

Abstract
Compstatin and its active peptide analogs can potentially be used for therapeutic purposes because their binding to C3 prohibits its conversion into C3b, thus inhibiting complement cascades in all three complement pathways. Mallik and Morikis built three quasi-dynamic pharmacophore models for compstatin peptide analogs before, but only nine compstatin peptide analogs were incorporated in their study and the most active compstatin analog had only medium inhibitory activity. Since then, many more compstatin analogs have been synthesized and their inhibitory activities tested. Furthermore, the x-ray structure of AcCompNH2-V4W-H9A bound to C3 has become available (PDB ID: 2QKI). In this paper, we utilized all the new information and built a new pharmacophore model using a distinct approach. Our model demonstrated good performance in a separate test set of 82 compstatin analogs: it accurately identified 70% of the analogs of medium or high inhibitory activities and misclassified only 8.5% of the analogs of low or no inhibitory activities. The results proved our pharmacophore model to be a filter of great sensitivity and specificity and showed great promise for future identification of small-molecule complement C3 inhibitors.
Introduction
The third component of complement, C3, is the key player in all three activation pathways of the complement system1. Complement cascades launch drastically after C3 is converted into C3b by a C3 convertase and C3b binds to the surface of a bacterium1. Inappropriate activation of a complement cascade results in host cell damage, which has been present in pathological conditions such as autoimmune diseases, adult respiratory syndrome, stroke, heart attack, post-xenotransplant rejection, cardiopulmonary bypass, dialysis and burn injuries1,2. Therefore, C3 is a target protein of much therapeutic interest.
Using a phage-displayed random peptide library, Sahu et al. identified a complement inhibitor, compstatin, which is a 13-residue cyclic peptide with the sequence: ICVVQDWGHHRCT-NH2 (abbreviated as CompNH2)3. The two cysteine residues of compstatin, C2 and C12, formed a disulfide bond. Unlike other complement inhibitors, compstatin and its active peptide analogs are unique and powerful in that they bind to C3, prohibit different C3 convertases from converting C3 to C3b and thus inhibit complement cascades in all three complement pathways.
Previously, Mallik and Morikis4 built three quasi-dynamic pharmacophore models for compstatin analogs. Their models were based on only nine compstatin peptide analogs and the most active compstatin analog in their training set had only a 45-fold inhibitory activity compared with compstatin. Since then, additional compstatin analogs have been synthesized and their inhibitory activities tested. Our understanding of the structure-activity relationships of comsptatin analogs has also dramatically increased5–13. In addition, potent compstatin analogs have been discovered: AcCompNH2-V4(1MeW)-H9A and AcCompNH2-V4(1MeW)-W7(5fW)-H9A, both of which had IC50 values of 0.2 µM (a 264-fold inhibitory activity compared with compstatin)11. Furthermore, the x-ray structure of AcCompNH2-V4W-H9A bound to C3c fragment of C3 has become available (PDB ID: 2QKI)12. With the availability of all these new findings, the development of a new pharmacophore model is called for.
In this work, we utilized all these new findings and built a new pharmacophore model for compstatin analogs. Specifically, six structural features that play critical roles in determining inhibitory activities were identified based on the structure-activity relationships of nine comsptatin analogs. In addition, the availability of the x-ray structure of AcCompNH2-V4W-H9A bound to C3 made it possible for us to predict the bound conformations of the other 91 compstatin analogs using state-of-the-art modeling tools. Subsequently, the predicted bound conformations of the two most active compstatin analogs, AcCompNH2-V4(1MeW)-H9A and AcCompNH2-V4(1MeW)-W7(5fW)-H9A, were utilized to build all six structural features into the pharmacophore model.
The sensitivity and specificity of this pharmacophore model were tested. Using a separate test set of 82 compstatin analogs, we demonstrated that our pharmacophore model was able to identify 70% of the analogs of medium or high inhibitory activities and misclassify only 8.5% of the analogs of low or no inhibitory activities. The superior results proved our pharmacophore model to be a filter of great sensitivity and specificity and showed great promise for future identification of small-molecule complement C3 inhibitors.
Methods
In this paper, our goal is the development and evaluation of a pharmacophore model for compstatin analogs. The development of our pharmacophore model consists of three steps. First, we identified the structural features that play a critical role in inhibitory activity based on the structure-activity relationships of compstatin analogs. Second, we predicted the bound conformations of all other compstatin analogs using as the template the x-ray structure of AcCompNH2-V4W-H9A bound to C3 (PDB ID: 2QKI). Thirdly, we built the pharmacophore model using the predicted bound conformations of the two most active compstatin analogs.
Identification of Critical Structural Features
Based on the previous experimental data on compstatin analogs, the following structure-activity relationships have been derived. We discuss them here, summarizing the important literature findings and focusing our investigation to known important structural features. First, it was found previously that the following Ala scans, V3A, Q5A, D6A, W7A and G8A, resulted in significant decreases in activities. This finding indicated that V3, Q5, D6, W7 and G8 were important residues for activities7. Second, the substitution of Val4 with Trp increased activity to 12-fold (24.364 compared with compstatin versus 2.961 compared with compstatin)6. It was proposed that the aromatic side chain caused the activity increase. Thirdly, the Q5N mutation also yielded a similar activity compared with parent compstatin (2.857 versus 1)8, indicating that the amide groups at the tail of both side chains were crucial for keeping activity. This has been confirmed by the modeled complex structures in which the CO and NH2 of both side chains acted as hydrogen bond acceptors and donors, respectively, to the surrounding C3 residues. In particular, the carbonyl oxygen on the parent compstatin Q5 side chain acted as a hydrogen bond acceptor to the backbone amide nitrogen on C3 M457. In addition, the amide nitrogen on the Q5 side chain acted as a hydrogen bond acceptor to the backbone carbonyl oxygen on C3 L455 as well as the [O−] anion on the C3 D491 side chain. For the Q5N mutant, the amide nitrogen on the parent compstatin N5 side chain acted as a hydrogen bond acceptor to the [O−] anion on the C3 D491 side chain. In Figure 1, illustrations of the interactions between comstatin analogs and C3 are shown. In addition, the D6P mutant only carried 1/10 of the parent compstatin activity (0.122 versus 1)9. Since both D6A and D6P mutations resulted in significant activity decreases, the carbonyl oxygen on the Asp side chain seemed to play an important role in retaining the activities. Indeed, the modeled complex structure showed that the carbonyl oxygen acted as a hydrogen bond acceptor to the amide nitrogen of C3 R459 side chain. Moreover, the mutation of V4W/H9A to V4W/W7(1MeW)/H9A resulted in a significant decrease in activity. The IC50 value for V4W/H9A was 1.2 µM while the activity of V4W/W7(1MeW)/H9A was too low to be measured experimentally11. It was proposed that the indole NH of W7 formed a hydrogen bond with the C3 residue and that mutating W7 to 1MeW disabled its hydrogen-bond donor capability completely. This was confirmed in modeled structures where the hydrogen bond was formed between the indole nitrogen of W7 and the backbone carbonyl oxygen of M457. Lastly, breaking the disulfide bond between C2 and C12 resulted in a significant decrease in the inhibitory activity (IC50 of Comp-linear: >600 µM), indicating the importance of disulfide bond in retaining activity. Indeed, the NMR studies conducted by Morikis et al. 8 showed that the disulfide bridge brings together the termini of compstatin thus contributing to the formation of the hydrophobic patch, which in turn is necessary for recognition and binding to C3.
Figure 1.
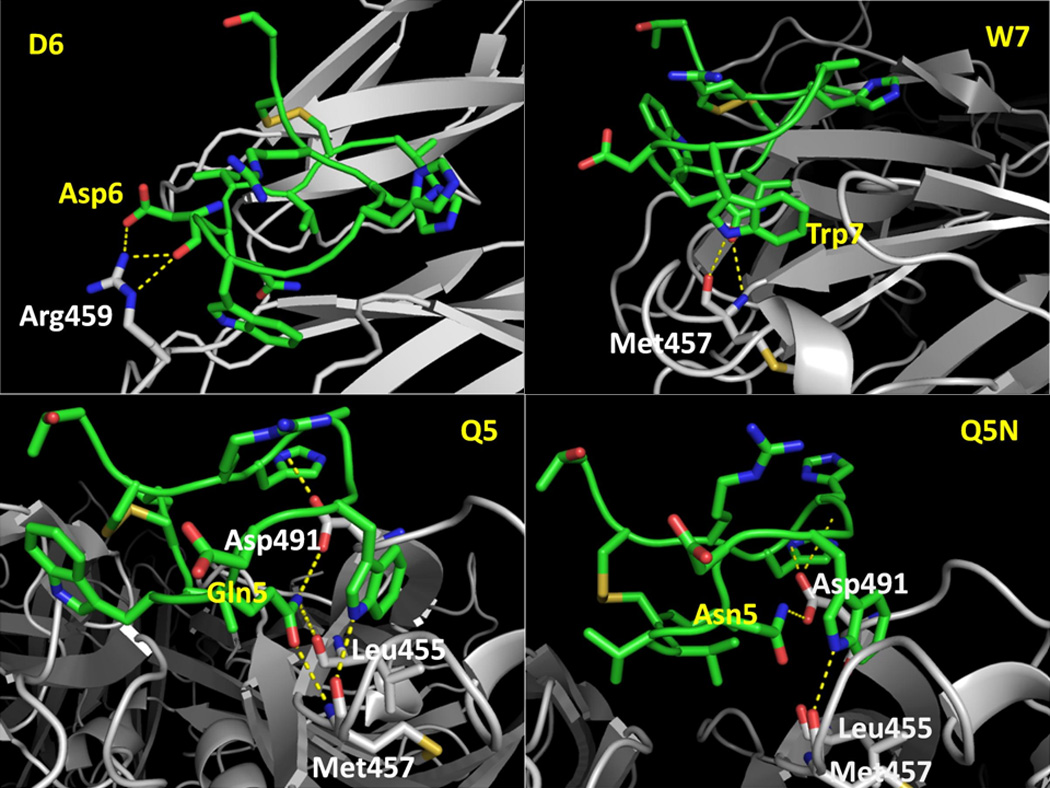
Snapshots of four comstatin analogs illustrating specific interactions that stabilize the interactions with C3. Compstatin is colored in green. Its residues are labeled in yellow. C3 is colored in grey. Its residues are labeled in white. The hydrogen bonds are shown in dotted yellow lines.
Structures of compstatin analogs
Natural amino acid compstatin peptide analogs
The bound conformation of AcCompNH2-V4W-H9A (PDB ID: 2QKI)12 was used as the template to build the bound conformations of all other compstatin analogs. Using the Rotamer Explorer function inside of MOE14, each individual compstatin peptide analog was computationally mutated and the most favorable rotamer of each mutated residue was adopted. The most favorable rotamer was determined based on the lowest DeltaG14. DeltaG is the sum of the change in free energy between the states where the protein and rotamer are separated and when they form a complex, plus the torsional strain of the rotamer. Subsequently, each individual peptide analog and its surrounding C3 residues (within 5 Å of distance) were selected for energy minimizations. In other words, each individual peptide analog and its surrounding C3 residues (within 5 Å of distance) were allowed to move while other residues were kept rigid in energy minimizations. The energies were minimized to a gradient of 0.001 using Amber99 force field and the generalized Born solvation model in MOE.
Compstatin peptide analogs with unnatural amino acid residues
The partial charges of the unnatural amino acid residues were derived separately using the RED program developed by Pigache et al.15. The structures of the unnatural amino acid residues were subsequently optimized using Gaussian (Hartree-Fock) with the 6-31G* basis set. The whole mutant peptides were constructed inside of MOE and the partial charges of unnatural amino acid residues were modified to those calculated by RED in the previous step. The peptide analog as well as the C3 residues within 5 Å of distance to the peptide analog were selected for energy minimizations. The energies were minimized to a gradient of 0.001 using Amber99 force field in MOE.
Construction of the Pharmacophore Model
Alignment of the minimized structures of most active peptide analogs
The pharmacophore model were developed using the minimized structures of the two most active compstatin peptide analogs, AcCompNH2-V4(1MeW)-H9A and AcCompNH2-V4(1MeW)-W7(5fW)-H9A. Both carried 261-fold activities compared with parent compstatin. Their minimized structures were aligned using the Homology/Align function of MOE with default parameters. Figure 2 shows the overlay of the two peptide analogs. As can be seen, the two peptide analogs were very well aligned except for the flexible side chains of Ile1, Asp6 and His10 where they had the largest discrepancies.
Figure 2.

The superposition of the six pharmacophore features over the aligned structures of the two most active compstatin analogs, AcCompNH2-V4(1MeW)-H9A and AcCompNH2-V4(1MeW)-W7(5fW)-H9A. Aro: Aromatic ring center. Acc: Hydrogen-bond acceptor. Don: Hydrogen-bond donor. Hyd: Hydrophobic region.
Building pharmacophore features
Each individual structural feature was built into a single pharmacophore feature using the Pharmacophore Modeling function of MOE. For W4, the aromatic ring on the side chain is important. Thus, an aromatic sphere centered at the center of mass of the indole ring was built. The default radius of 1.2 Å was accepted. For Q5, the amide group on the tail of the side chain plays an important role in keeping the activities. Specifically, the amide carbonyl and the amide nitrogen served as a hydrogen-bond donor and a hydrogen-bond acceptor, respectively. Therefore, we created two pharmacophore features on this residue: A hydrogen-bond acceptor of 0.8 Å in radius by default centered at the carbonyl oxygen and a hydrogen-bond donor of 0.8 Å in radius by default centered at the amide nitrogen. For D6, the carbonyl oxygen acted as a hydrogen bond acceptor to the surrounding C3 residue the amide nitrogen on the C3 R459 side chain. Thus, we built a feature on the carbonyl oxygen of the D6 side chain. The default feature assigned by MOE was hydrogen-bond donor of 2.2 Å radius. Since we are interested in using our pharmacophore model to find drug-like small molecules in the future and molecules with anions are not good drug candidates in general, we modified the feature to hydrogen-bond donor only. The default radius (0.8 Å) of this feature, was too small to cover the space where the of the two most active compstatin analogs occupied, we increased the radius to 2.2 Å For W7, we added a hydrogen-bond donor feature to the indole nitrogen of W7 given its importance in keep the activities. The default radius of 0.8 Å was adopted. Finally, we added a hydrophobe feature between the two sulfur atoms of C2 and C12. The default radius of 0.7 Å was kept.
Pre-processing the database of known compstatin analogs
In order to facilitate the subsequent pharmacophore searches, the minimized structures of compstatin peptide analogs were pre-processed using the Pharmacophore Preprocessor function inside of MOE with the scheme set to PCH. Running the preprocessor added an annotation field, PH4:PCH, to the minimized structure of each compstatin peptide analog. The annotation field is an encoding of the positions, labels, and indices of the annotation points of the compstatin peptide analogs.
Selection of test set
We incorporated most of the compstatin analogs published in the literature5–11 into our test set but excluded the following: (1) retro-inverso compstatin9 and three biotynylated analogs9 due to their significant differences from compstatin analog in the x-ray structure (PDB ID: 2QKI); (2) three D6P9 and one T13P10 mutants because it was not possible to generate appropriate conformers for them. Furthermore, we removed the compstatin analogs used in the pharmacophore modeling process (listed in Table 1). The remaining 82 comsptatin analogs were used as our test set. They are listed in Table 2.
Table 1.
The nine compstatin analogs used in the pharmacophore modeling. Relative activity is the activity relative to compstatin.
| Name | Sequence | IC50 (µM) | Relative Activity |
|---|---|---|---|
| AcCompNH2-V4(1MeW)/H9A | Ac-ICV(1MeW)QDWGAHRCT-NH2 | 0.205 | 261.463 |
| AcCompNH2-V4(1MeW)/W7(5fW)/H9A | Ac-ICV(1MeW)QD(5fW)GAHRCT-NH2 | 0.205 | 261.463 |
| AcCompNH2-V4W | Ac-ICVWQDWGHHRCT-NH2 | 2.2 | 24.364 |
| AcCompNH2-Q5N | Ac-ICVVNDWGHHRCT-NH2 | 4.2 | 2.857 |
| CompCC-W7A | CVVQDAGHHRC | 182 | 0.066 |
| CompCC-D6A | CVVQAWGHHRC | 257 | 0.047 |
| CompCC-Q5A | CVVADWGHHRC | 910 | 0.013 |
| AcCompNH2-V4W/W7(1MeW)/H9A | Ac-ICVWQD(1MeW)GAHRCT-NH2 | NA | - |
| Comp-linear | IAVVQDWGHHRAT | >600 | - |
Table 2.
The 82 compstatin analogs in the test set. The data were compiled from references 2–8. The redundant compstatin analogs used to compare different experiments were listed in this table but were not counted as new analogs.
| Name | Sequence | IC50 (µM) | Relative Activity | |
|---|---|---|---|---|
| 1 | Comp | ICVVQDWGHHRCT | 12 | 1 |
| 2 | CompCC | CVVQDWGHHRC | 33 | 0.364 |
| 3 | CompCC-V3A | CAVQDWGHHRC | 1200 | 0.01 |
| 4 | CompCC-V4A | CVAQDWGHHRC | 67 | 0.179 |
| 5 | CompCC-G8A | CVVQDWAHHRC | >1200 | - |
| 6 | CompCC-H9A | CVVQDWGAHRC | 15 | 0.8 |
| 7 | CompCC-H10A | CVVQDWGHARC | 74 | 0.162 |
| 8 | CompCC-R11A | CVVQDWGHHAC | 70 | 0.171 |
| Linear analogs | ||||
| 9 | Comp-NH2 | ICVVQDWGHHRCT-NH2 | 12 | 1 |
| 10 | Comp-C2A/C12A | IAVVQDWGHHRAT-NH2 | >600 | - |
| 11 | Comp-cleavedRC | ICVVQDWGHHRCT-NH2 | >300 | - |
| Acetylated Compstatin | ||||
| 12 | 12 AcCompNH2 | Ac-ICVVQDWGHHRCT-NH2 | 3 | |
| Deletion analogs | ||||
| 13 | CompNH2-del1I | CVVQDWGHHRCT-NH2 | 25 | 0.480 |
| 14 | CompNH2-del[1I,13T] | CVVQDWGHHRC-NH2 | 33 | 0.364 |
| 15 | CompNH2-del[1I,3V-8G] | CHHRCT-NH2 | >600 | - |
| 16 | CompNH2-del[1I,3V-7W] | CGHHRCT-NH2 | >600 | - |
| 18 | CompNH2-del[1I,3V-6D] | CWGHHRCT-NH2 | >600 | - |
| 19 | CompNH2-del[1I,3V-5Q] | CDWGHHRCT-NH2 | >600 | - |
| 20 | CompNH2-del[1I,3V-4V] | CQDWGHHRCT-NH2 | >600 | - |
| 21 | CompNH2-del[1I,3V] | CVQDWGHHRCT-NH2 | >600 | - |
| 22 | CompNH2-del[1I,8G-11R,13T] | CVVQDWC-NH2 | >600 | - |
| 23 | CompNH2-del[1I,10H-11R,13T] | CVVQDWGHC-NH2 | >600 | - |
| beta-turn analogs | ||||
| 24 | AcCompNH2-Q5G | Ac-ICVVGDWGHHRCT-NH2 | 567 | 0.021 |
| 25 | AcCompNH2-W7F | Ac-ICVVQDFGHHRCT-NH2 | >400 | - |
| Table 2.I | ||||
| 26 | AcCompNH2-H9A | Ac-ICVVQDWGAHRCT-NH2 | 2.9 | 4.138 |
| 27 | AcCompNH2-V4A/H9A/T13I | Ac-ICVAQDWGAHRCI-NH2 | 4 | 3 |
| AcCompNH2-W7F | Ac-ICVVQDFGHHRCT-NH2 | >400 | - | |
| 28 | AcCompNH2-Q5G/D6A/W7A | Ac-ICVVGAAGHHRCT-NH2 | >100 | - |
| Table 2.II | ||||
| 29 | AcCompNH2-V3L | Ac-ICLVQDWGHHRCT-NH2 | 10 | 1.2 |
| 30 | AcCompNH2-V3L/Q5N | Ac-ICLVNDWGHHRCT-NH2 | 8.3 | 1.446 |
| 31 | CompNH2-R11K | ICVVQDWGHHKCT-NH2 | 20.2 | 0.594 |
| 32 | AcCompNH2-R11S | Ac-ICVVQDWGHHSCT-NH2 | 25 | 0.48 |
| 33 | AcCompNH2-Q5N/R11A | Ac-ICVVNDWGHHACT-NH2 | 60 | 0.2 |
| 34 | AcCompNH2-H9A/R11A | Ac-ICVVQDWGAHACT-NH2 | 9.9 | 1.212 |
| 35 | AcCompNH2-H9A/R11dR | Ac-ICVVQDWGAHdRCT-NH2 | >1000 | - |
| 36 | AcCompNH2-T13I | Ac-ICVVQDWGHHRCI-NH2 | 3.2 | 3.75 |
| Elisa competition binding experiment | ||||
| AcCompNH2 | Ac-ICVVQDWGHHRCT-NH2 | 5.6 | 3 | |
| 37 | AcCompNH2-I1L/V4W/H9R/R11Q/T13F | Ac-LCVWQDWGRHQCF-NH2 | 131 | 0.128 |
| 38 | AcCompNH2-I1L/H9W/T13G | Ac-LCVVQDWGWHRCG-NH2 | 5.4 | 3.111 |
| 39 | AcCompNH2-I1M/V4H/R9F/T13F | Ac-MCVHQDWGGHRCF-NH2 | 85.2 | 0.197 |
| Inhibition of complement mediated rabbit Erythrocyte lysis experiment |
||||
| CompNH2 | ICVVQDWGHHRCT-NH2 | 12 | 1 | |
| AcCompNH2 | Ac-ICVVQDWGHHRCT-NH2 | 4.5 | 2.667 | |
| AcCompNH2-H9A | Ac-ICVVQDWGAHRCT-NH2 | 2.9 | 4.138 | |
| AcCompNH2-I1L/V4W/H9R/R11Q/T13F | Ac-LCVWQDWGRHQCF-NH2 | 254 | 0.047 | |
| AcCompNH2-I1L/H9W/T13G | Ac-LCVVQDWGWHRCG-NH2 | 2.9 | 4.138 | |
| AcCompNH2-I1M/V4H/H9G/T13F | Ac-MCVHQDWGGHRCF-NH2 | 87.4 | 0.137 | |
| 40 | AcCompNH2-I1R | Ac-RCVVQDWGHHRCT-NH2 | 8 | 1.5 |
| 41 | AcCompNH2-I1D | Ac-DCVVQDWGHHRCT-NH2 | 22 | 0.545 |
| Table 2.III | ||||
| CompNH2 | ICVVQDWGHHRCT-NH2 | 53.6 | 1 | |
| AcCompNH2 | Ac-ICVVQDWGHHRCT-NH2 | 18.1 | 2.961 | |
| AcCompNH2-H9A | Ac-ICVVQDWGAHRCT-NH2 | 12.4 | 4.322 | |
| 42 | AcCompNH2-V4T | Ac-ICVTQDWGHHRCT-NH2 | 68.3 | 0.785 |
| 43 | AcCompNH2-V4S | Ac-ICVSQDWGHHRCT-NH2 | 50.9 | 1.053 |
| 44 | AcCompNH2-V4H | Ac-ICVHQDWGHHRCT-NH2 | 10.5 | 5.105 |
| 45 | AcCompNH2-V4F | Ac-ICVFQDWGHHRCT-NH2 | 10.2 | 5.255 |
| 46 | AcCompNH2-V4Y/H9A | Ac-ICVYQDWGAHRCT-NH2 | 3.8 | 14.889 |
| 47 | AcCompNH2-V4W/H9W | Ac-ICVWQDWGWHRCT-NH2 | 3.1 | 17.290 |
| 48 | 48 AcComp-V4W/H9A | Ac-ICVWQDWGAHRCT | 2 | 26.8 |
| 49 | AcCompNH2-V4W/H9A | Ac-ICVWQDWGAHRCT-NH2 | 1.2 | 44.667 |
| Table 2.IV | ||||
| CompNH2 | ICVVQDWGHHRCT-NH2 | 53.6 | 1 | |
| 50 | AcCompNH2-V4(Cha)/H9A | Ac-ICV(Cha)QDWGAHRCT | 47.8 | 1.138 |
| 51 | AcCompNH2-V4(Dht)/H9A | Ac-ICV(Dht)QDWGAHRCT | 10.2 | 5.255 |
| 52 | AcCompNH2-V4(Yphs)/H9A/T13I | Ac-ICV(Yphs)QDWGAHRCI-NH2 | 9.6 | 5.583 |
| 53 | AcCompNH2-V4W/H9(Abu) | Ac-ICVWQDWG(Abu)HRCT-NH2 | 1.5 | 35.733 |
| 54 | AcComp-V4(2Ig1)/H9A | Ac-ICV(2Ig1)QDWGAHRCT | 1.5 | 35.733 |
| 55 | AcCompNH2-V4(2Ig1)/H9A | Ac-ICV(2Ig1)QDWGAHRCT-NH2 | 1.4 | 38.286 |
| 56 | AcComp-V4(Bta)/H9A | Ac-ICV(Bta)QDWGAHRCT | 0.8 | 67 |
| 57 | AcCompNH2-V4(Bta)/H9A | Ac-ICV(Bta)QDWGAHRCT-NH2 | 0.8 | 67 |
| 58 | AcComp-V4(Bpa)/H9A | Ac-ICV(Bpa)QDWGAHRCT | 1.1 | 48.727 |
| 59 | AcCompNH2-V4(Bpa)/H9A | Ac-ICV(Bpa)QDWGAHRCT-NH2 | 0.6 | 89.333 |
| 60 | AcComp-V4(1Nal)/H9A | Ac-ICV(1Nal)QDWGAHRCT | 1.8 | 29.778 |
| 61 | AcComp-V4(2Nal)/H9A | Ac-ICV(2Nal)QDWGAHRCT | 1.4 | 38.286 |
| 62 | AcCompNH2-V4(2Nal)/H9A | Ac-ICV(2Nal)QDWGAHRCT-NH2 | 0.5 | 107.2 |
| Table 2.V | ||||
| 63 | AcCompNH2-V4dY/H9A/T13I | Ac-ICVdYQDWGAHRCI-NH2 | >1000 | - |
| 64 | AcCompNH2-V4dW/H9A/T13I | Ac-ICVdWQDWGAHRCI-NH2 | >1000 | - |
| 65 | AcComp-V4W/H9A/R11dR | Ac-ICVWQDWGAHdRCT | >1000 | - |
| 66 | AcCompNH2-Q5dQ/delT13 | Ac-ICVVdQDWGHHRC-NH2 | >1000 | - |
| 67 | AcCompNH2-D6dD/delT13 | Ac-ICVVQdDWGHHRC-NH2 | >1000 | - |
| 68 | AcCompNH2-W7dW/delT13 | Ac-ICVVQDdWGHHRC-NH2 | >1000 | - |
| 69 | AcComp-V3dV/V4W/H9A | Ac-ICdVWQDWGAHRCT | >1000 | - |
| 70 | AcComp-V4W/H9A/H10dH | Ac-ICVWQDWGAdHRCT | 136.2 | 0.394 |
| 71 | AcComp-V4W/H9dA | Ac-ICVWQDWGdAHRCT | 132.4 | 0.405 |
| 72 | AcComp-I1dI/V4W/H9A | Ac-dICVWQDWGAHRCT | 23.1 | 2.320 |
| 73 | AcComp-V4W/H9A/T13dT | Ac-ICVWQDWGAHRCdT | 2.7 | 19.852 |
| CompNH2 | ICVVQDWGHHRCT-NH2 | 53.6 | 1 | |
| AcCompNH2-V4W/H9A | Ac-ICVWQDWGAHRCT-NH2 | 1.2 | 44.667 | |
| 74 | Comp-G(-1)/V4W/H9A/N14 | GICVWQDWGAHRCTN | 1.2 | 44.667 |
| 75 | Comp-G(-1)/V4(6fW)/W7(6fW)/H9A/N14 | GICV(6fW)QD(6fW)GAHRCTN | 0.43 | 124.651 |
| 76 | Comp-G(-1)/V4(5OHW)/W7(5OHW)/H9A/N14 | GICV(5OHW)QD(5OHW)GAHRCTN | 33 | 1.624 |
| 77 | Comp-G(-1)/V4(7azaW)/W7(7azaW)/H9A/N14 | GICV(7azaW)QD(7azaW)GAHRCTN | 122 | 0.439 |
| CompNH2 | ICVVQDWGHHRCT-NH2 | 53.6 | 1 | |
| AcCompNH2-V4W/H9A | Ac-ICVWQDWGAHRCT-NH2 | 1.2 | 44.667 | |
| 78 | AcCompNH2-V4(5fW)/H9A | Ac-ICV(5fW)QDWGAHRCT-NH2 | 1.74 | 30.805 |
| 79 | AcCompNH2-V4(5MeW)/H9A | Ac-ICV(5MeW)QDWGAHRCT-NH2 | 0.87 | 61.609 |
| 80 | AcCompNH2-V4(2Nal)/H9A | Ac-ICV(2Nal)QDWGAHRCT-NH2 | 0.545 | 98.35 |
| 81 | AcCompNH2-V4W/W7(5fW)/H9A | Ac-ICVWQD(5fW)GAHRCT-NH2 | 0.446 | 120.279 |
| 82 | AcCompNH2- V4W/W7(5MeW)/H9A | Ac-ICVWQD(5MeW)GAHRCT-NH2 | NA | - |
Evaluation of the pharmacophore model
We evaluated both the sensitivity and specificity of our pharmacophore model. Specifically, sensitivity was measured by the ability of the model to identify the compstatin analogs of medium and high inhibitory activities while specificity was measured by the ability of the model to identify only the compstatin analogs of medium and high inhibitory activities and nothing else. We defined the relative activity of 10 compared with comsptatin to be the cut-off. The compstatin analogs having 10-fold or higher inhibitory activities compared with compstatin were considered of medium or high inhibitory activities.
Table 1 lists the nine compstatin analogs used in pharmacophore modeling process. four of them had 10-fold or higher inhibitory activities compared with compstatin. Table 2 lists the 82 compstatin analogs in the test set. Among them, 20 had 10-fold or higher inhibitory activities compared with compstatin. The Pharmacophore Search function of MOE was used to scan both sets using our pharmacophore model. The Use Annotation Field was automatically set to PH4:PCH by MOE. The other default options of MOE were used.
Results & Discussion
Taking advantage of the availability of extensive structure-activity relationships of compstatin analogs, an x-ray structure of AcCompNH2-V4W-H9A bound to the C3c fragment of C3 (PDB ID: 2QKI) and the two most active compstatin analogs known today, AcCompNH2-V4(1MeW)-H9A and AcCompNH2-V4(1MeW)-W7(5fW)-H9A), we built a new pharmacophore model for compstatin analogs. The model consists of six structural features, all of which play critical roles in determining inhibitory activities. Table 3 lists these six pharmacophore features in detail. Figure 2 shows the superposition of the six pharmacophore features over the aligned structures of the two most active compstatin analogs.
Table 3.
The six pharmacophore features
| ID | Feature | Center | Radius (Å) |
|---|---|---|---|
| F1 | Aromatic Ring | Center of mass of W4 indole ring | 1.2 |
| F2 | Hydrogen-bond Acceptor | Carbonyl oxygen on the Q5 side chain | 0.8 |
| F3 | Hydrogen-bond Donor | Amide nitrogen on the Q5 side chain | 0.8 |
| F4 | Hydrogen-bond Acceptor | Carbonyl oxygen on the D6 side chain | 2.2 |
| F5 | Hydrogen-bond Donor | Indole nitrogen of W7 | 0.8 |
| F6 | Hydrophobe | Centroid of two Sγ atoms of C2 and | 0.7 |
The sensitivity and specificity of this pharmacophore model were tested. We showed that this model was able to identify only compstatin analogs of medium and high inhibitory activities and nothing else from the set of nine compstatin analogs used in the pharmacophore modeling. In addition, using a separate test set of 82 compstatin analogs, we demonstrated that our pharmacophore model was able to identify 14 (14/20 or 70%) of the analogs of medium and high inhibitory activities and misclassify only five (5/62 or 8.5%) of the analogs of low or no inhibitory activities. The superior results proved our pharmacophore model to be a filter of great sensitivity and specificity and showed great promise for future identification of small-molecule complement C3 inhibitors.
To understand the relative importance of each individual pharmacophore feature on the sensitivity and specificity of the model, the following test was performed. Each pharmacophore feature was removed individually and the test set was scanned using the reduced pharmacophore model consisting of only the remaining five pharmacophore features. The test result is shown in Table 4. The removal of any one pharmacophore feature caused only a minor increase in the number of true positives but a dramatic increase in the number of false positives. Specifically, removal of either F1 or F6 resulted in the largest false positive rates of 74% and 66%, respectively. Therefore, the aromatic ring on the fourth residue and the hydrophobe on the disulfide bond were the most critical pharmacophore features in ensuring the specificity of the model.
Table 4.
The relative importance of each individual pharmacophore feature on the sensitivity and specificity of the model, as indicated by the success rate and the false positive rate after the feature was removed.
| ID | Feature | Success rate after the feature was removed | False positive rate after the feature was removed |
|---|---|---|---|
| F1 | Aromatic Ring | 17/20 (85%) | 46/62 (74%) |
| F2 | Hydrogen-bond Acceptor | 15/20 (75%) | 22/62 (35%) |
| F3 | Hydrogen-bond Donor | 16/20 (80%) | 19/62 (31%) |
| F4 | Hydrogen-bond Acceptor | 15/20 (75%) | 18/62 (29%) |
| F5 | Hydrogen-bond Donor | 15/20 (75%) | 27/62 (44%) |
| F6 | Hydrophobe | 18/20 (90%) | 41/62 (66%) |
In a separate quantitative structure-activity relationship (QSAR) study of compstatin analogs, Mulakala et al. identified four important physico-chemical and geometrical properties of the analogs that correlated strongly with activity16: the number of aromatic bonds and the hydrophobicity of the fourth residue, hydrophobic patch size near the disulfide bond and the solvent accessible surface area occupied by nitrogen atoms of basic amino acid residues. Independently, we also identified the aromatic ring of the fourth residue (F1) and the hydrophobe on the disulfide bond (F6) to be the most critical pharmacophore features in determining inhibitory activity as demonstrated by the removal test results. Lastly, the solvent accessible surface area occupied by nitrogen atoms of basic amino acid residues was related to the charges on the nitrogen atoms of basic amino acid residues. Because we built our pharmacophore model with the ultimate goal of using the model to identify small-molecule C3 inhibitors, we intentionally ignored any charge-related features in our pharmacophore modeling process (charged groups are generally removed from consideration in small-molecule drug design because of their potential penetration into the blood-brain barrier). Our removal test results also suggested the hydrogen acceptor group (F2) and hydrogen donor group (F3) on the fifth residue, hydrogen acceptor group on the sixth residue (F4) and the hydrogen bond donor (F5) on the seventh residues to be important in determining activity.
The work presented in this paper is simply the first step towards our ultimate goal of identifying small-molecule C3 inhibitors in the future. Small-molecule inhibitors are highly desirable because they possess many advantages (such as small size, low price, oral availability, ability to cross membranes, and straightforward synthesis15) over their peptide counterparts. The results demonstrated in our work complement the QSAR model we developed14 and showed great promise in future identification of small-molecule C3 inhibitors. In fact, we have set forth in this direction and have searched the ZINC16 database using our pharmacophore model. By further prioritizing the initial pharmacophore search hits using docking simulations, we have successfully identified several small molecules that are potential potent C3 small-molecule inhibitors. Other pharmacophore modeling techniques such as allowing partial matches or adding the binding site shape will be applied to the existing pharmacophore model to further increase its sensitivity and specificity in searching for small-molecule C3 inhibitors.
Acknowledgments
T.-L. Chiu would like to thank Drs. Yuk Sham and Angelo Pugliese for useful discussions. We want to thank Dr. Tony Hill for re-producing Figure 2 using pymol. This work was supported by NIH Grants GM-069736, GM-62134 and AI-068730.
Abbreviations
- C3
The third component of complement
- C3b
The proteolytically activated form of C3
- Ac
acetylated N terminus
- CompNH2
Comsptatin
- 1MeW
1-methyl-tryptophan
- 5fW
5-fluorotryptophan
References
- 1.Ricklin D, Lambris JD. Complement targeted Therapeutics. Nature Biotechnology. 2007;25:1265–1275. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sahu A, Lambris JD. Complement inhibitors: a resurgent concept in anti-inflammatory therapeutics. Immunopharmacology. 2000;49:133–148. doi: 10.1016/s0162-3109(00)80299-4. [DOI] [PubMed] [Google Scholar]
- 3.Sahu A, Kay BK, Lambris JD. Inhibition of human complement by a C3-binding peptide isolated from a phage-displayed random peptide library. J. Immunol. 1996;157:884–891. [PubMed] [Google Scholar]
- 4.Mallik B, Morikis D. Development of a quasi-dynamic pharmacophore model for anti-complement peptide analogues. J. Am. Chem. Soc. 2005;127:10967–10976. doi: 10.1021/ja051004c. [DOI] [PubMed] [Google Scholar]
- 5.Katragadda M, Lambris JD. Expression of compstatin in Escherichia coli: incorporation of unnatural amino acids enhances its activity. Protein Expr. Purif. 2006;47:289–295. doi: 10.1016/j.pep.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 6.Mallik B, Katragadda M, Spruce LA, Carafides C, Tsokos CG, Morikis D, Lambris JD. Design and NMR characterization of active analogues of compstatin containing non-natural amino acids. J. Med. Chem. 2005;48:274–286. doi: 10.1021/jm0495531. [DOI] [PubMed] [Google Scholar]
- 7.Morikis D, Assa-Munt N, Sahu A, Lambris JD. Solution structure of Compstatin, a potent complement inhibitor. Protein Sci. 1998;7:619–627. doi: 10.1002/pro.5560070311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morikis D, Roy M, Sahu A, Troganis A, Jennings PA, Tsokos GC, Lambris JD. The structural basis of compstatin activity examined by structure-function-based design of peptide analogs and NMR. J. Biol. Chem. 2002;277:14942–14953. doi: 10.1074/jbc.M200021200. [DOI] [PubMed] [Google Scholar]
- 9.Sahu A, Soulika AM, Morikis D, Spruce L, Moore WT, Lambris JD. Binding kinetics, structure-activity relationship, and biotransformation of the complement inhibitor compstatin. J. Immunol. 2000;165:2491–2499. doi: 10.4049/jimmunol.165.5.2491. [DOI] [PubMed] [Google Scholar]
- 10.Soulika AM, Morikis D, Sarrias MR, Roy M, Spruce LA, Sahu A, Lambris JD. Studies of structure-activity relations of complement inhibitor compstatin. J. Immunol. 2003;171:1881–1890. doi: 10.4049/jimmunol.171.4.1881. [DOI] [PubMed] [Google Scholar]
- 11.Katragadda M, Magotti P, Sfyroera G, Lambris JD. Hydrophobic effect and hydrogen bonds account for the improved activity of a complement inhibitor, compstatin. J. Med. Chem. 2006;49:4616–4622. doi: 10.1021/jm0603419. [DOI] [PubMed] [Google Scholar]
- 12.Janssen B, Els Halff JC, Lambris JD, Gros P. Structure of compstatin in complex with complement component c3c reveals a new mechanism of complement inhibition. J Biol Chem. 2007;282:29241–29247. doi: 10.1074/jbc.M704587200. [DOI] [PubMed] [Google Scholar]
- 13.Ricklin D, Lambris JD. Compstatin – A Complement Inhibitor on its Way to Clinical Application. Adv Exp Med Biol. 2008 doi: 10.1007/978-0-387-78952-1_20. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MOE (The Molecular Operating Environment) Version 2006.08, C. C. G. I. Montreal, Quebac, Canada: http://www.chemcomp.com/ [Google Scholar]
- 15.Pigache A, Cieplak AP, Dupradeau F-Y. Automatic and highly reproducible RESP and ESP charge derivation: Application to the development of programs RED and X RED; 227th ACS National Meeting; Anaheim, CA, USA: 2004. [Google Scholar]
- 16.Mulakala C, Lambris JD, Kaznessis Y. A simple, yet highly accurate, QSAR model captures the complement inhibitory activity of compstatin. Bioorg. Med. Chem. 2007;15:1638–1644. doi: 10.1016/j.bmc.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pillarisetti S. Are peptides the future? Small molecule versus peptides. Curr. Pharm. Biotechnol. 2006;7:225–227. doi: 10.2174/138920106777950799. [DOI] [PubMed] [Google Scholar]
- 16.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]