, cIAP2, functions as a ubiquitin-protein ligase and promotes in vitro monoubiquitination of caspases 3 and 7. J Biol Chem. 2000;275:26661–4. doi: 10.1074/jbc.C000199200. [DOI] [PubMed] [Google Scholar]
- 16.White K, Grether ME, Abrams JM, Young L, Farrell K, Steller H. Genetic control of programmed cell death in Drosophila. Science. 1994;264:677–83. doi: 10.1126/science.8171319. [DOI] [PubMed] [Google Scholar]
- 17.Grether ME, Abrams JM, Agapite J, White K, Steller H. The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes & Dev. 1995;9:1694–1708. doi: 10.1101/gad.9.14.1694. [DOI] [PubMed] [Google Scholar]
- 18.Chen P, Nordstrom W, Gish B, Abrams JM. grim, a novel cell death gene in Drosophila. Genes Dev. 1996;10:1773–1782. doi: 10.1101/gad.10.14.1773. [DOI] [PubMed] [Google Scholar]
- 19.Wing JP, Karres JS, Ogdahl JL, Zhou L, Schwartz LM, Nambu JR. Drosophila sickle Is a Novel grim-reaper Cell Death Activator. Curr Biol. 2002;12:131–5. doi: 10.1016/s0960-9822(01)00664-9. [DOI] [PubMed] [Google Scholar]
- 20.Srinivasula SM, Datta P, Kobayashi M, Wu JW, Fujioka M, Hegde R, Zhang Z, Mukattash R, Fernandes-Alnemri T, Shi Y, Jaynes JB, Alnemri ES. sickle, a Novel Drosophila Death Gene in the reaper/hid/grim Region, Encodes an IAP-Inhibitory Protein. Curr Biol. 2002;12:125–30. doi: 10.1016/s0960-9822(01)00657-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christich A, Kauppila S, Chen P, Sogame N, Ho SI, Abrams JM. The Damage-Responsive Drosophila Gene sickle Encodes a Novel IAP Binding Protein Similar to but Distinct from reaper, grim, and hid. Curr Biol. 2002;12:137–40. doi: 10.1016/s0960-9822(01)00658-3. [DOI] [PubMed] [Google Scholar]
- 22.Wu JW, Cocina AE, Chai J, Hay BA, Shi Y. Structural analysis of a functional DIAP1 fragment bound to grim and hid peptides. Mol Cell. 2001;8:95–104. doi: 10.1016/s1097-2765(01)00282-9. [DOI] [PubMed] [Google Scholar]
- 23.Harvey A, AP B, LK M. Doom, a product of the Drosophila mod(mdg4) gene, induces apoptosis and binds to baculovirus inhibitor-of-apoptosis proteins. Mol Cell Biol. 1997;17(5):2835–2843. doi: 10.1128/mcb.17.5.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 25.Chai J, Du C, Wu JW, Kyin S, Wang X, Shi Y. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature. 2000;406:855–62. doi: 10.1038/35022514. [DOI] [PubMed] [Google Scholar]
- 26.Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, Savopoulos J, Gray CW, Creasy CL, Dingwall C, Downward J. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a Reaper-like motif. J Biol Chem. 2001;15:15. doi: 10.1074/jbc.M109784200. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell. 2001;8:613–21. doi: 10.1016/s1097-2765(01)00341-0. [DOI] [PubMed] [Google Scholar]
- 28.Verhagen AM, Silke J, Ekert PG, Pakusch M, Kaufmann H, Connolly LM, Day CL, Tikoo A, Burke R, Wrobel C, Moritz RL, Simpson RJ, Vaux DL. HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J Biol Chem. 2002;277:445–54. doi: 10.1074/jbc.M109891200. [DOI] [PubMed] [Google Scholar]
- 29.Itoh N, Nagata S. A novel protein domain required for apoptosis. Mutational analysis of human Fas antigen. J Biol Chem. 1993;268:10932–7. [PubMed] [Google Scholar]
- 30.Daniel PT, Krammer PH. Activation induces sensitivity toward APO-1 (CD95)-mediated apoptosis in human B cells. J Immunol. 1994;152:5624–32. [PubMed] [Google Scholar]
- 31.Schmitz I, Kirchhoff S, Krammer PH. Regulation of death receptor-mediated apoptosis pathways. Int J Biochem Cell Biol. 2000;32:1123–36. doi: 10.1016/s1357-2725(00)00048-0. [DOI] [PubMed] [Google Scholar]
- 32.Nagata S. Fas ligand-induced apoptosis. Annu Rev Genet. 1999;33:29–55. doi: 10.1146/annurev.genet.33.1.29. [DOI] [PubMed] [Google Scholar]
- 33.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 34.Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–12. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 35.Yang X, Chang HY, Baltimore D. Autoproteolytic activation of pro-caspases by oligomerization. Mol Cell. 1998;1:319–25. doi: 10.1016/s1097-2765(00)80032-5. [DOI] [PubMed] [Google Scholar]
- 36.Zhou L, Song Z, Tittle J, Marinescu VD, Steller H. Piecing up cell death machinary in Drosophila. Cold Spring Harbor Meeting on Programmed Cell Death. 1999 [Google Scholar]
- 37.Zhou L, Song Z, Tittel J, Steller H. HAC-1, a Drosophila homolog of APAF-1 and CED-4 functions in developmental and radiation-induced apoptosis. Mol Cell. 1999;4:745–55. doi: 10.1016/s1097-2765(00)80385-8. [DOI] [PubMed] [Google Scholar]
- 38.Kanuka H, Sawamoto K, Inohara N, Matsuno K, Okano H, Miura M. Control of the cell death pathway by Dapaf-1, a Drosophila Apaf-1/CED-4- related caspase activator. Mol Cell. 1999;4:757–69. doi: 10.1016/s1097-2765(00)80386-x. [DOI] [PubMed] [Google Scholar]
- 39.Rodriguez A, Oliver H, Zou H, Chen P, Wang X, Abrams JM. Dark is a Drosophila homologue of Apaf-1/CED-4 and functions in an evolutionarily conserved death pathway. Nat Cell Biol. 1999;1:272–9. doi: 10.1038/12984. [DOI] [PubMed] [Google Scholar]
- 40.Igaki T, Kanuka H, Inohara N, Sawamoto K, Nunez G, Okano H, Miura M. Drob-1, a Drosophila member of the Bcl-2/CED-9 family that promotes cell death. Proc Natl Acad Sci U S A. 2000;97:662–7. doi: 10.1073/pnas.97.2.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brachmann CB, Jassim OW, Wachsmuth BD, Cagan RL. The Drosophila bcl-2 family member dBorg-1 functions in the apoptotic response to UV-irradiation. Curr Biol. 2000;10:547–50. doi: 10.1016/s0960-9822(00)00474-7. [DOI] [PubMed] [Google Scholar]
- 42.Hu S, Yang X. dFADD, a novel death domain-containing adapter protein for the Drosophila caspase DREDD. J Biol Chem. 2000;275:30761–4. doi: 10.1074/jbc.C000341200. [DOI] [PubMed] [Google Scholar]
- 43.Georgel P, Naitza S, Kappler C, Ferrandon D, Zachary D, Swimmer C, Kopczynski C, Duyk G, Reichhart JM, Hoffmann JA. Drosophila immune deficiency (IMD) is a death domain protein that activates antibacterial defense and can promote apoptosis. Dev Cell. 2001;1:503–14. doi: 10.1016/s1534-5807(01)00059-4. [DOI] [PubMed] [Google Scholar]
- 44.Jones G, Jones D, Zhou L, Steller H, Chu Y. Deterin, a new inhibitor of apoptosis from Drosophila melanogaster. J Biol Chem. 2000;275:22157–65. doi: 10.1074/jbc.M000369200. [DOI] [PubMed] [Google Scholar]
- 45.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 46.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, A Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 47.Adrain C, Creagh EM, Martin SJ. Apoptosis-associated release of Smac/DIABLO from mitochondria requires active caspases and is blocked by Bcl-2. Embo J. 2001;20:6627–36. doi: 10.1093/emboj/20.23.6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi Y. Mechanisms of Caspase Activation and Inhibition during Apoptosis. Mol Cell. 2002;9:459–70. doi: 10.1016/s1097-2765(02)00482-3. [DOI] [PubMed] [Google Scholar]
- 49.Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y, Alnemri ES. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature. 2001;410:112–6. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 50.Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, Shi Y. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408:1008–12. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 51.Zhivotovsky B, Joseph B, Orrenius S. Tumor radiosensitivity and apoptosis. Exp Cell Res. 1999;248:10–7. doi: 10.1006/excr.1999.4452. [DOI] [PubMed] [Google Scholar]
- 52.Bishay K, Ory K, Lebeau J, Levalois C, Olivier MF, Chevillard S. DNA damage-related gene expression as biomarkers to assess cellular response after gamma irradiation of a human lymphoblastoid cell line. Oncogene. 2000;19:916–23. doi: 10.1038/sj.onc.1203405. [DOI] [PubMed] [Google Scholar]
- 53.Ouhtit A, Muller HK, Davis DW, Ullrich SE, McConkey D, Ananthaswamy HN. Temporal events in skin injury and the early adaptive responses in ultraviolet-irradiated mouse skin. Am J Pathol. 2000;156:201–7. doi: 10.1016/S0002-9440(10)64720-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lapucci A, Donnini M, Papucci L, Witort E, Tempestini A, Bevilacqua A, Nicolin A, Brewer G, Schiavone N, Capaccioli S. AUF1 is a bcl-2 ARE-binding protein involved in bcl-2 mRNA destabilization during apoptosis. J Biol Chem. 2002;20:20. doi: 10.1074/jbc.M201377200. [DOI] [PubMed] [Google Scholar]
- 55.Holcik M, Yeh C, Korneluk RG, Chow T. Translational upregulation of X-linked inhibitor of apoptosis (XIAP) increases resistance to radiation induced cell death. Oncogene. 2000;19:4174–7. doi: 10.1038/sj.onc.1203765. [DOI] [PubMed] [Google Scholar]
- 56.Ito T, Deng X, Carr B, May WS. Bcl-2 phosphorylation required for anti-apoptosis function. J Biol Chem. 1997;272:11671–3. doi: 10.1074/jbc.272.18.11671. [DOI] [PubMed] [Google Scholar]
- 57.Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol. 1999;19:8469–78. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science. 2000;288:874–7. doi: 10.1126/science.288.5467.874. [DOI] [PubMed] [Google Scholar]
- 59.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-Dependent Formation of Apaf-1/Caspase-9 Complex Initiates an Apoptotic Protease Cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 60.Amundson SA, Bittner M, Chen Y, Trent J, Meltzer P, Fornace AJ., Jr Fluorescent cDNA microarray hybridization reveals complexity and heterogeneity of cellular genotoxic stress responses. Oncogene. 1999;18:3666–72. doi: 10.1038/sj.onc.1202676. [DOI] [PubMed] [Google Scholar]
- 61.Burns TF, Bernhard EJ, El-Deiry WS. Tissue specific expression of p53 target genes suggests a key role for KILLER/DR5 in p53-dependent apoptosis in vivo. Oncogene. 2001;20:4601–12. doi: 10.1038/sj.onc.1204484. [DOI] [PubMed] [Google Scholar]
- 62.Gobe GC, Harmon B, Leighton J, Allan DJ. Radiation-induced apoptosis and gene expression in neonatal kidney and testis with and without protein synthesis inhibition. Int J Radiat Biol. 1999;75:973–83. doi: 10.1080/095530099139737. [DOI] [PubMed] [Google Scholar]
- 63.Belka C, Rudner J, Wesselborg S, Stepczynska A, Marini P, Lepple-Wienhues A, Faltin H, Bamberg M, Budach W, Schulze-Osthoff K. Differential role of caspase-8 and BID activation during radiation- and CD95-induced apoptosis. Oncogene. 2000;19:1181–90. doi: 10.1038/sj.onc.1203401. [DOI] [PubMed] [Google Scholar]
- 64.Granville DJ, Shaw JR, Leong S, Carthy CM, Margaron P, Hunt DW, McManus BM. Release of cytochrome c, Bax migration, Bid cleavage, and activation of caspases 2, 3, 6, 7, 8, and 9 during endothelial cell apoptosis. Am J Pathol. 1999;155:1021–5. doi: 10.1016/S0002-9440(10)65202-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001;15:1481–6. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman B, Reed JC. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–1805. [PubMed] [Google Scholar]
- 68.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 69.Gong B, Almasan A. Apo2 ligand/TNF-related apoptosis-inducing ligand and death receptor 5 mediate the apoptotic signaling induced by ionizing radiation in leukemic cells. Cancer Res. 2000;60:5754–60. [PubMed] [Google Scholar]
- 70.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–8. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 71.Conradt B, Horvitz HR. The TRA-1A sex determination protein of C. elegans regulates sexually dimorphic cell deaths by repressing the egl-1 cell death activator gene. Cell. 1999;98:317–27. doi: 10.1016/s0092-8674(00)81961-3. [DOI] [PubMed] [Google Scholar]
- 72.Guan RJ, Moss SF, Arber N, Krajewski S, Reed JC, Holt PR. 30 KDa phosphorylated form of Bcl-2 protein in human colon. Oncogene. 1996;12:2605–9. [PubMed] [Google Scholar]
- 73.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 74.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–4. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 75.Deng X, Xiao L, Lang W, Gao F, Ruvolo P, May WS., Jr Novel role for JNK as a stress-activated Bcl2 kinase. J Biol Chem. 2001;276:23681–8. doi: 10.1074/jbc.M100279200. [DOI] [PubMed] [Google Scholar]
- 76.Schurmann A, Mooney AF, Sanders LC, Sells MA, Wang HG, Reed JC, Bokoch GM. p21-activated kinase 1 phosphorylates the death agonist bad and protects cells from apoptosis. Mol Cell Biol. 2000;20:453–61. doi: 10.1128/mcb.20.2.453-461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–28. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 78.Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–43. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- 79.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–31. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 80.Dunkern TR, Fritz G, Kaina B. Ultraviolet light-induced DNA damage triggers apoptosis in nucleotide excision repair-deficient cells via Bcl-2 decline and caspase-3/-8 activation. Oncogene. 2001;20:6026–38. doi: 10.1038/sj.onc.1204754. [DOI] [PubMed] [Google Scholar]
- 81.Pritchard DM, Print C, O’Reilly L, Adams JM, Potten CS, Hickman JA. Bcl-w is an important determinant of damage-induced apoptosis in epithelia of small and large intestine. Oncogene. 2000;19:3955–9. doi: 10.1038/sj.onc.1203729. [DOI] [PubMed] [Google Scholar]
- 82.Pritchard DM, Potten CS, Korsmeyer SJ, Roberts S, Hickman JA. Damage-induced apoptosis in intestinal epithelia from bcl-2-null and bax-null mice: investigations of the mechanistic determinants of epithelial apoptosis in vivo. Oncogene. 1999;18:7287–93. doi: 10.1038/sj.onc.1203150. [DOI] [PubMed] [Google Scholar]
- 83.Lee JU, Hosotani R, Wada M, Doi R, Kosiba T, Fujimoto K, Miyamoto Y, Tsuji S, Nakajima S, Nishimura Y, Imamura M. Role of Bcl-2 family proteins (Bax, Bcl-2 and Bcl-X) on cellular susceptibility to radiation in pancreatic cancer cells. Eur J Cancer. 1999;35:1374–80. doi: 10.1016/s0959-8049(99)00134-3. [DOI] [PubMed] [Google Scholar]
- 84.Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–50. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- 85.Honarpour N, Gilbert SL, Lahn BT, Wang X, Herz J. Apaf-1 deficiency and neural tube closure defects are found in fog mice. Proc Natl Acad Sci U S A. 2001;98:9683–7. doi: 10.1073/pnas.171283198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhou l, Steller H. Distinct pathways mediate UV-induced apoptosis in Drosophila embryo. 2002 doi: 10.1016/s1534-5807(03)00085-6. submitted. [DOI] [PubMed] [Google Scholar]
- 87.Moroni MC, Hickman ES, Denchi EL, Caprara G, Colli E, Cecconi F, Muller H, Helin K. Apaf-1 is a transcriptional target for E2F and p53. Nat Cell Biol. 2001;3:552–8. doi: 10.1038/35078527. [DOI] [PubMed] [Google Scholar]
- 88.Muller H, Bracken AP, Vernell R, Moroni MC, Christians F, Grassilli E, Prosperini E, Vigo E, Oliner JD, Helin K. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 2001;15:267–85. doi: 10.1101/gad.864201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shinoura N, Sakurai S, Asai A, Kirino T, Hamada H. Over-expression of APAF-1 and caspase-9 augments radiation-induced apoptosis in U-373MG glioma cells. Int J Cancer. 2001;93:252–61. doi: 10.1002/ijc.1327. [DOI] [PubMed] [Google Scholar]
- 90.Manji GA, Hozak RR, LaCount DJ, Friesen PD. Baculovirus inhibitor of apoptosis functions at or upstream of the apoptotic suppressor P35 to prevent programmed cell death. J Virol. 1997;71:4509–16. doi: 10.1128/jvi.71.6.4509-4516.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sasaki H, Sheng Y, Kotsuji F, Tsang BK. Down-regulation of X-linked inhibitor of apoptosis protein induces apoptosis in chemoresistant human ovarian cancer cells. Cancer Res. 2000;60:5659–66. [PubMed] [Google Scholar]
- 92.Bergmann A, Agapite J, McCall K, Steller H. The Drosophila Gene hid Is a Direct Molecular Target of Ras-Dependent Survival Signaling. Cell. 1998;95:331–341. doi: 10.1016/s0092-8674(00)81765-1. [DOI] [PubMed] [Google Scholar]
- 93.Brodsky MH, Nordstrom W, Tsang G, Kwan E, Rubin GM, Abrams JM. Drosophila p53 binds a damage response element at the reaper locus. Cell. 2000;101:103–13. doi: 10.1016/S0092-8674(00)80627-3. [DOI] [PubMed] [Google Scholar]
- 94.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 95.Rehemtulla A, Hamilton CA, Chinnaiyan AM, Dixit VM. Ultraviolet radiation-induced apoptosis is mediated by activation of CD-95 (Fas/APO-1) J Biol Chem. 1997;272:25783–6. doi: 10.1074/jbc.272.41.25783. [DOI] [PubMed] [Google Scholar]
- 96.Bouvard V, Zaitchouk T, Vacher M, Duthu A, Canivet M, Choisy-Rossi C, Nieruchalski M, May E. Tissue and cell-specific expression of the p53-target genes: bax, fas, mdm2 and waf1/p21, before and following ionising irradiation in mice. Oncogene. 2000;19:649–60. doi: 10.1038/sj.onc.1203366. [DOI] [PubMed] [Google Scholar]
- 97.Caricchio R, Reap EA, Cohen PL. Fas/Fas ligand interactions are involved in ultraviolet-B-induced human lymphocyte apoptosis. J Immunol. 1998;161:241–51. [PubMed] [Google Scholar]
- 98.Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, Krammer PH. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med. 1998;188:2033–45. doi: 10.1084/jem.188.11.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gutierrez del Arroyo A, Gil-Lamagniere C, Lazaro I, de Marco MC, Layunta I, Silva A. Involvement of p53 and interleukin 3 in the up-regulation of CD95 (APO-1/Fas) by X-ray irradiation. Oncogene. 2000;19:3647–55. doi: 10.1038/sj.onc.1203662. [DOI] [PubMed] [Google Scholar]
- 100.Gross S, Knebel A, Tenev T, Neininger A, Gaestel M, Herrlich P, Bohmer FD. Inactivation of protein-tyrosine phosphatases as mechanism of UV-induced signal transduction. J Biol Chem. 1999;274:26378–86. doi: 10.1074/jbc.274.37.26378. [DOI] [PubMed] [Google Scholar]
- 101.Bender K, Blattner C, Knebel A, Iordanov M, Herrlich P, Rahmsdorf HJ. UV-induced signal transduction. J Photochem Photobiol B. 1997;37:1–17. doi: 10.1016/s1011-1344(96)07459-3. [DOI] [PubMed] [Google Scholar]
- 102.Ivanov VN, Ronai Z. p38 protects human melanoma cells from UV-induced apoptosis through down-regulation of NF-kappaB activity and Fas expression. Oncogene. 2000;19:3003–12. doi: 10.1038/sj.onc.1203602. [DOI] [PubMed] [Google Scholar]
- 103.Kasibhatla S, Brunner T, Genestier L, Echeverri F, Mahboubi A, Green DR. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa B and AP-1. Mol Cell. 1998;1:543–51. doi: 10.1016/s1097-2765(00)80054-4. [DOI] [PubMed] [Google Scholar]
- 104.Aragane Y, Kulms D, Metze D, Wilkes G, Poppelmann B, Luger TA, Schwarz T. Ultraviolet light induces apoptosis via direct activation of CD95 (Fas/APO-1) independently of its ligand CD95L. J Cell Biol. 1998;140:171–82. doi: 10.1083/jcb.140.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Belka C, Schmid B, Marini P, Durand E, Rudner J, Faltin H, Bamberg M, Schulze-Osthoff K, Budach W. Sensitization of resistant lymphoma cells to irradiation-induced apoptosis by the death ligand TRAIL. Oncogene. 2001;20:2190–6. doi: 10.1038/sj.onc.1204318. [DOI] [PubMed] [Google Scholar]
- 106.Yount GL, Afshar G, Ries S, Korn M, Shalev N, Basila D, McCormick F, Haas-Kogan DA. Transcriptional activation of TRADD mediates p53-independent radiation-induced apoptosis of glioma cells. Oncogene. 2001;20:2826–35. doi: 10.1038/sj.onc.1204393. [DOI] [PubMed] [Google Scholar]
- 107.Maeda T, Hao C, Tron VA. Ultraviolet Light (UV) Regulation of the TNF Family Decoy Receptors DcR2 and DcR3 in Human Keratinocytes. J Cutan Med Surg. 2001;5:294–8. doi: 10.1007/s102270000030. [DOI] [PubMed] [Google Scholar]
- 108.Degenhardt K, Sundararajan R, Lindsten T, Thompson C, White E. Bax and Bak independently promote cytochrome c release from mitochondria. J Biol Chem. 2002;8:8. doi: 10.1074/jbc.M109939200. [DOI] [PubMed] [Google Scholar]
- 109.Aravind L, V, Dixit M, Koonin EV. Apoptotic molecular machinery: vastly increased complexity in vertebrates revealed by genome comparisons. Science. 2001;291:1279–84. doi: 10.1126/science.291.5507.1279. [DOI] [PubMed] [Google Scholar]
- 110.Kulms D, Schwarz T. Molecular mechanisms of UV-induced apoptosis. Photodermatol Photoimmunol Photomed. 2000;16:195–201. doi: 10.1034/j.1600-0781.2000.160501.x. [DOI] [PubMed] [Google Scholar]
- 111.Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–52. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 112.Holmberg C, Helin K, Sehested M, Karlstrom O. E2F-1-induced p53-independent apoptosis in transgenic mice. Oncogene. 1998;17:143–55. doi: 10.1038/sj.onc.1201915. [DOI] [PubMed] [Google Scholar]
- 113.Tang D, Wu D, Hirao A, Lahti JM, Liu L, Mazza B, Kidd VJ, Mak TW, Ingram AJ. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J Biol Chem. 2002;30:30. doi: 10.1074/jbc.M111598200. [DOI] [PubMed] [Google Scholar]
- 114.Jin S, Fan F, Fan W, Zhao H, Tong T, Blanck P, Alomo I, Rajasekaran B, Zhan Q. Transcription factors Oct-1 and NF-YA regulate the p53-independent induction of the GADD45 following DNA damage. Oncogene. 2001;20:2683–90. doi: 10.1038/sj.onc.1204390. [DOI] [PubMed] [Google Scholar]
- 115.Reinke V, Lozano G. The p53 targets mdm2 and Fas are not required as mediators of apoptosis in vivo. Oncogene. 1997;15:1527–34. doi: 10.1038/sj.onc.1201316. [DOI] [PubMed] [Google Scholar]
- 116.Fuchs EJ, McKenna KA, Bedi A. p53-dependent DNA damage-induced apoptosis requires Fas/APO-1-independent activation of CPP32beta. Cancer Res. 1997;57:2550–4. [PubMed] [Google Scholar]
- 117.Zheng TS, Hunot S, Kuida K, Momoi T, Srinivasan A, Nicholson DW, Lazebnik Y, Flavell RA. Deficiency in caspase-9 or caspase-3 induces compensatory caspase activation. Nat Med. 2000;6:1241–7. doi: 10.1038/81343. [DOI] [PubMed] [Google Scholar]
- 118.Perez CA, Brady LW. Overview. In: Perez CA, Brady LW, editors. Principles and practice of radiation oncology. 2. new york: J.B. Lippincott; 1992. [Google Scholar]
- 119.Tron VA, Trotter MJ, Tang L, Krajewska M, Reed JC, Ho VC, Li G. p53-regulated apoptosis is differentiation dependent in ultraviolet B-irradiated mouse keratinocytes. Am J Pathol. 1998;153:579–85. doi: 10.1016/S0002-9440(10)65600-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ideker T, Galitski T, Hood L. A new approach to decoding life: systems biology. Annu Rev Genomics Hum Genet. 2001;2:343–72. doi: 10.1146/annurev.genom.2.1.343. [DOI] [PubMed] [Google Scholar]