

Abstract
Dithiocarbamates (DTCs) can be formed by the in situ condensation of polar alkylamines with CS2, and assembled into dithiocarbamate-anchored monolayers (DAMs) on Au substrates in aqueous solutions. Primary and secondary amines can both be used to prepare DTCs, but have significant differences in their reactivities and product stabilities. Ultraviolet absorption spectroscopy provides a convenient method for monitoring in situ DTC formation as well as the formation of potential byproducts. The kinetics of DAM assembly on Au substrates as measured by second harmonic generation (SHG) indicated first-order rate processes and saturation coverages similar to those of alkanethiols on Au. However, the rate of adsorption did not change with DTC concentration in a manner expected of Langmuir kinetics, and is attributed to the competitive adsorption of alkylammonium counterions to the freshly oxidized Au substrate. These analyses establish a practical range of conditions for preparing DAMs from polar amines using in situ DTC formation.
Introduction
The directed assembly of organic surfactants into monolayers is ubiquitous in the design of functionalized surfaces.1 Among the numerous methods developed for preparing monolayer films, processes based on chemisorption are widely favored by virtue of their simplicity and breadth of application. Chemisorption provides directionality to self-assembled monolayers (SAMs), enabling one to engineer surface properties based on molecular design principles. Chemisorption also imparts a certain degree of stability to SAMs, and this has promoted their use in a wide assortment of applications in surface science and nanotechnology. As an example, bioanalytical sensors that utilize Au substrates (e.g., surface plasmon resonance and quartz crystal microbalance) are often passivated with alkanethiols appended to biomolecular recognition elements, as well as oligoethylene glycol chains to prevent nonspecific adsorption.2 Chemisorptive surfactants are also widely used to functionalize metal nanoparticles for biological applications or nanoscale self-assembly.3
The phenomenal versatility of chemisorptive SAMs is limited only by their compatibility with the environments of their intended applications. In the case of alkanethiol-based SAMs on Au, the quality of the monolayer can be compromised by surfactant exchange,4,5 oxidative and thermal desorption,6,7 and exposure to ultraviolet light.8 The relative rate and impact of such environmental factors on monolayer degradation is a topic of some debate; nevertheless, several studies have now established some limits of stability for alkanethiol-based SAMs on Au. In the context of biological applications, the durability of SAMs in aqueous cell culture media and other physiologically relevant conditions has been estimated to be on the order of a week, with longer exposures resulting in a significant loss of performance.9,10 Multivalent thiols may provide additional stability against surface desorption, but this assumption is not always valid: for example, chemisorbed divalent ligands based on thioctic acid have been found to be less stable under tensile stress than monovalent thiols.11 A general method of surface functionalization with improved stability against oxidation or desorption, while retaining the simplicity of self-assembly, is clearly a desirable goal.
Our studies on the surface functionalization of Au have led us to propose dithiocarbamate-anchored monolayers (DAMs) as a robust alternative to alkanethiol-based SAMs under ambient conditions.12 Dithiocarbamates (DTCs) have been widely used for decades as chelating ligands in coordination chemistry,13 but their application as chemisorptive surfactants has been largely overlooked until recently.14,15,16,17,18,19 DTCs feature a carbodithioate (−CS2) group whose intramolecular S-S distance is nearly equal to that of adjacent bonding sites on Au surfaces, suggesting a simple form of epitaxial adsorption (see Figure 1).20 Furthermore, the chemical structure of simple DTCs allow for the formation of stable monolayers without significant interchain interactions. A recent scanning tunneling microscopy study of diethyl DTC on Au(111) revealed densely packed DAMs with moderate local order.17a
Figure 1.
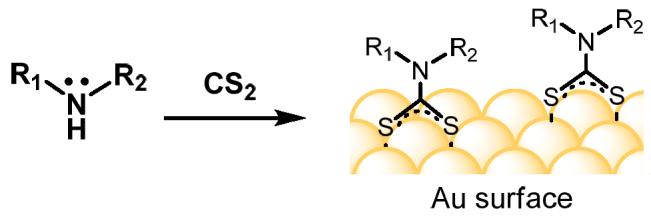
Dithiocarbamate-anchored monolayers (DAMs) by in situ condensation of amines and CS2.
A wide variety of DTCs can be prepared in situ by the condensation of CS2 with nucleophilic amines, using mildly basic conditions and polar solvents.13,21 This simple condensation can be conducted either offline or in the presence of Au substrates, permitting DAMs to be assembled from amines in a single step. DTCs can even be formed and attached onto Au surfaces in water, providing a useful complement to bioconjugation methods developed for amines. Lastly, we have found DAMs assembled from N,N-dialkyl DTCs to resist displacement by polar thiols such as mercaptoethanol.12 This feature has special relevance for surfaces in physiological settings, where biogenic thiols such as cysteine and glutathione can act as competing adsorbates and contribute toward surface degradation and biofouling.
Here we investigate DAM assembly in aqueous solutions via in situ DTC formation, and establish the scope and conditions for DAM formation using water-soluble amine derivatives based on oligoethylene glycol and oligopeptides (see Figure 2). Several of these structures have been featured in recent studies involving antifouling surfaces22 or the selective penetration of cell membranes.23 The kinetics and saturation coverage of DAM assembly were measured by second harmonic generation (SHG) analysis, and collated with atomic surface densities measured by x-ray photoelectron spectroscopy (XPS). The adsorption of DTCs proceeded with first-order kinetics and achieved surface coverages comparable to that of alkanethiols, but notable differences were observed with respect to rate and adsorbate concentration. We attribute this to the chemical reactivity of the substrate itself, namely the presence of a transient but reactive Au oxide formed during surface cleaning.
Figure 2.
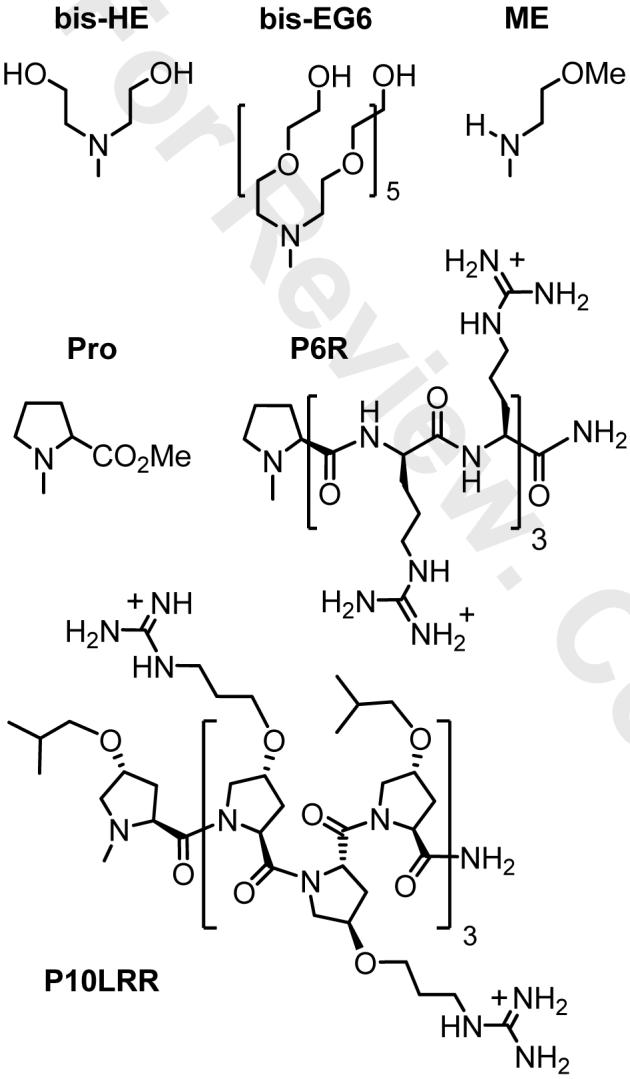
Parent structure of some amines and DTCs. HE = 2-hydroxyethyl; EG6 = hexaethyleneglycol; ME = 2-methoxyethyl; Pro = prolyl methyl ester; P6R and P10LRR = oligoprolines (see text for details).
Experimental Section
Materials
Au-coated glass substrates (1 × 1 cm2) were purchased from Reichart and cleaned just prior to use by immersion in freshly prepared piranha solution (5 parts 18 M H2SO4, 2 parts 30% H2O2) for 3 minutes, thoroughly rinsing with deionized water, then dried under a stream of nitrogen. Deionized water was obtained from an ultrafiltration system (Milli-Q, Millipore) with a measured resistivity above 18 MΩ·cm, and passed through a 0.22-μm filter to remove particulate matter. CS2 was freshly distilled from CaH2 before use.
Proline methyl ester (Pro) and (3-mercaptopropyl)hexaethylene glycol (EG6-SH) were prepared according to literature procedures.22,24 Oligopeptides with N-terminal prolines (P10LRR and P6R) were prepared by solid-phase synthesis as previously described.23 Bis-EG6 amine was expediently synthesized by Pd-mediated reductive dimerization (see Scheme 1).25 EG6 azide26 (0.34 g, 1.11 mmol) and 10% Pd on C (0.051 g, 15 wt%) were treated with 5 mL methanol and purged with H2, then stirred for 6 h at 40 °C. The reaction mixture was filtered through Celite and concentrated to yield a mixture of EG6 amine and bis-EG6 amine (0.28 g). Bis-EG6 amine could be isolated by alumina column chromatography (5% CH3OH in CH2Cl2) as a light yellow oil (91 mg, 30% yield). 1H NMR (300 MHz, CD3OD): δ 3.60-3.45 (m, 44H), 2.79 (t, 4H); 13C NMR (300 MHz, CD3OD): δ 73.68, 71.80-71.30, 70.08. ESI-MS: m/z calcd for C24H52NO12 [M+H]+: 546.35; found 546.32.
Scheme 1.
DTC formation
Most DTCs were prepared in situ as alkylammonium salts, with CS2 as the limiting reagent to minimize the formation of side products. In a typical procedure, a 25-mM solution of DTC was prepared using two equivalents of amine dissolved in methanol, with one equivalent serving as base. The solution was degassed for several minutes with argon then treated with one equivalent of CS2, agitated with a vortex mixer for 30 seconds, and left to sit for 20 min at room temperature in a capped vial. Solutions of P6R-DTC and P10LRR-DTC were prepared using the corresponding oligopeptides as the limiting agents. Oligopeptides in degassed MeOH (P6R: 8 mM; P10LRR: 4 mM) were treated with three equivalents of CS2 and one equivalent of Et3N to serve as base. The solutions were agitated as above and left to sit for 30 min at room temperature, then diluted with deionized water to 250 μM. Molar extinction coefficients (λmax=290 nm) were measured by UV absorption spectroscopy using a Cary-50 spectrophotometer with a cell path length of 1 cm. In the case of P10LRR-DTC, UV absorption peaks continued to increase after dilution to 250 μM, with no further change after 15 h. Serial dilutions of the stock solution were prepared using deionized water (pH 6) or aqueous phosphate buffer (pH 10).
Surface analyses
SHG studies were performed using the discrete retardance nonlinear optical ellipsometer described previously.27 Clean Au substrates were suspended vertically in a cylindrical glass cell initially filled with deionized water. The samples were analyzed in the reflection configuration with the p-polarized fundamental beam at a 45° angle of incidence. The native SHG response from the clean Au substrate was monitored for a minimum of 30 minutes to ensure a constant baseline value. The beam was momentarily blocked while the solution was quickly exchanged with an aqueous DTC solution, and the SHG response was then monitored until no further changes in signal were observed. The data were normalized and corrected for baseline drift, and processed using standard data analysis software (KaleidaGraph).
XPS data were obtained by a Kratos Ultra DLD spectrometer using monochromatic Al Kα radiation (hν = 1486.58 eV). The XPS spectrometer was equipped with an RF plasma generator (Evactron® C, XEI Scientific, Inc.) for in situ sample cleaning and analysis without exposure to air. Samples were mounted on a double-sided adhesive Cu conductive tape. The survey and high-resolution spectra were collected at a fixed analyzer pass energy of 160 and 20 eV, respectively. The binding-energy scale of the XPS instrument was calibrated using Au 4f7/2 = 84.00 eV and Cu 2p3/2 = 932.67 eV.28 The charge reference was calibrated against the Au 4f7/2 peak set at 84.00 eV; the standard deviation in peak position was ±0.05 eV. A Kratos charge neutralizer was used to achieve a full width at half-maximum (FWHM) resolution of the Au 4f7/2 peak of 0.65-0.75 eV. XPS data were analyzed with commercially available software (CasaXPS, version 2313Dev64) with individual peaks fitted to a Gaussian/Lorenzian function. The atomic concentrations of the elements in the near-surface region were estimated after the subtraction of a Shirley type background, taking into account the corresponding Scofield atomic sensitivity factors and inelastic mean free path (IMFP) of photoelectrons. The peak areas without IMFP correction could be used to calculate coverage because IMFP attenuation is included in the calculation method (see Supporting Information).
Results and Discussion
In situ DTC formation
The conditions for converting alkylamines into DTCs are remarkably mild and straightforward. Relatively simple amines treated with CS2 in a 2:1 ratio will typically produce DTC alkylammonium salts within minutes and in nearly quantitative yield, as determined by 1H and 13C NMR spectroscopy (the 13C chemical shift of the carbodithioate unit is 210-220 ppm).29 For larger or more complex structures in which the amine is the limiting agent, DTCs can still be produced in significant yields by treatment with excess CS2 under mildly basic conditions (see below). The latter condition is useful for conjugating amine-terminated macromolecules onto the surfaces of anisotropic Au nanoparticles in aqueous suspensions, for their targeted delivery to tumor cells and for preventing nonspecific adsorption and uptake.30
While the in situ condensation of amines and CS2 can proceed in a variety of solvents, we have found this reaction to be most efficient and reliable in polar organic solvents at initial CS2 concentrations of 10-100 mM. For this reason, we used conditions optimized for DTC formation in MeOH or DMF, but subsequently performed serial dilutions in water for quantitative SHG studies. These aqueous solutions contain much less than 1% organic solvent. It must be emphasized that in situ DTC formation and DAM assembly can be conducted entirely in water if desired, albeit at some expense of reaction efficiency.
DTCs produce two characteristic absorption peaks at 260 and 290 nm, providing a convenient method for measuring solution concentrations.31 Molar extinction coefficients were established by serial dilution of the stock solutions with deionized water or buffered aqueous solution to low micromolar concentrations (see Figure 3 and Table 1). The DTC solutions were also monitored by UV absorption spectroscopy for stability at two different pH values as well as exposure to air. Decomposition was characterized by a loss of absorption intensity at 260 and 290 nm, and also by the appearance of a broad UV peak centered at 330 nm (see Figure 4). Earlier studies have correlated the latter with the formation of trithiocarbonate (CS32-), a byproduct which often appears at high pH.31,32
Figure 3.

UV absorption spectra of bis-HE-DTC, formed in situ in methanol and diluted with aqueous phosphate buffer (pH 10). Twofold dilutions were performed from an initial concentration of 125 μM.
TABLE 1.
Molar extinction coefficients of DTCs, diluted in aqueous solutions (λmax = 290 nm)
| DTC | ε(M−1 cm−1) |
|---|---|
| bis-HE | 8.84 ×103a |
| 8.45 ×103b | |
| HE | 6.45 ×103a |
| ME | 8.47 ×103a |
| bis-EG6 | 8.63 ×103b,c |
| Pro | 8.21 ×103b |
| P6R | 8.19 ×103a |
| P10LRR | 8.21 ×103d |
Diluted with deionized water (pH 6).
Diluted with phosphate buffer (pH 10).
Stock solution prepared from either MeOH or DMF.
Assumed from Pro-DTC data; see text for details.
Figure 4.

UV absorption spectra of DTC solutions, before and after a 6-hour exposure to air (solid and dashed curves, respectively). (a) Bis-HE-DTC (pH 6); (b) HE-DTC (pH 6); (c) ME-DTC (pH 6 and 10).
Significant differences were observed in the formation and stability of N,N-disubstituted DTCs derived from secondary amines and monosubstituted DTCs derived from primary amines. For example, bis-HE-DTC was formed quantitatively by the in situ condensation of diethanolamine and CS2,21 and observed to be stable at pH 6 or pH 10 for at least several days when protected from air. Furthermore, bis-HE-DTC was only mildly sensitive to air oxidation, with less than 10% change in absorbance intensity after a 6-hour exposure (see Figure 4a). In contrast, HE-DTC (derived from ethanolamine and CS2) was not fully formed under comparable conditions and experienced significant degradation upon exposure to air, in accord with earlier studies on the decomposition of monoalkyl DTCs (see Figure 4b).33 Not all primary amines fared as poorly; ME-DTC could be formed from 2-methoxyethylamine with comparable efficiency as bis-HE-DTC and was relatively stable in air at pH 6, but was still susceptible to air oxidation at pH 10 (see Figure 4c). DTCs prepared from other secondary amines such as bis(hexaethyleneglycol)amine (bis-EG6) and proline methyl ester (Pro) could also be formed in nearly quantitative yields (see Supporting Information), and exhibited similar levels of stability to air oxidation as bis-HE-DTC. Overall, we found secondary amines to be more reliable substrates than primary amines for in situ DTC formation.
For situations in which the amine serves as the limiting reagent, in situ DTC formation can be conducted with a stoichiometric amount of Et3N as base and a slight excess of CS2. Proline-terminated heptapeptide P6R (8 mM in methanol) was treated with CS2 (24 mM) and converted quantitatively to its corresponding DTC within 30 min, as determined by comparison of its molar extinction coefficient with that of Pro-DTC (see Table 1). The amine and CS2 concentrations are comparable to those used in recent studies involving the functionalization of Au nanorods in basic aqueous solutions (pH 9.5).30 However, in situ DTC formation did not reach completion when lower concentrations were used: in the case of decapeptide derivative P10LRR, a condensation performed with amine and CS2 concentrations of 4 and 12 mM respectively stopped at 44% conversion after 24 h, again based on the measured extinction coefficient for Pro-DTC (see Table 1). It is worth mentioning that the guanidine moieties of P6R and P10LRR have negligible reactivity toward CS2, which was confirmed by a control study involving simple alkylguanidines.34 These results suggest some practical lower limits in reagent concentrations for in situ DTC formation.
Kinetics of DAM assembly and saturation coverage
Alkanethiols and DTCs are both capable of assembling into monolayers on Au by chemisorption, but their rate and mechanism of self-assembly cannot be assumed to be identical as they may be influenced by differences in chemical behavior. Alkanethiols are well known to be mobile on Au surfaces after their chemisorption, enabling them to assemble with thermodynamic control into crystalline, close-packed monolayers.2,35 A widely accepted model of alkanethiol SAM assembly features an initial Langmuir adsorption, followed by a slower reorganization step.36 The first-order Langmuir adsorption equation can be expressed simply as:
| (1) |
where θ is the fractional surface coverage between 0 and 1, θsat is the saturation coverage, c is the molar concentration of the adsorbate, and k is the adsorption rate constant. First-order kinetics should also be appropriate for describing the adsorption of DTCs on Au, which has the added benefit of low desorption at unbiased potentials, allowing experimental measurements to be interpreted simply as the forward rate constant. We were particularly interested to measure the kinetics of DAM assembly in aqueous solutions, and to determine whether their saturation coverage would be comparable to that of alkanethiol-based SAMs.
SHG is a straightforward method of measuring surface adsorption kinetics, and has been used to characterize the self-assembly of alkanethiols on Au at micromolar concentrations in various solvents.37 The normalized SHG intensity ISHG is proportional to the square of the sum of nonlinear susceptibilities at the substrate, adsorbate, and interface (χsub, χads, and χint respectively); the latter term is highly sensitive to changes in surface states due to chemisorptive bonding. In such cases, the intensity decreases as a function of θ
| (2) |
until a saturation value Isat is attained, such that R = 1−(Isat)1/2.
The SHG analysis uses the simplifying assumption that Isat is achieved (i.e. θ/θsat = 1) when the surface binding sites are fully occupied and no further chemisorption is possible. In actuality, Isat reaches a plateau when adsorption becomes self-limiting due to molecular crowding. The true extent of saturation coverage can be addressed by complementing SHG with XPS, by measuring the atomic populations of adsorbates on Au after Isat is achieved. In the case of alkanethiols, the saturation plateau of initial Langmuir adsorption has been correlated with 80-90% of the packing density achieved in a close-packed SAM.36
DAMs were assembled by introducing dilute aqueous solutions of DTC alkylammonium salts to freshly oxidized Au-coated glass substrates, which were soaked in deionized water prior to surfactant exposure. Changes in normalized ISHG were measured as a function of exposure time to adsorbates at micromolar concentrations, and were consistent with first-order adsorption kinetics (see Table 2 and Figure 5). The saturation SHG intensities for a given adsorbate did not decrease at higher concentrations, indicating that the chemical potential for monolayer assembly was constant within the range of experimental conditions. Control studies performed in the absence of CS2 (amines only) did not produce any significant modulations in SHG, confirming that changes in χint were sensitive to chemisorption but not physisorption (see below). The adsorption kinetics of DTCs was also compared with that of EG6-SH, a water-soluble thiol used to prepare protein-resistant SAMs on Au surfaces.22,38 EG6-SH was selected for its low self-affinity in water, which might otherwise introduce significant deviations from Langmuir adsorption kinetics.
TABLE 2.
SHG analysis of DTC and thiol adsorption on Au
| adsorbate | c (μm)a | Isat | K(M−1 s−1) |
|---|---|---|---|
| bis-HE-DTC | 10 | 0.325 | 250 |
| 5 | 0.387 | 546 | |
| 2.5 | 0.331 | 87 | |
| Pro-DTC | 10 | 0.532 | 490 |
| 5 | 0.563 | 890 | |
| 2.5 | 0.545 | 486 | |
| bis-EG6-DTC | 5 a | 0.303 | 1165 |
| 5 b | 0.277 | 1237 | |
| 5 c | 0.259 | 836 | |
| P10LRR-DTC | 50 d | 0.277 | 3 |
| ME-DTC e | 5 | 0.124 | 319 |
| EG6-SH | 5 | 0.358 | n/af |
| 2.5 | 0.414 | 4205 | |
| 1.25 | 0.418 | 4725 |
Diluted with deionized water (pH 6).
Diluted with pH 7.5 buffer.
Diluted with pH 9.5 buffer.
Estimated from extinction coefficient of Pro-DTC.
XPS suggests formation of byproducts during SHG analysis.
Insufficient data was collected, due to the speed with which saturation was achieved.
Figure 5.

SHG analysis of DTC adsorption onto Au in water (pH 6). (a-c) bis-HE-DTC; (d-f) Pro-DTC. Least-squares fit to first-order kinetics defined by red curves.
DAMs assembled from bis-HE-DTC were evaluated by XPS to determine the surface density of DTCs upon saturation of the SHG signal. Integrated peak ratios of elemental S/N and S/Au were measured at a normal photoemission angle, and found to be 2.09 and 0.077 respectively. The S/N ratio is consistent with the chemical composition of the DTC adsorbates, and the S/Au ratio is close to the literature value of 0.084 for close-packed butanethiol SAMs on Au(111).39 However, the S/Au ratio is not a reliable indicator of surface saturation, as it is influenced by the sampling conditions as well as by differences between instruments. An alternative approach for measuring saturation coverage is based on the adlayer approximation model introduced by Fadley, which uses the differential intensities from the substrate and adsorbed species and takes full account of sampling geometry and detection efficiency (see Supporting Information for details).40 This method can quantify the fractional coverage of monolayer (ML) based on the ratio of adatoms or adsorbates to surface atoms in a Au(111) plane, but requires that the overlayer be very thin so that electron emission from the substrate is unattenuated. In this regard, the DAM based on bis-HE DTC is ideal: the short HE chains allow the N 1s, S 2p, and Au 4f signal intensities to be collected with minimal extinction.
Measurements of adlayer coverage at normal and 60° photoemission angles are remarkably similar, with mean values of 0.46 and 0.45 ML derived from N 1s and S 2p peak intensities, respectively. This coverage is significantly greater than that of close-packed alkanethiol SAMs, which are assumed to be 0.33 ML for a superlattice on Au(111).35,41 A simple packing analysis suggests that DAMs can provide complete surface passivation at 0.40 ML when each sulfur atom occupies a threefold hollow site on the Au surface (see Figure 6). Alternatively, the bidentate DTCs can occupy twofold bridging sites with herringbone packing, resulting in an adlayer coverage of 0.50 ML. Experimental error prevents the unambiguous assignment of a single surface structure to the bis-HE DTC adlayer, and may suggest the coexistence of both bridging motifs. On the other hand, deviations from substrate planarity may cause an underestimation of the effective DAM thickness, with subsequent overestimation of the experimental ML values. If this factor is taken into account, then the DAM structure is more likely dominated by DTC chemisorption to threefold hollow sites.
Figure 6.

Side and top views of close-packed monolayers on Au(111): alkanethiol SAM with structure (0.33 ML, left), DAM occupying threefold hollow sites (0.40 ML, center), and DAM occupying twofold bridging sites (0.50 ML, right). The dashed ellipses delineate the space available to accommodate molecular cross sections.
Remarkably, the rate of DTC adsorption did not change with concentration as anticipated for Langmuir adsorption kinetics (see Eq. 1), despite the good fit of the SHG data to a first-order process (see Figure 5). In the cases of bis-HE-DTC and Pro-DTC, the decreases in ISHG over time at 5 μM were essentially identical to those at 10 μM, causing a doubling of the calculated rate constants (see Table 2). In contrast, the adsorption of EG6-SH onto Au proceeded much more rapidly than any of the DTC adsorbates, and obeyed Langmuir kinetics in a concentration-dependent fashion. These stark differences suggest that the DTC counterions and the method of surface preparation may have an important influence on the unusual kinetics of DAM assembly.42 Subjecting the Au substrate to rigorous cleaning treatments with piranha solution or oxygen plasma is known to produce a metastable oxide layer, which is sufficiently long-lived under ambient conditions to permit its characterization by XPS (see Figure 7a).43 Freshly oxidized Au surfaces are both electron-rich and highly reactive, enabling the electrostatic adsorption of the alkylammonium counterions to compete with DTC chemisorption during formation of the initial adlayer (see Figure 7b). The Au surface is gradually neutralized by oxidation of the organic adsorbates, which may then be displaced by free DTC ligands in solution with a subsequent decrease in SHG. Recent studies have shown that Au surfaces prepared under conditions similar to ours are capable of oxidizing amines,44 but the oxidation proceeds at a modest rate. This suggests that amine adsorption and oxidation may be limiting the rate of DAM formation, when using DTC alkylammonium salts above a threshold concentration. In comparison, the oxidation of thiols is rapid and has very little effect on the Langmuir kinetics of surface adsorption. Furthermore, only minute quantities of reducible surfactant are needed to neutralize the oxidized Au surface, as attested by the unimpeded adsorption kinetics of EG6-SH at micromolar concentrations (see Table 2). These observations support the notion that DAM assembly can be retarded by the competitive adsorption and slow displacement of organic counterions, resulting in significant deviations from classical Langmuir kinetics.
Figure 7.
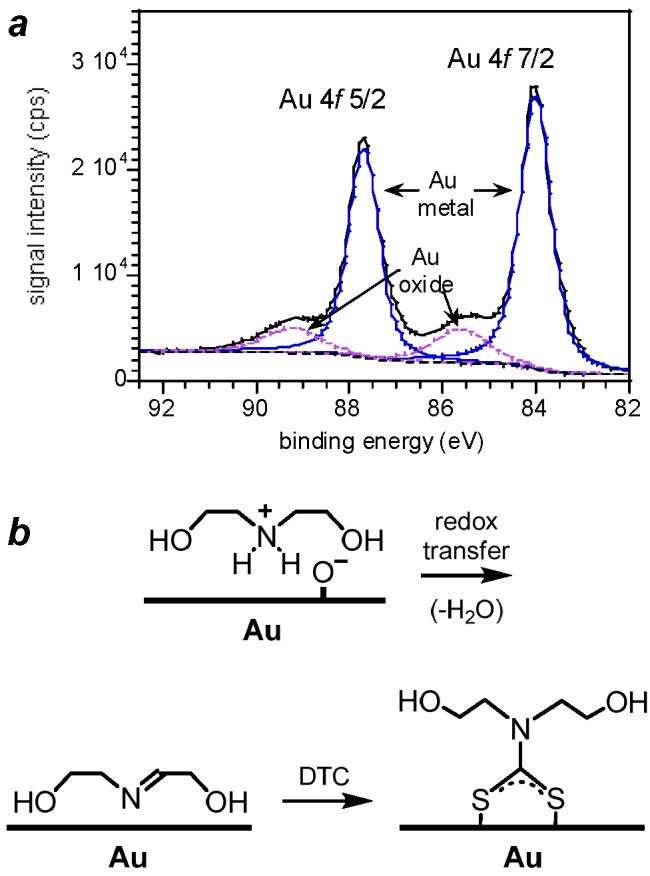
(a) XPS spectrum of the Au 4f region of Au substrate cleaned by in situ plasma treatment. Signals were collected at a 60° takeoff angle. Deconvolution of the Au 4f signal (solid black) enabled peak assignments to Au metal (solid blue) and Au-oxide (dashed purple) after subtraction of the background signal (dashed black). (b) Competitive adsorption of alkylammonium counterion onto oxidized Au substrate, followed by redox transfer and displacement by DTC ligand.
Competitive cation adsorption may also explain some other unusual behavior in the kinetics of DAM assembly using DTCs formed in situ. One interesting observation is that the DTC adsorption rate increases with respect to molecular size; DAMs assembled from bis-HE-DTC (mw 180) formed twice as slowly as those assembled from bis-EG6-DTC (mw 620) under equivalent conditions. This can be attributed to chemical differences between DTC counterions: the conjugate acid of diethanolamine (formed in situ with bis-HE-DTC) is small and an effective hydrogen bond donor, whereas the conjugate acid of bis-EG6-amine is much larger and a less effective donor, so its nonspecific adsorption is expected to be relatively slow. Another unexpected result is that the DTCs derived from oligopeptides P6R and P10LRR did not form DAMs on freshly oxidized Au substrates at 5 or 10 μM, despite clear evidence for in situ DTC formation (see Table 1). In this case the DTC counterion (conjugate acid of triethylamine) is a weak hydrogen bond donor; however, P6R and P10LRR each display six guanidinium cations, and can be expected to adsorb electrostatically onto the oxidized Au surface. These functional groups are not readily oxidized, and their polyvalent adsorption increases their resistance to desorption and surfactant exchange.
Interestingly, chemisorption was observed upon exposing Au substrates to a 50 μM solution of P10LRR-DTC. A first-order decrease in SHG response was recorded, albeit at a much slower rate relative to other DTCs. XPS revealed an elemental N/S ratio of 5.5, much lower than the expected ratio of 15 for P10LRR-DTC. Partial DAM formation was confirmed by calculating adlayer coverages based on N 1s and S 2p peak intensities at normal and 60° photoemission angles, which produced mean values of 0.07 and 0.22 ML, respectively. The very slow chemisorption rate and the disparity in ML values based on N 1s and S 2p peak intensities do not support a high density of DTCs on Au, but rather suggest the coadsorption of other sulfur-containing species, likely generated by the slow hydrolysis of CS2 left over from in situ DTC formation.
Finally, we measured the adsorption kinetics of ME-DTC in order to compare the efficiencies of mono- and disubstituted DTCs in DAM assembly. The first-order rate constant using ME-DTC at 5 μM was in the same range as that determined for bis-HE-DTC but the Isat value was exceptionally low (see Table 2), beyond that expected even for 100% saturation. XPS analysis indicates that ME-DTC does not assemble into a more densely packed adlayer than disubstituted bis-HE-DTC, despite the differences in cross section; indeed, the mean estimate of saturation coverage based on the N 1s peak is only 0.27 ML. Furthermore, the substrate exposed to ME-DTC produces a S/N peak ratio of 3.62. The high sulfur count again indicates the substantial coadsorption of sulfur-containing byproducts, likely generated in this case by the decomposition of ME-DTC during SHG analysis.
Conclusions
The in situ condensation of alkylamines and CS2 into DTCs offers a practical and versatile method for DAM assembly on Au surfaces. Simple DTCs assemble into DAMs with saturation coverage at micromolar concentrations in aqueous solutions. Both secondary and primary amines can be used to form DTCs, although the latter is more prone to solution decomposition and the generation of chemisorptive byproducts. For more complex amines, some care should be taken to reduce competitive counterion adsorption to ensure efficient DAM assembly. The alkylammonium counterion can impede DAM formation if freshly oxidized Au substrates are used. One recommendation is to ensure that the Au substrates are in an unoxidized state, prior to exposure to organic DTC salts. This can be achieved simply by allowing the oxidized substrate sufficient time to be reduced back into elemental Au, or more expediently by using electrochemical means or treatment with a mild and nonadsorptive reducing agent.42,45 Substrates with freshly evaporated Au may also be used to circumvent the issue of surface oxidation. Additional studies addressing surface potential, counterions, and the structures of the DTC adsorbates will provide further insights into the parameters which influence DAM assembly.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge financial support from the National Institutes of Health (EB-001777) and the National Science Foundation (CHE-0078923, CHE-0243496, and CHE-0640549), and thank XEI Scientific for the loan of the Evactron plasma cleaning system.
Supporting Information Available: Additional UV absorption spectra characterizing in situ DTC formation, SHG data on the adsorption kinetics of DAM assembly, and XPS data describing the density of surface adsorbates. These information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ulman A. Chem. Rev. 1996;96:1533–1554. doi: 10.1021/cr9502357. [DOI] [PubMed] [Google Scholar]
- 2.(a) Flink S, van Veggel FCJM, Reinhoudt DN. Adv. Mater. 2000;12:1315–1328. [Google Scholar]; (b) Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM. Chem. Rev. 2005;105:1103–1170. doi: 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- 3.(a) Storhoff JJ, Mirkin CA. Chem. Rev. 1999;99:1849–1862. doi: 10.1021/cr970071p. [DOI] [PubMed] [Google Scholar]; (b) Templeton AC, Wuelfing MP, Murray RW. Acc. Chem. Res. 2000;33:27–36. doi: 10.1021/ar9602664. [DOI] [PubMed] [Google Scholar]; (c) Drechsler U, Erdogan B, Rotello VM. Chem. Eur. J. 2004;10:5570–5579. doi: 10.1002/chem.200306076. [DOI] [PubMed] [Google Scholar]; (b) Wei A. Chem. Commun. 2006:1581–1591. doi: 10.1039/b515806k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlenoff JB, Li M, Ly H. J. Am. Chem. Soc. 1995;117:12528–12536. [Google Scholar]
- 5.(a) Demers LM, Mirkin CA, Mucic RC, Reynolds RA, Letsinger RL, Elghanian R, Viswanadham G. Anal. Chem. 2000;72:5535–5541. doi: 10.1021/ac0006627. [DOI] [PubMed] [Google Scholar]; (b) Castelino K, Kannan B, Majumdar A. Langmuir. 2005;21:1956–1961. doi: 10.1021/la047943k. [DOI] [PubMed] [Google Scholar]
- 6.Schoenfisch MH, Pemberton JE. J. Am. Chem. Soc. 1998;120:4502–4513. [Google Scholar]
- 7.Dasog M, Scott RWJ. Langmuir. 2007;23:3381–3387. doi: 10.1021/la0627415. [DOI] [PubMed] [Google Scholar]
- 8.(a) Tarlov MJ, Burgess DRF, Gillen G. J. Am. Chem. Soc. 1993;115:5305–5306. [Google Scholar]; (b) Ryan D, Parviz BA, Linder V, Semetey V, Sia SK, Su J, Mrksich M, Whitesides GM. Langmuir. 2004;20:9080–9088. doi: 10.1021/la048443u. [DOI] [PubMed] [Google Scholar]
- 9.Mrksich M, Dike LE, Tien J, Ingber DE, Whitesides GM. Exp. Cell Res. 1997;235:305–313. doi: 10.1006/excr.1997.3668. [DOI] [PubMed] [Google Scholar]
- 10.Flynn NT, Tran TN, Cima MJ, Langer R. Langmuir. 2003;19:10909–10915. [Google Scholar]
- 11.Langry KC, Ratto TV, Rudd RE, McElfresh MW. Langmuir. 2005;21:12064–12067. doi: 10.1021/la0513555. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y, Pérez-Segarra W, Shi Q, Wei A. J. Am. Chem. Soc. 2005;127:7328–7329. doi: 10.1021/ja050432f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Coucouvanis D. Progress in Inorganic Chemistry. Vol. 11. John Wiley and Sons; New York: 1970. pp. 233–371. [Google Scholar]; (b) Heard PJ. In: Progress in Inorganic Chemistry. Karlin KD, editor. Vol. 53. John Wiley and Sons; New York: 2005. pp. 1–70. [Google Scholar]; (c) Hogarth G. In: Progress in Inorganic Chemistry. Karlin KD, editor. Vol. 53. John Wiley and Sons; New York: 2005. pp. 71–560. [Google Scholar]
- 14.Arndt T, Schupp H, Schrepp W. Thin Solid Films. 1989;178:319–326. [Google Scholar]
- 15.(a) Almirall E, Fragoso A, Cao R. Electrochem. Commun. 1999;1:10–13. [Google Scholar]; (b) Cao R, Jr., Diaz A, Cao R, Otero A, Cea R, Serra C. J. Am. Chem. Soc. 2007;129:6927–6930. doi: 10.1021/ja068349v. [DOI] [PubMed] [Google Scholar]
- 16.Li Z-Y, Kosov DS. J. Phys. Chem. B. 2006;110:9893–9898. doi: 10.1021/jp0610665. [DOI] [PubMed] [Google Scholar]
- 17.(a) Morf P, Raimondi F, Nothofer H-G, Schnyder B, Yasuda A, Wessels JM, Jung TA. Langmuir. 2006;22:658–663. doi: 10.1021/la052952u. [DOI] [PubMed] [Google Scholar]; (b) Weinstein RD, Richards J, Thai SD, Omiatek DM, Bessel CA, Faulkner CJ, Othman S, Jennings GK. Langmuir. 2007;23:2887–2891. doi: 10.1021/la062905h. [DOI] [PubMed] [Google Scholar]; (c) Long DP, Troisi A. J. Am. Chem. Soc. 2007;129:15303–15310. doi: 10.1021/ja074970z. [DOI] [PubMed] [Google Scholar]
- 18.Metal nanoparticles: Wessels JM, Nothofer H-G, Ford WE, von Wrochem F, Scholz F, Vossmeyer T, Schroedter A, Weller H, Yasuda A. J. Am. Chem. Soc. 2004;126:3349–3356. doi: 10.1021/ja0377605.. Vickers MS, Cookson J, Beer PD, Bishop PT, Thiebaut B. J. Mater. Chem. 2006;16:209–215.. Tong MC, Chen W, Sun J, Ghosh D, Chen S. J. Phys. Chem. B. 2006;110:19238–19242. doi: 10.1021/jp0631174.
- 19.Semiconductor nanoparticles: Kamat PV, Dimitrijevic NM. J. Phys. Chem. 1989;93:4259–4263.. Dubois F, Mahler B, Dubertret B, Doris E, Mioskowski C. J. Am. Chem. Soc. 2007;129:482–483. doi: 10.1021/ja067742y..
- 20.Colorado R, Villazana RJ, Lee TR. Langmuir. 1998;14:6337–6340. [Google Scholar]
- 21.(a) Woelfel WC. Anal. Chem. 1948;20:722–724. [Google Scholar]; (b) Critchfield FE, Johnson JB. Anal. Chem. 1956;28:430–436. [Google Scholar]
- 22.Vanderah DJ, Parr T, Silin V, Meuse CW, Gates RS, La H, Valincius G. Langmuir. 2004;20:1311–1316. doi: 10.1021/la035829g. [DOI] [PubMed] [Google Scholar]
- 23.(a) Fillon YA, Anderson JP, Chmielewski J. J. Am. Chem. Soc. 2005;127:11798–11803. doi: 10.1021/ja052377g. [DOI] [PubMed] [Google Scholar]; (b) Geisler IM, Chmielewski J. Bioorg. Med. Chem. Lett. 2007:2765–2768. doi: 10.1016/j.bmcl.2007.02.077. [DOI] [PubMed] [Google Scholar]
- 24.Yamada T, Isono N, Inui A, Miyazawa T, Kuwata S, Watanabe H. Bull. Chem. Soc. Jpn. 1978;51:1897–1898. [Google Scholar]
- 25.Pd-catalyzed reductive dimerizations have been reported previously. For some recent examples, see: Lange M, Pettersen AL, Undheim K. Tetrahedron. 1998;54:5745–5752.. An IH, Seong H, Ahn KH. Bull. Korean Chem. Soc. 2004:420–422.
- 26.Zych AJ, Iverson BL. J. Am. Chem. Soc. 2000;122:8898–8909. [Google Scholar]
- 27.Dehen CJ, Everly RM, Plocinik RM, Hedderich HG, Simpson GJ. Rev. Sci. Instrum. 2007;78:013106. doi: 10.1063/1.2400011. [DOI] [PubMed] [Google Scholar]
- 28.Seah MP. Surf. Interface Anal. 1989;14:488. [Google Scholar]
- 29.van Gaal HLM, Diesveld JW, Pijpers FW, van der Linden JGM. Inorg. Chem. 1979;18:3251–3260. [Google Scholar]
- 30.(a) Huff TB, Hansen MN, Zhao Y, Cheng J-X, Wei A. Langmuir. 2007;23:1596–1599. doi: 10.1021/la062642r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huff TB, Tong L, Zhao Y, Hansen MN, Cheng J-X, Wei A. Nanomedicine. 2007;2:125–132. doi: 10.2217/17435889.2.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tong L, Zhao Y, Huff TB, Hansen MN, Wei A, Cheng J-X. Adv. Mater. 2007;19:3136–3141. doi: 10.1002/adma.200701974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee AWM, Chan WH, Chiu CML, Tang KT. Anal. Chim. Acta. 1989;218:157–160. [Google Scholar]
- 32.Wertheim E. J. Am. Chem. Soc. 1926;48:826–830. [Google Scholar]
- 33.(a) Joris SJ, Aspila KI, Chakrabarti CL. Anal. Chem. 1970;42:647–651. [Google Scholar]; (b) Humeres E, Debacher NA, de S. Sierra M, Franco JD, Schutz A. J. Org. Chem. 1998;63:1598–1603. [Google Scholar]
- 34.Shi J, Wei A. Unpublished results.
- 35.Dubois LH, Nuzzo RG. Annu. Rev. Phys. Chem. 1992;43:437–463. [Google Scholar]
- 36.(a) Bain CD, Troughton EB, Tao YT, Evall J, Whitesides GM, Nuzzo RG. J. Am. Chem. Soc. 1989;111:321–335. [Google Scholar]; (b) Hähner G, Woell C, Buck M, Grunze M. Langmuir. 1993;9:1955–1958. [Google Scholar]
- 37.(a) Dannenberger O, Buck M, Grunze M. J. Phys. Chem. B. 1999;103:2202–2213. [Google Scholar]; (b) Tokumitsu S, Liebich A, Herrwerth S, Eck W, Himmelhaus M, Grunze M. Langmuir. 2002;18:8862–8870. [Google Scholar]
- 38.Vanderah DJ, La H, Naff J, Rubinson VSA. J. Am. Chem. Soc. 2004;126:13639–13641. doi: 10.1021/ja047744n. [DOI] [PubMed] [Google Scholar]
- 39.(a) Hutt DA, Leggett GJ. Langmuir. 1997;13:3055–3058. [Google Scholar]; (b) Li Z, Lieberman M, Hill W. Langmuir. 2001;17:4887–4894. [Google Scholar]
- 40.Fadley CS. In: Electron spectroscopy: Theory, techniques and applications. Baker AD, Brundle CR, editors. Vol. 2. Academic Press; New York: 1978. pp. 1–156. [Google Scholar]
- 41.Roper MG, Jones RG. Phys. Chem. Chem. Phys. 2008;10:1336–1346. doi: 10.1039/b715682k. [DOI] [PubMed] [Google Scholar]
- 42.(a) Ron H, Matlis S, Rubinstein I. Langmuir. 1998;14:1116–1121. [Google Scholar]; (b) Ron H, Rubinstein I. J. Am. Chem. Soc. 1998;120:13444–13452. [Google Scholar]
- 43.(a) Tsai H, Hu E, Perng K, Chen M, Wu J-C, Chang Y-S. Surface Sci. 2003;537:L447–450. [Google Scholar]; (b) Ono LK, Cuenya BR. J. Phys. Chem. C. 2008;112:4676–4686. [Google Scholar]
- 44.(a) Zhu B, Angelici RJ. Chem. Commun. 2007:2157–2159. doi: 10.1039/b700555e. [DOI] [PubMed] [Google Scholar]; (b) Pina CD, Falletta E, Rossi M. Topics Catal. 2007;44:325–329. [Google Scholar]
- 45.Rubenstein and coworkers have shown that simple alcohols such as ethanol are sufficient to reduce the Au oxide layer. However, control measurements in our laboratories indicate a decrease in SHG signal when freshly cleaned Au substrates are immersed in EtOH. It is currently unknown whether the time-dependent change in ISHG is due to oxide reduction or a nonspecific chemisorptive process
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.