

Abstract
Microglial cells respond to herpes simplex virus (HSV)-1 by producing proinflammatory cytokines and chemokines. Following this inflammatory burst, these cells undergo apoptotic cell death. We have recently demonstrated that both virus-induced immune mediator production and apoptosis were induced through Toll-like receptor (TLR) 2 signaling. Based upon these findings, we hypothesized that inhibition of TLR2 signaling may serve as a means to alleviate excessive neuroinflammation. In the present study, we cloned four vaccinia virus (VV) proteins which have been reported to disrupt either TLR signaling or NF-κB activation, and overexpressed them in HEK293T cells stably expressing murine TLR2, as well as in primary murine microglia. Using an NF-κB-driven luciferase reporter gene assay, we show that upon stimulation with HSV and Listeria monocytogenes, all four vaccinia proteins inhibited TLR2 signaling, with different levels of inhibition in the TLR2-expressing cell line and primary microglia. We found similar results when microglial cells were stimulated with the TLR4 ligand LPS and the TLR9 ligand CpG ODN. Taken together, these data provide evidence that these VV proteins can function as inhibitors of TLR signaling in primary microglial cells.
Keywords: Microglia, Toll-like receptors, vaccinia virus, NF-κB, HSV
Introduction
Microglial cells are the resident macrophages of the brain that perform surveillance functions to maintain the integrity of central nervous system (CNS). In response to a wide range of invading pathogens, microglia mount innate immune responses that can be characterized by the production of cytokines and chemokines, up-regulation of cell surface molecules, and expansion of local immune responses (Streit, 2004). During the early onset of an infection, microglia rapidly become activated and robustly produce proinflammatory cytokines and chemokines. Production of these immune mediators may result in the infiltration of various leukocytes across the blood-brain barrier to sites of infection (Aloisi, 2001). The effective functioning of microglial cells is critical for controlling neuroinflammation and alleviating neuropathogenesis.
The Toll-like receptor (TLR) family of cell surface proteins recognize pathogen-associated molecular patterns (PAMP) on the surface of pathogens and trigger intracellular signals to generate innate immune responses (Akira, 2006). A number of recent studies have demonstrated that TLRs expressed by microglial cells are critical in identifying and generating innate immune responses against bacterial and viral pathogens in the CNS (reviewed in Aravalli, 2007). In a recent report, TLR2 KO neonatal mice were found to be less susceptible to HSV-1, suggesting that TLR2 plays an important role in herpes encephalitis (Kurt-Jones, 2004). We have previously shown that murine microglial cells respond to HSV-1 by producing a large number of proinflammatory immune mediators in a TLR2-dependent manner (Aravalli, 2005). This immune mediator production was suppressed in human microglia pretreated with IL-10 prior to HSV infection (Marques, 2004). Pretreatment with IL-10 also reduced NF-κB activation by 50% in these cells. HSV infection has also been shown to activate signaling in TLR2- and TLR9-expressing cell lines (Hochrein, 2004; Krug, 2004). Furthermore, microglia have been shown to recognize the major Gram-negative bacterial cell wall component lipopolysaccharide (LPS) through TLR4 (Jung, 2005) and bacterial DNA containing motifs of CpG dinucleotides triggered innate immune responses in murine microglia through TLR9 (Dalpke, 2002). In prototypical TLR signaling, following pathogen recognition, the signal is transmitted through one of the TLR adaptors into the cytoplasm leading to the activation of NF-κB which culminates in the generation of immune responses.
The ubiquitous NF-κB/Rel family of transcription factors regulates expression of genes involved in diverse cellular processes such as proliferation, inflammation, immune responses and apoptosis. Since the activation of NF-κB is an integral part of TLR signaling pathways, it is not surprising that a number of microbial pathogens have developed mechanisms to modulate these pathways (Finlay, 2006). At least four proteins encoded by vaccinia virus (VV) that interfere either directly or indirectly with NF-κB activation have been identified in studies using cell lines. Among them, A46R targets multiple TLR adaptors including MyD88, Mal, TRIF, and TRAM and interferes with activation of NF-κB and MAP kinases (Bowie, 2000; Stack, 2005). Another VV protein, A52R associates with IRAK2 and TRAF6 and blocks NF-κB activation signaled through multiple TLRs (Bowie, 2000; Harte, 2003). The third VV protein, N1L inhibits NF-κB activation by a variety of stimuli including IL-1β, TNF-α, as well as agonists for TLR2, TLR3 and TLR4 (DiPerna, 2004). Fourthly, K1L has been shown to inhibit NF-κB activation in rabbit kidney RK-13 cells by preventing IκBα degradation (Shisler, 2004). In the present study, we tested the ability of these proteins to inhibit TLR signaling in primary murine microglial cells. Results presented here show that these viral proteins inhibit signaling from TLR2, TLR4, and TLR9 stimulated by L. monocytogenes and HSV, LPS, and CpG oligodeoxynucleotides (ODN), respectively. This is the first report to demonstrate endogenous inhibition of TLR signaling in primary microglial cells.
Materials and Methods
Preparation of microglial cultures
Microglial cell cultures were purified from wild-type and TLR2 KO mice (Jackson Laboratories, Bar Harbor, ME) using a method described previously with minor modifications (Chao, 1993). Briefly, cerebral cortical cells from 1-d-old mice were dissociated after a 30 min trypsinization (0.25%) and plated in 75-cm2 Falcon culture flask in DMEM (Sigma-Aldrich, St. Louis, MO) containing 10% heat-inactivated FBS (Hyclone Laboratories, Logan, UT) and penicillin/streptomycin (Sigma-Aldrich). The medium was replenished 1 and 4 d after plating. On d 8 of culture, flasks were shaken for 20 min at a speed of 180 rpm in an orbital shaker to remove unattached cells. On d 12 of culture, microglia floating in the media were collected by aspiration, pooled, centrifuged and seeded at appropriate densities after counting. The cells were washed twice with fresh medium 1 h after seeding to remove non-adherent cells. Microglia prepared this way stain 95-98% positive with Mac-1 antibody (Roche Applied Science, Indianapolis, IN). Cells were infected with the highly neurotropic HSV-1 strain 17syn+, propagated and purified on rabbit skin fibroblasts, at multiplicity of infection (MOI) of 2.
Cloning of VV TLR inhibitors
DNA obtained from the VV Western Reserve strain was used to clone four viral gene products: A46R, A52R, N1L and K1L using PCR. Primers used for amplification were: A46R: Forward: 5′-CAT GCC ATG GCG TTT GAT ATC AGT-3′ and Reverse: 5′-CAT GCC ATG GAT GGC GTT TGA TAT-3′; A52R: Forward: 5′-CAT GCC ATG GAC ATA AAG ATA GAT-3′ and Reverse: 5′-GTG GAA ATG TCA TAG GCT AGC TAG-3′; N1L: Forward: 5′-CAG GTC ATG AGG ACT CTA CTT ATT-3′ and Reverse: 5′- CTA GCT AGC TTA TTT TTC ACC ATA-3′; K1L: Forward: 5′-CAG GAT ATC ATG GAT CTG TCA CGA-3′ and Reverse: 5′-CTA GCT AGC TTA GTT TTT CTT TAC AC -3′. PCR was performed on a Gradient 40 Robocycler (Stratagene, La Jolla, CA) using Pfu polymerase (Stratagene) with the following conditions: initial denaturation at 95°C for 2 min 30 sec, followed by 30 cycles of 95°C for 1 min, annealing at 60°C for 1 min and elongation at 72°C for 3 min. Following PCR amplification, viral gene products were purified using a 0.8% agarose gel and were cloned into pORF5-mIL10 (InvivoGen) by replacing the mIL-10 ORF with each VV ORF. This vector carries the murine IL-10 ORF under the control of a composite binary promoter comprised of the elongation factor 1α (EF-1α) and the 5′ untranslated region of the human eukaryotic initiation factor 4g (eIF-4g). The expression vectors thus generated were termed pORF5-A46R, pORF5-A52R, pORF5-N1L and pORF5-K1L. Expression of these viral proteins was confirmed using Western blot analysis.
Construction of stable cell line expressing murine TLR2
HEK293T cells were transfected with the pUNO-mTLR2 plasmid (InvivoGen), which expresses murine TLR2 under the control of a composite promoter comprising of the eukaryotic elongation factor-1α (EF-1α) core promoter and the R segment and part of the U5 sequence of the human T-cell leukemia virus type 1 long terminal repeat, using the Fugene 6 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s recommendations. The transfectants were selected in RPMI medium containing penicillin/streptomycin and 10 μg/ml blasticidin (InvivoGen). The transfected cells multiplied and formed single clones in 6 well plates. After 2 d, six clones were picked up and were further grown for 7 d in growth medium under blasticidin selection. Cells from each clone were then harvested and the expression of TLR2 was analyzed by quantitative real time PCR using primers specific for murine TLR2. Murine TLR2 primer sequences used in the study were: Forward: 5′-TGC TTT CCT GCT GGA GAT TT-3′ and Reverse: 5′-TGT AAC GCA ACA GCT TCA GG-3′. The clone that showed highest expression of mTLR2 by real-time PCR was selected for further experiments.
Luciferase assay
HEK293T cells, as well as wild-type and TLR2 KO microglia, were transfected with 1 μg pNiFty2-Luc plasmid (InvivoGen) expressing an NF-κB-driven firefly luciferase reporter gene. FuGene 6 was used for transfection of the 293T cells. Primary microglia are post-mitotic cells which are extremely difficult to transfect using standard methods. In this study, they were successfully transfected using the mouse macrophage nucleofection kit (Amaxa Biosystems, Gaithersburg, MD) and the program Y-01 on the nucleofector I device (Amaxa). Although the transfection efficiency using nucleofection was still low (<10%), luciferase expression occured only in cells that took up the pNiFty2-Luc plasmid. Following nucleofection, the cells were plated in 12-well plates and incubated overnight at 37°C. To stimulate TLR signaling, TLR2 ligands HSV 17+ syn at a MOI of 2 and 0.01% heat-killed L. monocytogenes (InvivoGen), TLR4 ligand LPS (100 ng/ml), and 5 μg/ml of TLR9 ligand phosphodiester CpG oligonucleotide ODN1826 (InvivoGen) were added for 5 h. The cells were then lysed and luciferase activity was measured using Bright-Glo luciferase assay substrate (Promega, Madison, WI) on the IVIS® Imaging System (Xenogen Corporation, Alameda, CA). Expression levels of the luciferase reported gene were quantified using Living Image® software (Xenogen).
Results
The VV protein A46R interferes with TLR signaling by targeting multiple TLR adaptors (Bowie, 2000; Stack, 2005), whereas A52R blocks TLR4-induced NF-κB activation by associating with the downstream molecule IRAK2 (Harte, 2003). Although the involvement of N1L and K1L in TLR signaling has not specifically been demonstrated, they have been shown to inhibit NF-κB activation by preventing release from its inhibitior IκB (DiPerna, 2004; Shisler, 2004). Since NF-κB activation is a critical step in the TLR pathway, it is likely that these viral proteins also inhibit TLR signaling. To test this hypothesis, we have amplified ORFs encoding each of these viral proteins and cloned them into pORF5 vector downstream of a strong ubiquitous EF-1α/eF-4g hybrid promoter. In addition, we have constructed a stable 293T-mTLR2 cell line that expresses murine TLR2. To test the activation of NF-κB that occurs as a result of TLR stimulation, we have devised an experimental strategy that relies upon a highly sensitive luciferase gene assay (Fig 1). The amount of luciferase produced in this experimental setting directly correlates with the level of NF-κB activation within the cells. Expression of VV proteins from each plasmid constructs was verified by Western blot analysis following transfection into 293T-mTLR2 cells (Fig. 2A). Additionally, these proteins, with the exception of A46R, were also able to inhibit the low basal level of NF-κB activation that occurred in the absence of TLR ligands (Fig. 2A).
Fig 1.

Experimental design. Induction of TLR signaling by HSV, LPS, and CpG ODN1826 leads to the activation of IRAK2 and NF-κB. NF-κB is then released from its inhibitor IκB and translocates into the nucleus, where it binds to five NF-κB binding sites on the pNiFty2-Luc plasmid, and activates transcription to produce luciferase. VV A46R targets multiple TLR adaptors including MyD88, Mal, and TRAM, and A52R associates with IRAK-1 and TRAF6 to disrupt downstream signaling. VV N1L and K1L proteins prevent the release of NF-κB from IκB. Expression of these proteins from their corresponding pORF5-VV plasmid leads to the inhibition of NF-κB activation and decreased luciferase expression from pNiFty2-Luc.
Fig 2.
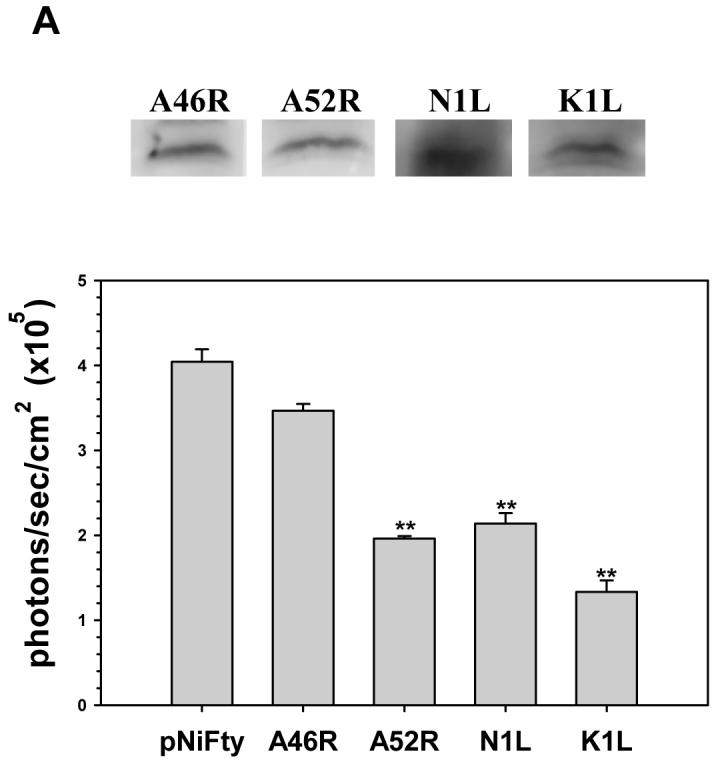

Inhibition of TLR2-mediated NF-κB activation by VV A46R, A52R, N1L and K1L proteins in 293T cells. A stable 293T cell line expressing the murine TLR2 protein was constructed (293T-mTLR2). Expression plasmids carrying the open-reading frames of VV proteins A46R, A52R, N1L, or K1L were transiently co-transfected into this stable cell line, together with pNiFty2-Luc. A) Expression of the VV proteins in 293T-mTLR2 cells from the vector constructs was analysis by Western blot using antibodies specific for each protein (upper panel). The ability of these VV proteins to inhibit NF-κB activation in these cells was tested by performing the luciferase assay as outlined in Fig. 1. B) Following transient transfection, 293T-TLR2 cells were incubated overnight at 37° C and were then stimulated with heat-killed Listeria monocytogenes (HKLM). Cells were harvested at 5 h post-stimulation and the amount of luciferase produced was quantified using the bright glow substrate (Promega). Data presented are mean values + SD and are representative of at least three independent experiments. Statistical analysis was performed using student t test. *P <0.05; **P<0.01
Inhibition of TLR2 signaling induced by L. monocytogenes
In these experiments, 1 μg of each pORF5-VV plasmid was co-transfected into 293T-mTLR2 cells together with 1 μg of the NF-κB-driven reporter gene containing plasmid pNiFty2-Luc. After overnight incubation at 37 °C, cells were treated with heat-killed L. monocytogenes (HKLM), which has previously been shown to trigger signaling from TLR2 (Torres, 2004), for 5 h. Cells were harvested and the expression levels of luciferase in the transfected cells was measured by luciferase assay. As shown in Fig 2B, NF-κB activation was severely impaired in cells expressing each of these viral proteins when compared to cells expressing pNiFty2-Luc alone, a result which demonstrates that all four viral proteins were able to inhibit HKLM-induced TLR2 signaling in 293T-mTLR2 cells.
NF-κB activation in wild-type and TLR2 KO murine microglial cells
We have shown previously that HSV infection triggers signaling through TLR2 in murine microglia (Aravalli, 2005). In order to deduce the activation levels of NF-κB in these cells during HSV infection, 1 × 106 microglial cells from both wild type and TLR2 KO mice were transfected with pNiFty2-Luc plasmid and, after overnight incubation, they were infected with HSV at a MOI of 2. At 5 h p.i., activation of NF-κB was measured using a luciferase reporter gene assay. As shown in Fig. 3A, HSV infection stimulated TLR2 signaling in wild type microglial cells as evidenced by high level expression of luciferase. In comparison, luciferase expression in TLR2 KO microglia was markedly reduced.
Fig 3.

A) Activation of NF-κB in primary murine microglia following infection with HSV 17syn+. Purified microglial cells (1 × 106) from wild-type and TLR2 knockout mice were transiently transfected with pNiFty2-Luc plasmid by electroporation using the mouse macrophage nucleofector kit (Amaxa). The cells were incubated overnight at 37°C and were then infected with HSV at an MOI of 2. They were harvested at 5 h p.i. and the amount of luciferase produced as a result of NF-κB activation was quantified using the bright glow substrate. B) Inhibition of TLR2-mediated, HSV-induced NF-κB activation in primary murine microglia by VV proteins. Plasmids carrying the open-reading frames of A46R, A52R, K1L, or N1L were co-transfected along with pNiFty2-Luc into primary murine microglial cells (1 × 106) by electroporation. The cells were incubated overnight at 37°C and infected with HSV at an MOI of 2. Cells were harvested at 5 h p.i. and the amount of luciferase produced was quantified using bright glow substrate. Data presented are mean values + SD and are representative of at least three independent experiments. Statistical analysis was performed using student t test. *P <0.05; **P<0.01
VV proteins inhibit TLR2 signaling in primary murine microglia
Having demonstrated the inhibition of TLR2 signaling by VV proteins in 293T cells (Fig 2B), we went on to examine their ability to inhibit TLR2 signaling in primary murine microglial cells. 1 μg of plasmid DNA carrying the open-reading frames of VV proteins A46R, A52R, K1L and N1L, and 1 μg of pNiFty2-Luc, were separately co-transfected into primary murine microglial cells by nucleofection. Following incubation overnight at 37° C, the microglia were infected with HSV at an MOI of 2. At 5 h p.i., cells were harvested and the activation of NF-κB was measured by luciferase reporter gene assay. Similar to the experiment with the 293T cell line, all four VV proteins inhibited TLR2 signaling in primary microglial cells (Fig 3B). The expression of luciferase in cells expressing viral proteins was markedly lower than in cells expressing pNiFty2-Luc alone.
Expression of VV proteins in microglia inhibits TLR4 and TLR9 signaling
As MyD88 is a common adaptor protein for all TLRs, except TLR3, it is possible that A46R could also inhibit signaling from these TLRs as well. Similarly, since A52R, N1L, and K1L proteins inhibit IRAK and NF-κB, they might possess the potential to block signaling from other TLRs, as these downstream molecules are involved (Akira, 2006). To test these hypotheses, 1 × 106 microglial cells were transfected with 1 μg pNiFty2-luc and 1 μg pORF5-VV containing open-reading frame for each VV protein by nucleofection. Following overnight incubation at 37° C, the cells were stimulated with the TLR4 and TLR9 ligands LPS and CpG ODN, respectively. After a 5 h treatment with ligands, cells were harvested and luciferase expression was determined. Luciferase expression was inhibited in cells expressing N1L and K1L demonstrating that these viral proteins could inhibit signaling from TLR4 and TLR9 as well (Fig. 4).
Fig 4.

Inhibition of TLR4- and TLR9-mediated NF-κB activation in primary murine microglia by VV proteins. Plasmids encoding the open-reading frames of A46R, A52R, K1L, or N1L were cotransfected along with pNiFty2-Luc into primary murine microglial cells (1 × 106) by electroporation. The cells were incubated overnight at 37°C and were subsequently treated with the TLR4 ligand LPS and the TLR9 ligand CpG ODN1826 for 5h. Cells were harvested and the amount of luciferase produced was quantified using the bright glow substrate. Data presented are mean values + SD and are representative of at least three independent experiments. Statistical analysis was performed by student t test. *P <0.05; **P<0.01
Discussion
Successful evasion of host immune responses by microbial pathogens may result in severe infection and disease. Virulence strategies used by pathogens are diverse, and involve various mechanisms such as inhibition of cytokines and chemokines, blocking apoptosis, and interfering with cellular pathways such as TLR signaling (Alcami, 2003; Finlay, 2006). In recent years the engagement of TLRs has been shown to mediate cellular signaling pathways which lead to the generation of innate immune responses in a number of experimental systems, including the CNS (Aravalli, 2007). We show here a mechanism for inhibiting TLR expression in primary microglial cells. Our data demonstrate that expression of four VV proteins from vector constructs inside the cell successfully inhibited signaling from TLR2, TLR4, and TLR9 in murine microglia.
VV is a large poxvirus that can infect a broad range of mammalian species. It encodes two proteins: A46R and A52R, that were previously shown to inhibit TLR signaling in cell lines, share amino acid sequence similarity with the Toll/IL-1 receptor (TIR) domain (Bowie, 2000; Harte, 2003). When expressed in mammalian cells, these proteins have been shown to inhibit TLR- and IL-1-mediated NF-κB activation through a mechanism involving myeloid differentiation factor 88 (MyD88), a TIR domain-containing adaptor molecule essential for most types of TLR signaling (Bowie, 2000; Stack, 2005). The expression of these proteins during infection represents a mechanism by which VV subverts the innate immune response. Viral deletion mutants in these genes are attenuated in murine infection models (Stack, 2005). Interestingly, all four VV proteins were able to inhibit NF-κB activation in microglia with differential effects, which could be attributed to the nature of the stimulus. For instance, K1L inhibits the degradation of IαBα (Shisler, 2004) and N1L was suggested to target downstream molecules in the NF-κB pathway (DiPerna, 2004). While K1L was more effective than N1L in inhibiting NF-κB when cells were stimulated with CpG ODN, the opposite effect was observed with LPS (Fig 4). Recently it was shown that stimulus-specific induction of TLR2/4/9 signaling will induce IκBζ to interact with the p50 subunit of NF-κB (Yamazaki, 2005). This IκBζ/p50 dimer binds to a κB site in the promoter region of proinflammatory cytokines such as IL-6 and induces their expression. Similar to K1L-IαBα interactions, VV proteins tested in this study may interact with IκBζ to inhibit NF-κB activation. Additionally, the inhibition of NF-κB activation by VV proteins observed in microglia could be underestimated, due to cytokine release from untransfected cells triggering NF-κB activation in transfected cells via alternate pathways. In comparison, the inhibition of Listeria-induced TLR2 signaling by VV proteins in the 293T cell line could be more pronounced because of higher transfection efficiency, lack of other TLR expression which would eliminate possible redundancy, as well as lack of other bystander effects such as TNF and other cytokine pathways that induce NF-κB activation. However, caution should be used when interpreting overexpression studies as overexpression of foreign proteins could cause non-specific disruptions to other cellular functions.
Microglia and other brain macrophages are the principle regulators of neuroinflammation within the central nervous system (CNS). Our recent studies have demonstrated that HSV infection of microglia stimulates production of a large number of cytokines and chemokines through a TLR2-dependent mechanism (Aravalli, 2005). We also showed that production of these mediators was attenuated by treatment with anti-inflammatory cytokines, and that this inhibition was associated with decreased HSV-induced activation of NF-κB (Marques, 2004). Following this burst of immune mediator production, microglial cells rapidly undergo apoptosis (Aravalli, 2006). In this study, we attempted to inhibit TLR signaling using a novel strategy involving the expression of VV proteins within the cells prior to stimulation with a TLR ligand. We found that overexpression of these proteins clearly inhibited NF-κB activation, a finding which implies that microglial cells expressing these proteins would also produce proinflammatory immune mediators at reduced levels. Therefore, these viral proteins could possibly be used to modulate TLR signaling and potentially to alleviate excessive neuroinflammation. Further studies using these viral TLR inhibitors are expected to pave the way towards the eventual modulation of microglial cell immune responses with the ultimate goal of improving treatments for CNS infections and neuroinflammatory diseases.
Acknowledgements
This work was supported by United States Public Health Service Award MH066703. The authors thank Dr. Andrew Bowie (Trinity College, Ireland) for A46R and A52R antibodies, Dr. Geoffrey Smith (Imperial College London, UK) for the N1L antibody, and Dr. Joanna Shisler (University of Illinois, Urbana) for the K1L antibody.
References
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Alcami A. Viral mimicry of cytokines, chemokines and their receptors. Nature Rev Immunol. 2003;3:36–50. doi: 10.1038/nri980. [DOI] [PubMed] [Google Scholar]
- Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- Aravalli RN, Hu S, Rowen TN, Gekker G, Lokensgard JR. Differential apoptotic signaling in primary glial cells infected with herpes simplex virus 1. J Neurovirol. 2006;12:501–510. doi: 10.1080/13550280601064921. [DOI] [PubMed] [Google Scholar]
- Aravalli RN, Hu S, Rowen TN, Palmquist J, Lokensgard JR. Cutting Edge: TLR2-mediated production of proinflammatory cytokines and chemokines by microglial cells in response to herpes simplex virus. J Immunol. 2005;175:4189–4193. doi: 10.4049/jimmunol.175.7.4189. [DOI] [PubMed] [Google Scholar]
- Aravalli RN, Peterson PK, Lokensgard JR. Toll-like receptors in defense and damage of the central nervous system. J Neuroimmune Pharmacol. 2007 doi: 10.1007/s11481-007-9071-5. in press. [DOI] [PubMed] [Google Scholar]
- Bowie A, Kiss-Toth E, Symons JA, Smith GL, Dower SK, O’Neill LA. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc Natl Acad Sci U S A. 2000;97:10162–10167. doi: 10.1073/pnas.160027697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao CC, Molitor TW, Hu S. Neuroprotective role of IL-4 against activated microglia. J Immunol. 1993;151:1473–1481. [PubMed] [Google Scholar]
- Dalpke AH, Schafer MK, Frey M, Zimmermann S, Tebbe J, Weihe E, Heeg K. Immunostimulatory CpG-DNA activates murine microglia. J Immunol. 2002;168:4854–4863. doi: 10.4049/jimmunol.168.10.4854. [DOI] [PubMed] [Google Scholar]
- DiPerna G, Stack J, Bowie AG, Boyd A, Kotwal G, Zhang Z, Arvikar S, Latz E, Fitzgerald KA, Marshall WL. Poxvirus protein N1L targets the I-kappaB kinase complex, inhibits signaling to NF-kappaB by the tumor necrosis factor superfamily of receptors, and inhibits NF-kappaB and IRF3 signaling by toll-like receptors. J Biol Chem. 2004;279:36570–36578. doi: 10.1074/jbc.M400567200. [DOI] [PubMed] [Google Scholar]
- Finlay BB, McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell. 2006;124:767–782. doi: 10.1016/j.cell.2006.01.034. [DOI] [PubMed] [Google Scholar]
- Harte MT, Haga IR, Maloney G, Gray P, Reading PC, Bartlett NW, Smith GL, Bowie A, O’Neill LA. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J Exp Med. 2003;197:343–351. doi: 10.1084/jem.20021652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochrein H, Schlatter B, O’Keefe M, Wagner C, Schmitz F, Schiemann M, Bauer S, Wagner H. Herpes simplex virus type-1 induces IFN-α production via toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci U S A. 2004;101:11416–11421. doi: 10.1073/pnas.0403555101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung DY, Lee H, Jung BY, Ock J, Lee MS, Lee WH, Suk K. TLR4, but not TLR2, signals autoregulatory apoptosis of cultured microglia: a critical role of IFN-beta as a decision maker. J Immunol. 2005;174:6467–6476. doi: 10.4049/jimmunol.174.10.6467. [DOI] [PubMed] [Google Scholar]
- Krug A, Luker GD, Barchet W, Lieb DA, Akira S, Colonna M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through Toll-like receptor 9. Blood. 2004;103:1433–1437. doi: 10.1182/blood-2003-08-2674. [DOI] [PubMed] [Google Scholar]
- Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci USA. 2004;101:1315–1320. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques CP, Hu S, Sheng W, Cheeran MC, Cox D, Lokensgard JR. Interleukin-10 attenuates production of HSV-induced inflammatory mediators by human microglia. Glia. 2004;47:358–366. doi: 10.1002/glia.20045. [DOI] [PubMed] [Google Scholar]
- Shisler JL, Jin XL. The vaccinia virus K1L gene product inhibits host NF-κB activation by preventing IκBα degradation. J Virol. 2004;78:3553–3560. doi: 10.1128/JVI.78.7.3553-3560.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack J, Haga IR, Schroder M, Bartlett NW, Maloney G, Reading PC, Fitzgerald KA, Smith GL, Bowie AG. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J Exp Med. 2005;201:1007–1018. doi: 10.1084/jem.20041442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Mrak RE, Griffin WST. Microglia and neuroinflammation: a pathological perspective. J Neuroinflamation. 2004;1:14. doi: 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres D, Barrier M, Bihl F, Quesniaux VJ, Maillet I, Akira S, Ryffel B, Erard F. Toll-like receptor 2 is required for optimal control of Listeria monocytogenes infection. Infect Immun. 2004;72:2131–2139. doi: 10.1128/IAI.72.4.2131-2139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki S, Muta T, Matsuo S, Takeshige K. Stimulus-specific induction of a novel nuclear factor-κB regulator, IκB-ζ, via Toll/Interleukin-1 receptor is mediated by mRNA stabilization. J Biol Chem. 2005;280:1678–1687. doi: 10.1074/jbc.M409983200. [DOI] [PubMed] [Google Scholar]