

Abstract
Purpose
To survey families with clinical evidence of autosomal dominant retinitis pigmentosa (adRP) for mutations in genes known to cause adRP.
Methods
Two hundred adRP families, drawn from a cohort of more than 400 potential families, were selected by analysis of pedigrees. Minimum criteria for inclusion in the adRP cohort included either evidence of at least three generations of affected individuals or two generations with evidence of male-to-male transmission. Probands from each family were screened for mutations in 13 genes known to cause adRP: CA4, CRX, FSCN2, IMPDH1, NRL, PRPF3 (RP18), PRPF8 (RP13), PRPF31 (RP11), RDS, RHO, ROM1, RP1, and RP9. Families without mutations in autosomal genes and in which an X-linked mode of inheritance could not be excluded were tested for mutations in ORF 15 of X-linked RPGR. Potentially pathogenic variants were evaluated based on a variety of genetic and computational criteria, to confirm or exclude pathogenicity.
Results
A total of 82 distinct, rare (nonpolymorphic) variants were detected among the genes tested. Of these, 57 are clearly pathogenic based on multiple criteria, 10 are probably pathogenic, and 15 are probably benign. In the cohort of 200 families, 94 (47%) have one of the clearly pathogenic variants and 10 (5%) have one of the probably pathogenic variants. One family (0.5%) has digenic RDS-ROM1 mutations. Two families (1%) have a pathogenic RPGR mutation, indicating that families with apparent autosomal transmission of RP may actually have X-linked genetic disease. Thus, 107 families (53.5%) have mutations in known genes, leaving 93 whose underlying cause is still unknown.
Conclusions
Together, the known adRP genes account for retinal disease in approximately half of the families in this survey, mostly Americans of European origin. Among the adRP genes, IMPDH1, PRPF8, PRPF31, RDS, RHO, and RP1 each accounts for more than 2% of the total; CRX, PRPF3, and RPGR each accounts for roughly 1%. Disease-causing mutations were not found in CA4, FSCN2, NRL, or RP9. Because some mutations are frequent and some regions are more likely to harbor mutations than others, more than two thirds of the detected mutations can be found by screening less than 10% of the total gene sequences. Among the remaining families, mutations may lie in regions of known genes that were not tested, mutations may not be detectable by PCR-based sequencing, or other loci may be involved.
Retinitis pigmentosa (RP) is the most common form of inherited retinopathy, with a prevalence of approximately 1 in 3500.1 From linkage mapping, positional cloning, and candidate gene screening, at least 35 unique loci have been identified for nonsyndromic forms of RP. The underlying genes for 26 of these loci have been reported (RetNet; http://sph.uth.tmc.edu/RetNet/ provided in the public domain by the University of Texas Houston Health Science Center, Houston, TX). For autosomal dominant RP (adRP), which accounts for 20% to 40% of all cases, 14 genes have been identified (Table 1). Population surveys of subsets of these genes suggest that the known adRP genes may account for between one fourth to one half of cases or families (Table 1).
Table 1.
Autosomal and X-Linked Genes Known to Cause Dominant Retinitis Pigmentosa
| Symbol | Protein | Location | Prevalence | Population | References |
|---|---|---|---|---|---|
| CA4 (RP17) | Carbonic anhydrase 4 | 17q23.2 | - | 2 | |
| CRX | Cone-rod homeobox transcription factor | 19q13.32 | 2/206 (1%) 0/148 (0%) |
American Spanish |
3, 4 |
| FSCN2 | Fascin 2 | 17q25.3 | 4/120 (3%) | Japanese | 5, 6 |
| GUCA1B | Guanylate cyclase activating protein 1B | 6p21.1 | 0/200 (0%) 3/63 (5%) |
British Japanese |
7, 8 |
| IMPDH1 (RP10) | Inosine monophosphate dehydrogenase 1 | 7q32.1 | 2/60 (3%) 7/183 (4%) 2/96 (2%) 6/190 (2%) |
American American Japanese North American |
9-11* |
| NRL | Neural retina leucine zipper | 14q11.2 | 4/189 (2%) 3/200 (2%) 0/96 (0%) 1/148 (1%) |
American British Japanese Spanish |
4, 12-14† |
| PRPF3 (RP18) | Pre-mRNA processing factor 3 | 1q21.2 | 1/96 (1%) 1/150 (1%) |
Japanese Spanish |
15, 16† |
| PRPF8 (RP13) | Pre-mRNA splicing factor C8 | 17p13.3 | 4/190 (2%) 0/96 (0%) 5/150 (3%) |
American Japanese Spanish |
16, 17†,‡ |
| PRPF31 (RP11) | Pre-mRNA splicing factor 31 | 19q13.42 | 4/96 (4%) 3/150 (2%) |
Japanese Spanish |
16‡ |
| RDS | Peripherin 2 | 6p21.2 | 17/206 (8%) 5/96 (5%) 2/148 (1%) |
American Japanese Spanish |
3, 4, 18† |
| RHO | Rhodopsin | 3q22.1 | 53/206 (26%) 2/96 (2%) 19/90 (20%) 29/148 (20%) |
American Japanese North American Spanish |
3, 4, 19, 20† |
| ROM1 | Rod outer membrane protein 1 | 11q12.3 | 0/173 (0%) 2/224 (1%) |
American North American |
21-23 |
| RP1 | RP1 protein | 8q12.1 | 8/206 (4%) 21/266 (8%) 1/96 (1%) 5/148 (3%) |
American British Japanese Spanish |
3, 4, 24-26† |
| RP9 (PAP1) | Pim-1 associated protein | 7p14.3 | - | 27 | |
| RPGR | Retinitis pigmentosa GTPase regulator | Xp11.4 | - | 28, 29 |
Gene symbols are those approved by the HUGO Nomenclature Committee (http://www.gene.ucl.ac.uk/nomenclature/) followed by the alternate symbol, if any, in parenthesis. Genes are listed alphabetically by symbol here and in the text. Alternate symbols are not used further, except to distinguish the pre-mRNA splicing proteins. Map locations are based on the assembled human genome (May 2004 build) using the UCSC Genome Browser (http://genome.ucsc.edu/). Affected/total (percent) is unrelated adRP probands or families, with probable or definite pathogenic mutations, based on surveys of more than 50 individuals, as can best be deduced from the publication. Note: additional prevalence values are reported by Ramesar et al.30 and Ziviello et al.31
Wada Y, et al. IOVS 2003; 44: ARVO E-Abstract 2306.
Wada Y et al. IOVS 2004; 45: ARVO E-Abstract 2456.
De Erkenez AC et al. IOVS 2002;43: ARVO E-Abstract 791.
The Laboratory for Molecular Diagnosis of Inherited Eye Diseases was established in 1994 to determine the disease-causing genes and mutations in patients and families with RP. The Diagnostic Laboratory is a joint project of the School of Public Health and the Hermann Eye Center at the University of Texas Health Science Center in Houston. To date, more than 400 adRP families or probands have been ascertained by clinicians submitting samples to the Diagnostic Laboratory. From these families, we selected a set of 200 with a diagnosis of adRP and pedigree evidence to support this mode of inheritance. In this report, we summarize a systematic screen of these families for mutations in genes known to cause adRP.
We screened the 200 families for mutations in 13 of the 14 currently known adRP genes (Table 1). The thirteen genes have an expected prevalence of 2% or greater or an unknown prevalence. We did not screen for mutations in GUCA1B because previous surveys suggest that it accounts for much less than 1% of adRP in white subjects.7 Also, for three genes, PRPF3, PRPF8, and RP1, sequencing was limited to one exon each because previous surveys failed to find mutations outside of these exons15,16,32 (De Erkenez AC, et al. IOVS 2002;43:ARVO E-Abstract 791).
In addition, we tested selected families for mutations in open reading frame (ORF) 15 of the X-linked RP gene RPGR. Although X-linked RP usually affects males only, some RPGR mutations, particularly in ORF 15, can cause disease in “carrier” females with a simulation of autosomal dominant inheritance.28,33,34 Females are usually more mildly affected than males in these families, but the pedigree may be misconstrued as adRP. Thus some “adRP” families without mutations in known adRP genes may, in fact, have X-linked RP.
A major difficulty in screening for mutations in dominant-acting genes is to distinguish rare, benign sequence changes from pathogenic mutations.35 Often, the number of affected individuals available for testing is not large enough to establish pathogenicity by segregation alone. Ideally, functional studies could distinguish benign from pathogenic protein changes. However, the genes implicated in dominant RP are a heterogeneous group with widely differing functions. Many are predominantly or exclusively photoreceptor-specific. Rhodopsin and GUCA1B are the only known adRP-associated proteins directly involved in phototransduction; but RDS and ROM1 maintain disc structure in photoreceptors, CRX and NRL are retinal transcription factors, and RP1 and RPGR localize to the connecting cilium of photoreceptors. In contrast, CA4 and IMPDH1 code for widely expressed “housekeeping” enzymes, and three other adRP-associated proteins—PRPF3, PRPF8, and PRPF31—are highly conserved, ubiquitously expressed components of RNA-splicing complexes. Mechanisms by which mutations lead to retinal disease are also highly varied (see Table 1 and RetNet).
Because of this functional heterogeneity, there is no general method for evaluating genetic variation within each gene and assessing pathogenicity of rare variants. To establish pathogenicity in this survey we used a combination of segregation information, general computational approaches and individualized evaluation based on the unique characteristics of each gene and protein.
Methods
Patients, Families, and Control Subjects
Among the approximately 650 families or probands enrolled in studies at the Laboratory for Molecular Diagnosis of Inherited Eye Diseases, over 400 have a diagnosis of dominant RP. From these, we selected 200 families with a diagnosis of adRP at submission and pedigree evidence of autosomal dominant transmission. Our criteria were either the presence of three or more generations, with both males and females among all affected family members, or at least two affected generations with male-to-male transmission. The requirements for three generations, including females, or for male-to-male transmission, reduced the likelihood of including X-linked families.
Probands and other family members were ascertained primarily in Texas, California, and Michigan as part of studies in collaboration with investigators at (1) the Anderson Vision Research Center, Retina Foundation of the Southwest (Dallas, TX; DGB), (2) the Cullen Eye Institute, Baylor College of Medicine (Houston, TX; RAL), (3) the Hermann Eye Center, the University of Texas Health Science Center at Houston (CAG, RSR), (4) the Jules Stein Eye Institute, UCLA School of Medicine (Los Angeles, CA; JRH), and (5) the Kellogg Eye Center, University of Michigan (Ann Arbor, MI; JRH). Additional clinicians who submitted samples are listed in the Acknowledgments. Diagnostic testing included a complete ophthalmic and fundus examination; measurement of visual acuity, visual fields and dark adaptation; and electroretinography. Pedigrees were constructed based on patient interviews. Ethnicity is that reported by the proband.
Pedigrees were analyzed to determine the likelihood of autosomal dominant inheritance relative to autosomal recessive or X-linked inheritance. Each pedigree was coded into LINKAGE format with an invariant genotype at a dummy locus for each individual.36 The likelihood at 0% recombination to the dummy locus was then determined for each family based on three models: autosomal dominant inheritance with 95% penetrance, autosomal recessive inheritance with 99% penetrance in homozygotes, and X-linked inheritance with 10% penetrance in heterozygous females and 99% penetrance in males. The disease allele frequency for each model was set to 0.001 and the a priori probability of each mode was assumed to be equal. Likelihood ratios for autosomal dominant versus recessive, and autosomal dominant versus X-linked, were then calculated.
White control DNAs were from 50 unrelated CEPH (Centre d’Etude du Polymorphisme Humain) parent pairs.37 DNAs from Caribbean controls (n = 47) were kindly provided by Edwin Stone (University of Iowa, Iowa City, Iowa).
Blood and DNA samples were provided by our clinical collaborators under approved protocols. If blood was provided, DNA was prepared with a commercial extraction kit (PureGene; Gentra, Minneapolis, MN).3,32
The study was performed in accordance with the Declaration of Helsinki and informed consent was obtained from all participants. The research was approved by the Committee for Protection of Human Subjects, University of Texas HSC, Houston, and by the respective human subjects’ review boards at each participating academic institution.
Mutation Testing
DNA testing of CA4, FSCN2, NRL, PRPF3, PRPF8, PRPF31, ROM1, and RP9 was entirely by cycle sequencing. Mutation screening of CRX, IMPDH1, RDS, RHO, and RP1 in earlier years was done by single-strand conformational analysis (SSCA).3,9,32,38 Variants detected by SSCA were sequenced to determine the underlying nucleotide change. All recent testing of these genes was performed by direct sequencing.
PCR primers, annealing temperatures, and amplimer-specific details are listed in the Supplementary Tables, available online at http://www.iovs.org/cgi/content/full/47/7/3052/DC1. In general, 30-50 ng of genomic DNA was amplified with Taq polymerase (AmpliTaq Gold; Applied Biosystems [ABI], Foster City, CA, or HotStarTaq; Qiagen, Valencia, CA) in a 12.5 μL reaction volume for 35 cycles.
SSCA was conducted by incorporating 1 μCi of [32P]dCTP during PCR amplification. Radiolabeled fragments were separated by electrophoresis overnight on acrylamide gels (0.6 × MDE; FMC Bioproducts, Cambrex BioSciences, Walkersville, MD). Dried gels were visualized by autoradiography. (See Bowne et al.,32 Bowne et al.,9 Sohocki et al.,38 and Sohocki et al.3)
DNA sequencing was performed in forward and reverse directions as described previously.9 PCR products were treated with exonuclease I and shrimp alkaline phosphatase (ExoSapIt; USB, Cleveland, OH) and sequenced bidirectionally with fluorescent dye-terminators (BigDye v1.1; ABI) using primers listed in the Supplementary Tables, available online at http://www.iovs.org/cgi/content/full/47/7/3052/DC1. Sequence reactions were purified with sephadex columns (Princeton Separations, Adelphia, NJ) and run on an automated capillary sequencer (either an ABI 310 or ABI 3100 Avant Genetic Analyzer). Sequence analysis was done using commercial software (either AutoAssembler or SeqScape software; ABI). (See Bowne et al.,32 Bowne et al.,9 and Sohocki et al.3)
Families in which a definitely pathogenic mutation was found were not routinely tested for further mutations. Thus, not all genes were sequenced in all families.
IMPDH1 Haplotyping
Published SNPs within 250,000 bp of the IMPDH1 gene were amplified by PCR and typed by DNA sequencing.39 Haplotypes for the region were reconstructed after typing multiple family members carrying the IMPDH1 Asp226Asn mutation.
Analysis of Variants
All nonpolymorphic variants were sequenced at least twice in proband samples for confirmation. When available, additional family members were also sequenced to determine the presence or absence of the variant.
Several Web-based analysis programs were used to determine the likelihood that specific variants are pathogenic. Splice-site variants were analyzed using the prediction program NNSPLICE 0.9 (http://www.fruitfly.org/seq_tools/splice.html).40 The Web-based program PolyPhen (Polymorphism Phenotyping, http://www.bork.embl-heidelberg.de/-PolyPhen/ provided in the public domain by the European Molecular Biology Laboratory, Heidelberg, Germany)41 was the starting point for analysis of amino acid substitutions. In addition, we used SIFT (Sorting Intolerant from Tolerant: http://blocks.fhcrc.org/sift/SIFT.html/ provided in the public domain by the Fred Hutchinson Cancer Research Center, Seattle, WA)42 as a measure of sequence conservation and Grantham scores as a measure of the chemical change in amino acids.43
The first step of the PolyPhen analysis was locating the sequences in the current protein databases to identify relevant annotation of protein structure and function. Individual amino acid changes were queried and, if the amino acid change was at an annotated site (e.g., a disulfide bond or a binding site), this information was used in the subsequent analysis. If the amino acid change was within a defined site, appropriate subprograms determined whether the amino acid change would affect the site (e.g., PHAT to characterize transmembrane domain changes or COILS to predict secondary structure).
Next, via BLAST, an alignment of homologous sequences was constructed to assess the level of sequence conservation at the site of the change. The PSIC (position-specific independent counts) subprogram calculated the “profile matrix” for the specific set of sequences and this matrix evaluated the amino acid change in question. The difference in profile scores for the two amino acids is a measure of how often (if ever) that substitution has occurred in the protein family.
The last stage of the PolyPhen analysis involved mapping the site of the amino acid change to the known three dimensional (3-D) structure of the protein by a BLAST search of the protein structure databases (PDB or PQS) and analyses of matches. Amino acid changes were assessed for their effect on the hydrophobic core of the protein, possible interactions with ligands, and other known features of the protein structure. The results of all these analyses were merged to produce a prediction of how damaging the amino acid substitution is likely to be. Variants evaluated by PolyPhen were classified as probable, possible, benign, or unknown.
Results
Probands and Families
The demographic and pedigree characteristics of the 200 adRP families included in this study are summarized in Table 2 (details in Supplementary Tables, http://www.iovs.org/cgi/content/full/47/7/3052/DC1). Ninety-three percent of the families had three or more affected generations, and 25% had five or more. Nearly 70% were known to have five or more affected family members (living or dead), and male-to-male transmission was observed in more than half. Skipped generations were reported in 24% of families. The likelihood ratio for autosomal dominant versus autosomal recessive inheritance was 103 or greater for each family, with only one exception—a small family with a skipped generation (data in Supplementary Tables). The likelihood ratio of autosomal dominant versus X-linked inheritance was at least 103 for more than 70% of families, and more than 15% had suggestive evidence for dominant inheritance (odds of 10-102). However, 12% of families had a likelihood ratio that does not exclude X-linkage or actually favors X-linked inheritance (odds of 1 to <10-2). Ethnicity was at least 73% white, but inclusion of Hispanics and some of the “unknown” families would bring the proportion of “Americans of European origin” close to 90%.
Table 2.
Summary Characteristics of Families
| Number (%) | |
|---|---|
| Generations of known affected | |
| 2 | 13 (6.5) |
| 3 | 72 (36.0) |
| 4 | 65 (32.5) |
| 5 or more | 50 (25.5) |
| Unknown | 0 |
| Number of known affected | |
| 2-4 | 64 (32.0) |
| 5-10 | 88 (44.0) |
| 11-20 | 37 (18.5) |
| >20 | 11 (5.5) |
| Unknown | 0 |
| Male-to-male transmission | |
| No | 91 (45.5) |
| Yes | 108 (54.0) |
| Unknown | 1 (0.5) |
| Skipped generations | |
| 0 | 150 (75.0) |
| 1-2 | 47 (23.5) |
| >2 | 2 (1.0) |
| Unknown | 1 (0.5) |
| Odds adRP v. XIRP | |
| ≥103 | 143 (71.5) |
| 10-102 | 33 (16.5) |
| 1-10-1 | 21 (10.5) |
| ≤10-2 | 3 (1.5) |
| Unknown | 0 |
| Ethnicity | |
| Asian | 7 (3.5) |
| African American | 6 (3.0) |
| White | 146 (73.0) |
| Hispanic | 14 (7.0) |
| Other | 4 (2.0) |
| Unknown | 23 (11.5) |
N = 200.
DNA samples from four or more affected family members were available from one-third of the families. Conversely, another third of the families were represented by a DNA sample from a single affected proband only. However, the “proband only” families are not demographically different from families with multiple samples.
Mutation Screening
Summary
The potential disease-causing variants found in the 13 adRP genes and ORF 15 of RPGR in the 200 families are listed in Table 3. Coded family numbers, specific mutations, and related data are in the Supplementary Tables, http://www.iovs.org/cgi/content/full/47/7/3052/DC1. Variants are ranked as pathogenic, probably pathogenic, or benign based on criteria described in the next section. The table includes polymorphic variants if they were not known to us before observation in an affected proband. However, the table does not include a systematic listing of the many known polymorphic variants found in these genes among the individuals screened.
Table 3.
Potentially Pathogenic Variants Identified in the adRP Cohort
| Mutation |
Families (n) | Scores |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Type Variant | Protein | Nucleotide | Seg. | Prior | Struct. | SIFT | Funct | PolyP. | Gran. | Comment | |
| CA4—carbonic anhydrase 4 | |||||||||||
| Pathogenic | None | ||||||||||
| Probably pathogenic | None | ||||||||||
| Benign | Thr198Met | 593 C→T | 1 | No | No | - | No | - | Possib. | 81 | .96/.04 in African control subjects |
| Val234Ile | 700 G→A | 1 | No | No | - | No | - | Benign | 29 | In one control | |
| Val237Leu | 709 G→T | 1 | No | No | - | No | - | Benign | 32 | .98/.02 in African control subjects | |
| Arg239Gln | 716 G→A | 1 | No | No | - | No | - | Benign | 43 | .95/.05 in African control subjects | |
| CRX—cone rod homeobox protein | |||||||||||
| Pathogenic | Arg41Gln | 122 G→A | 1 | 5 | Yes38,44 | - | Yes | - | Prob. | 43 | |
| Leu146_Pro149del | 436_447del | 1 | 15 | Yes38 | - | NA | - | NA | NA | ||
| Probably pathogenic | None | ||||||||||
| Benign | None | ||||||||||
| FSCN2—retinal fascin | |||||||||||
| Pathogenic | None | ||||||||||
| Probably pathogenic | None | ||||||||||
| Benign | Val17Ile | 49 G→A | 1 | No | No | - | No | - | Benign | 29 | In one white control subjects |
| Gly165Arg | 493 G→A | 1 | No | No | - | No | - | Benign | 125 | ||
| Pro231Ser | 691 C→T | 1 | No | No | - | No | - | Benign | 74 | ||
| Ala323Thr | 967 G→A | 1 | No | Yes45 | - | Yes | - | Possib. | 58 | .96/.04 in white control subjects | |
| Leu489Val | 1465 C→G | 1 | No | No | - | No | - | Benign | 32 | .98/.02 in white control subjects | |
| IMPDH1 (RP10)—inosine monophosphate dehydrogenase | |||||||||||
| Pathogenic | Asp226Asn | 676 G→A | 5 | 20+ | Yes9 | - | Yes | Yes | Prob. | 23 | Affects CBS domain9 |
| Probably pathogenic | None | ||||||||||
| Benign | None | ||||||||||
| NRL—neural retinal leucine zipper | |||||||||||
| Pathogenic | None | ||||||||||
| Probably pathogenic | None | ||||||||||
| Benign | Arg147Arg | 441 G→A | 2 | Prbn. | No | - | - | - | - | - | Silent substitution |
| Gln174Arg | 521 A→G | 1 | No | No | - | Yes | - | Prob. | 43 | ||
| PRPF3 (RP18)—pre-mRNA splicing factor 3 | |||||||||||
| Pathogenic | Pro493Ser | 1477 C→T | 1 | Prbn. | Yes15 | - | Yes | - | Prob. | 74 | |
| Thr494Met | 1481 C→T | 1 | Prbn. | Yes15 | - | Yes | - | Prob. | 81 | ||
| Probably pathogenic | None | ||||||||||
| Benign | None | ||||||||||
| PRPF8 (RP13)—pre-mRNA splicing factor 8 | |||||||||||
| Pathogenic | Phe2304Leu | 6912 C→G | 1 | 2 | Yes17 | - | Yes | - | Prob. | 22 | |
| Arg2310Gly | 6928 A→G | 1 | 2 | Yes17 | - | Yes | - | Prob. | 125 | ||
| Glu2331fs/ter 2358 | 6991delG | 2 | 2 | No | fs | NA | NA | NA | NA | ||
| Probably pathogenic | IVS 41/exon 42 junction | IVS41-4 G→A | 1 | Prbn. | No | as | NA | NA | NA | NA | |
| Ala2328Val | 6983 C→T | 1 | Prbn. | No | - | No | - | Benign | 64 | ||
| Benign | None | ||||||||||
| PRPF31 (RP11)—pre-mRNA splicing factor 31 | |||||||||||
| Pathogenic | Exon 2/IVS 2 junction | IVS2+1 G→A | 1 | Prbn. | No | as | NA | NA | NA | NA | |
| Gln74ter | 220 C→T | 1 | Prbn. | No | ps | NA | NA | NA | NA | ||
| Asn131fs/ter197 | 390delC | 1 | 3 | No | fs | NA | NA | NA | NA | ||
| Glu141ter | 421 G→T | 1 | Prbn. | No | ps | NA | NA | NA | NA | ||
| Met212fs/ter238 | 636delG | 1 | 2 | No | fs | NA | NA | NA | NA | ||
| Gly253fs/ter317 | 758_767del | 1 | Prbn. | No | fs | NA | NA | NA | NA | ||
| Exon 10/IVS 10 junction | 1049_IVS10+20del/insCCCCT | 1 | Prbn. | No | as | NA | NA | NA | NA | ||
| Exon 10/IVS 10 junction | IVS10+1 G→A | 1 | 5 | No | as | NA | NA | NA | NA | ||
| Glu325ter | 973 G→T | 1 | Prbn. | No | ps | NA | NA | NA | NA | ||
| Probably pathogenic | Ala291Pro | 871 G→C | 1 | Prbn. | No | - | NA | NA | Possib. | 27 | |
| Cys299Arg | 895 T→C | 1 | 4 | No | - | NA | NA | Prob. | 180 | 1 Nonpenetrant if pathogenic | |
| Benign | Gly272Val | 815 G→T | 1 | No | No | - | NA | NA | Prob. | 109 | Chinese variant |
| RDS—peripherin 2 | |||||||||||
| Pathogenic | Val206_Val209del | 616_627del | 1 | 4 | No | Yes | NA | NA | NA | NA | 4 Amino acids lost |
| Exon 2/IVS 2 junction | IVS2+3 A→T | 4 | 20+ | Yes3 | as | NA | NA | NA | NA | Was “1068+3 A→T” | |
| Arg46ter | 136 C→T | 1 | Prbn. | Yes46 | ps | NA | NA | NA | NA | ||
| Leu126Arg | 377 T→G | 1 | Prbn. | Yes* | - | Yes | - | Prob. | 102 | ||
| Arg172Gln | 516 G→T | 1 | Prbn. | Yes47 | - | No | - | Benign | 91 | ||
| Digenic Leu185Pro | 554 T→C | 1 | 3 | Yes48 | - | Yes | - | Prob. | 98 | Digenic with ROM1 Ala114fs | |
| Pro210Arg | 629 C→G | 1 | Prbn. | Yes49 | - | Yes | - | Prob. | 103 | ||
| Pro210Leu | 629 C→T | 1 | Prbn. | Yes50 | - | Yes | - | Prob. | 98 | ||
| Pro216Ser | 646 C→T | 1 | Prbn. | Yes51 | - | Yes | - | Prob. | 74 | ||
| Pro216Leu | 647 C→T | 2 | 6 | Yes18 | - | Yes | - | Prob. | 98 | ||
| Gly266Asp | 797 G→A | 2 | 7 | Yes* | - | Yes | Yes | Possib. | 94 | Transmembrane site | |
| Probably pathogenic | Tyr141Cys | 422 A→G | 1 | 4 | No | - | Yes | - | Prob. | 149 | Other mutation at 141 |
| Ser198Arg | 594 C→G | 1 | Prbn. | No | - | Yes | - | Prob. | 110 | ||
| Pro216Arg | 647 C→G | 1 | Prbn. | No | - | Yes | - | Prob. | 103 | Other mutations at 216 | |
| Benign | Gly137Ser | 409 G→A | 1 | No | No | - | No | - | Benign | 56 | Family has another pathogenic mutation |
| Leu45Phe | 133 C→T | 1 | No | Yes35 | - | Yes | - | Possib. | 22 | Common Caribbean variant | |
| RHO-rhodopsin | |||||||||||
| Pathogenic | Thr17Met | 50 C→T | 1 | Prbn. | Yes52 | - | Yes | - | Prob. | 81 | |
| Pro23His | 68 C→A | 20 | 20+ | Yes53 | - | Yes | - | Prob. | 77 | ||
| Leu57Arg | 170 C→T | 1 | Prbn. | Yes54 | - | Yes | Yes | Possib. | 102 | Transmembrane site | |
| Thr58Arg | 173 C→G | 1 | Prbn. | Yes55 | - | Yes | Yes | Possib. | 71 | Transmembrane site | |
| Gly106Arg | 318 G→A | 1 | 8 | Yes54 | - | Yes | - | Prob. | 125 | ||
| Gly106Trp | 316 G→T | 2 | 2 | Yes56 | - | Yes | - | Prob. | 184 | ||
| Cys110Phe | 329 G→T | 1 | Prbn. | Yes57 | - | Yes | Yes | Prob. | 205 | Disulfide bond | |
| Arg135Trp | 403 C→T | 4 | 8 | Yes56 | - | Yes | Prob. | - | 101 | ||
| Arg135Leu | 404 G→T 405 G→T | 3 | 20+ | Yes56 | - | Yes | Prob. | - | 102 | ||
| Ala164Val | 491 C→T | 1 | Prbn. | Yes57 | - | Yes | Yes | Benign | 64 | Transmembrane site | |
| Pro170Arg | 509 C→G | 1 | 7 | Yes3 | - | Yes | Yes | Possib. | 103 | Transmembrane site | |
| Pro171Ser | 511 C→T | 1 | Prbn. | Yes58 | - | Yes | Yes | Possib. | 74 | Transmembrane site | |
| Pro171Gln | 512 C→A | 1 | Prbn. | Yes59 | - | Yes | Yes | Possib. | 76 | Transmembrane site | |
| Glu181Lys | 541 G→A | 1 | Prbn. | Yes55 | - | Yes | Prob. | Prob. | 56 | ||
| Cys187Tyr | 560 G→A | 1 | 3 | Yes60 | - | Yes | Yes | Prob. | 194 | Disulfide bond | |
| Asp190Asn | 568 G→A | 2 | 2 | Yes61 | - | No | No | Benign | 23 | ||
| His211Arg | 632 A→G | 1 | Prbn. | Yes62 | - | Yes | Yes | Possib. | 29 | Transmembrane site | |
| Pro267Leu | 800 C→T | 1 | Prbn. | Yes52 | - | Yes | Yes | Possib. | 98 | Transmembrane site | |
| IVS 4/exon 5 junction | IVS4-1 G→A | 1 | 7 | Yes63 | as | NA | NA | NA | NA | ||
| Val345Met | 1033 G→A | 1 | Prbn. | Yes† | - | Yes | Yes | Prob. | 21 | BNG binding site | |
| Pro347Ala | 1039 C→G | 1 | Prbn. | Yes62 | - | Yes | Yes | Prob. | 27 | BNG binding site | |
| Pro347Thr | 1039 C→A | 1 | 11 | Yes19 | - | Yes | Yes | Prob. | 38 | BNG binding site | |
| Pro347Leu | 1040 C→T | 2 | 2 | Yes55 | - | Yes | Yes | Prob. | 98 | BNG binding site | |
| Probably pathogenic | Leu46Arg | 137 T→G | 1 | 4 | No | - | Yes | Yes | Possib. | 102 | Transmembrane site |
| Ser270Arg | 810 C→A | 1 | Prbn. | No | - | Yes | Yes | Possib. | 110 | Transmembrane site | |
| ter349Gln | 1045 T→C | 1 | 2 | No | ap | NA | Yes | NA | NA | 58 Amino acids added | |
| Benign | Thr70Met | 209 C→T | 1 | No | No | - | Yes | - | Benign | 81 | |
| ROM1-rod outer membrane protein | |||||||||||
| Pathogenic | Digenic Leu114fs/ter131 | 339insG | 1 | 3 | No | NA | NA | NA | NA | NA | Digenic with RDS Leu185Pro |
| Probably pathogenic | None | ||||||||||
| Benign | Arg229His | 686 G→A | 1 | Prbn. | Yes64 | - | No | - | Benign | 29 | Rare population variant |
| Tyr234Tyr | 702 C→T | 1 | Prbn. | No | NA | NA | NA | NA | NA | Silent substitution | |
| Met271Thr | 812 T→C | 1 | Prbn. | Yes64 | - | No | - | Benign | 81 | Rare population variant | |
| Arg287Trp | 859 C→T | 1 | No | No | - | Yes | - | Prob. | 101 | ||
| RP1-RP1 protein | |||||||||||
| Pathogenic | Leu762fs/ter777 | 2285_2289del | 3 | 4 | Yes26 | fs | NA | NA | NA | NA | |
| Arg677ter | 2029 C→T | 3 | 20+ | Yes32 | ps | NA | NA | NA | NA | ||
| Gly723ter | 2167 G→T | 1 | 2 | Yes‡ | ps | NA | NA | NA | NA | ||
| Probably pathogenic | None | ||||||||||
| Benign | His1034Arg | 3101 A→G | 1 | No | No | - | NA§ | - | NA§ | 29 | .99/.01 in CEPH |
| Leu1808Pro | 5423 T→C | 1 | Prbn. | Yes32 | - | NA§ | - | NA§ | 98 | Family has another pathogenic mutation | |
| RP9 (PAP1)-Pim-1 associated protein | |||||||||||
| Pathogenic | None | ||||||||||
| Probably pathogenic | None | ||||||||||
| Benign | Lys210Arg | 629 A→G | NA | No | No | - | NA§ | NA§ | NA§ | NA§ | .73/.23 in white control subjects |
| RPGR-X-linked retinitis pigmentosa GTPase regulator | |||||||||||
| Pathogenic | Arg195fs/ter229 | ORF15+558del | 1 | 6 | Yes65 | fs | NA | - | NA | NA | |
| Glu256fs/ter492 | ORF15+764_765del | 1 | 11 | No | fs | NA | - | NA | NA | ||
| Probably pathogenic | None | ||||||||||
| Benign | None | ||||||||||
Families, number of families in the cohort with this mutation; Scores, evidence used to evaluate pathogenicity: (1) Seg., segregating in family; No, discordant segregation in family and/or present in control subjects; Prbn., observed in proband only; numbers represent affected with variant if more than proband; (2) Prior, prior publications with independently ascertained families, if known. (3) Struct.: predicted consequences to protein structure; as, abnormal splicing; ap, abnormal protein; fs, frame shift; ps, premature stop; -, no evidence. (4) SIFT, sorting intolerant from tolerant (http://blocks.fhcrc.org/sift/SIFT.html)42 as a measure of sequence conservation; No, not conserved or conserved in a few species only; Yes, conserved in several or all species tested; NA, not applicable; (5) Funct., amino acid change in a biologically relevant functional site; -, no evidence; NA, not applicable; (6) PolyP., PolyPhen (polymorphism phenotyping, http://www.bork.emblheidelberg.de/-PolyPhen)41 ratings for amino acid substitutions; Prob., probably pathogenic; Possib., possibly pathogenic; Benign, no evidence of pathogenicity; NA, not applicable; (7) Gran., Grantham score43 for chemical change of amino acid substitutions; NA, not applicable. To measure the severity of amino acid substitutions we used the chemical difference matrix of Grantham, which estimates the distance between amino acids based on side-chain composition, polarity, and molecular volume. Benign amino acid substitutions tend to have distance scores <70, whereas disease-causing substitutions are more likely to have scores >70.66 For disease-causing substitutions in rhodopsin, Grantham distances average >91.67
Kajiwara K, et al. IOVS 1992; 33:ARVO Abstract 1396.
Stone EM, et al. IOVS 1993; 34:ARVO Abstract:1149.
Grimsby JL, et al. IOVS 2000; 41:ARVO Abstract:192.
NA, no structures available for PolyPhen and/or SIFT comparative analysis (RP1) or sequence too repetitive for analysis (PAP1).
A total of 82 distinct, rare (nonpolymorphic) variants were detected. Of these, 57 are clearly pathogenic based on multiple criteria, 10 are probably pathogenic, and 15 are benign. In the cohort of 200 families, 94 (47%) have one of the clearly pathogenic variants and 10 (5%) have one of the probably pathogenic variants. One family (0.5%) has digenic RDS-ROM1 mutations. Two families (1%) have a pathogenic RPGR mutation, indicating that families with apparent autosomal transmission may actually have X-linked RP.
Determining Pathogenicity
Assessing whether a novel variant in a known disease-causing gene is pathogenic is often challenging. The first considerations are whether the variant segregates with disease, has been found in other affected individuals or families, and is absent from appropriate control subjects. Segregation may not be definitive, though, because in many cases only a few family members are available for testing, and an unaffected individual with a pathogenic mutation may represent incomplete penetrance rather than failure to segregate. A mutation that introduces a radical change in the message or protein is probably pathogenic, but the extreme situation—a null allele—may not be pathogenic by itself. Finally, missense mutations are particularly problematic because the background variation in human proteins is so great.
We assessed each potentially pathogenic mutation by several criteria, with results summarized in Table 3. The table shows whether the variant segregates with disease, has been reported previously, or is present in control subjects. Also indicated is whether a mutation introduces a radical change in the message or protein. Finally, a scoring system was used to evaluate missense changes.
The scoring system used in Table 3 is based on several techniques to assess whether a particular amino acid substitution is pathogenic (see the Methods section and the legend to Table 3). The techniques range from a simple assessment of “chemical conservation” based on structural and biochemical properties of individual amino acids,43 to methods that consider context-dependent properties of an amino acid within a particular protein. The most advanced methods use several sequential analyses: alignment of related sequences to determine evolutionary conservation, database searching to determine the location of critical positions such as active sites or disulfide bonds, and analysis of 3-D models.41 These analyses are then combined with other data to predict pathogenicity.35,67
Gene-Specific Findings
CA4
Four missense changes were found in the CA4 gene, three in African-American or African-Caribbean families. Of the three African variants, each was polymorphic in African-Caribbean control subjects, and benign or “possible” by PolyPhen analysis. The remaining variant, in a white family, was also found in white control subjects and is benign by analysis. Consistent with other reports (Ciccodicola A, et al. IOVS 2005; 46:ARVO E-Abstract 1817), the prevalence of mutations in CA4 causing adRP in this population was less than 1% (95% confidence interval [CI] 0%-1.5%).
CRX
One missense mutation and one deletion, both reported earlier,3,38 were found in the CRX gene. Both segregated with disease in multiple affected individuals and both were predicted to be pathogenic. Affected members of both families had diminished rod and cone ERGs, but did not present with a diagnosis of cone-rod dystrophy, which was the original phenotype associated with CRX mutations.44,68 Affected members of the family with the CRX deletion had early-onset, rapid-progression adRP.38 No other nonpolymorphic variants were found in the cohort.
FSCN2
Five missense variants were found in the FSCN2 gene. Each of the five was found in controls and/or did not segregate with disease. Computational analysis indicates that they are benign or “possible.” Thus, no pathogenic variants were observed in this population and the prevalence of mutations was less than 1%. This contrasts with the higher prevalence found in Japanese adRP families—in part, the result of a single mutation.5
IMPDH1 (RP10)
Five families (2.5%) of the cohort have disease-causing mutations in IMPDH1. All the families have the same mutation, an aspartic acid to asparagine substitution at codon 226, described previously.9,32,39 To determine whether the observed mutation has arisen more than once or if all the families share a common ancestor, haplotype analysis of the IMPDH1 region was performed in four families in which multiple affected individuals were available. SNPs flanking the IMPDH1 gene were typed in affected family members and haplotypes were reconstructed (data not shown). Two distinct haplotypes were found in the four families, suggesting the Asp226Asn mutation has arisen independently more than once.
NRL
Two nonpolymorphic variants were observed in NRL. One, Arg147Arg, is a silent substitution. The other variant, Gln174Arg, was found in an unaffected family member, has a low Grantham score and is probably benign. Thus, the prevalence of NRL mutations in this population was less than 1%.
PRPF3 (RP18)
Two families (1%) in the cohort had non-polymorphic missense changes in PRPF3. Both substitutions have been reported in other adRP families,15 and both are probably pathogenic by PolyPhen analysis.
PRPF8 (RP13)
Six families in the cohort have five different potential disease-causing variants in PRPF8. Three are amino acid substitutions, one is a deletion leading to a frame-shift, and the other is a splice-site mutation. Two of the amino acid substitutions, Phe2304Leu and Arg2310Gly, have been reported before and are known to be pathogenic.17 The previously unreported substitution, Ala2328Val, is predicted to be benign by PolyPhen analysis and has only a modest Grantham score based on the small chemical distance between alanine and valine. The splice-site variant is more likely to be pathogenic, based on sequence conservation, but splice-site prediction programs suggest that the variant sequence may be a better splice signal than the wild-type sequence. For each of these variants, only a single affected individual was availablefor testing. We conclude that four families have definitely pathogenic mutations and two have probably pathogenic mutations; however, evidence for pathogenicity of the two novel mutations is equivocal.
PRPF31 (RP11)
Twelve families in the cohort have possible disease-causing variants in PRPF31. One variant, Gly272Val, does not cosegregate with disease in the family. Nine variants are nonsense mutations, frameshift mutations, or alterations to splice-site consensus sequences, and are highly likely to be pathogenic. The two remaining variants, Ala291Pro and Cys299Arg, are predicted to be pathogenic, but the first was observed in a single affected individual only. The second was found in four affected members of one family. The Cys299Arg variant was also found in one at-risk, unaffected family member, but this is consistent with the high incidence of nonpenetrance for PRPF31 mutations. None of the 12 variants was found in control subjects. We conclude that 11 families (5.5%) have disease-causing mutations in PRPF31.
RDS and Digenic RDS-ROM1
Twenty families of the cohort have 15 different, potential disease-causing variants in peripherin/RDS. Most variants are amino acid substitutions, but one causes a premature termination after amino acid 45, another is a 12-bp deletion that causes an in-frame deletion of amino acids 206 to 209, and a third affects an intronexon junction (see Table 3). We also found a benign amino acid substitution in RDS: Leu45Phe, which was reported previously as a common variant in Caribbean populations.35
Of the 15 potentially pathogenic variants, 11 have been reported previously in other families with adRP. One of these, Leu185Pro, was originally reported as a cause of adRP but was later shown to cause disease only when inherited along with a null mutation in ROM1, that is, to cause digenic RP.18,48 After finding the Leu185Pro mutation in three affected members of one family in the cohort, we sequenced the ROM1 gene in these individuals. All three have a 1-bp insertion in exon 1 of ROM1, causing a frameshift and premature termination at codon 131. This ROM1 mutation has been observed in another family with digenic RP.48
Four RDS variants have not been reported previously. One, a Gly137Ser missense change, did not segregate with disease in the proband’s family and is apparently benign. The remaining three, Tyr141Cys, Ser198Arg, and Pro216Arg, are probably pathogenic. Each is at a conserved site in the protein and the amino acid changes are significant by PolyPhen analysis and Grantham score.
Thus, 18 families in the cohort (9%) have pathogenic mutations in RDS, and an additional family (0.5%) has mutations in both RDS and ROM1 and thus digenic (or diallelic) disease.
RHO
Fifty-four families of the cohort have 27 different, potential disease-causing variants in rhodopsin. Of these, 23 have been reported previously in other families with adRP. The remaining four were found in only a single proband each within the cohort. Most variants are amino acid substitutions, but one is a change at an intron/exon splice site, and another changes the termination codon to glutamine and adds an additional 58 amino acids to the protein.
Analysis of the four novel variants observed in our cohort suggests that three are potentially disease causing. Both the Leu46Arg and the Ser270Arg variants change amino acids in the transmembrane regions of the protein and both changes are likely to be disruptive. The ter349Gln variant is highly likely to be pathogenic, producing a protein that is 58 amino acids longer than the normal protein. A similar mutation, ter349Glu, is known to be one of the most severe rhodopsin mutations.69 The fourth novel variant, Thr70Met, was not found in subsequent testing of the affected father and affected brother of the proband and, therefore, must be benign.
Thus, 53 families (26.5%) in this cohort have a pathogenic rhodopsin mutation, by far the largest fraction of the total.
ROM1
Except for the digenic allele described in the RDS section, no pathogenic variants in ROM1 were observed in the cohort. Three missense changes and a silent substitution were found. Two of the missense changes, Arg229His and Met271Thr, have been reported previously in unaffected control subjects,64 and the third, Arg287Trp, was present in an unaffected family member. The prevalence of nondigenic, dominant-acting ROM1 mutations in this population is less than 1%.
RP1
Seven families (3.5%) of the cohort have three different disease-causing mutations in RP1. All have been reported previously in multiple adRP families. Two, Arg677ter and Gly273ter, are nonsense mutations that should cause premature protein termination; the other is a 5-bp deletion that should cause a frameshift and premature termination. We also detected two amino acid substitutions that can be classified as benign, His1034Arg and Leu1808Pro. The first is novel but was also found in two unaffected control subjects. The second, Leu1808Pro, was reported previously as possibly pathogenic,32 but subsequent testing revealed an IMPDH1 mutation in the same family; the IMPDH1 mutation is certainly the cause of disease in this family. To date, no missense mutations in RP1 are known to be pathogenic.70
RP9
No potentially pathogenic mutations were observed in PAP1, the putative RP9 gene.27 In the early stages of designing and optimizing primers for sequencing this gene, we observed a heterozygous His137Leu variant (ACA→ACT), identical with one of the two reported pathogenic mutations.27 Subsequent sequencing and analysis showed this to be a “paralogous variant”—that is, the result of simultaneous amplification of two highly similar sequences: PAP1 at 7p14.2 and a PAP1-like gene 20 kb distal to PAP1. By redesigning primers, we were able to eliminate this artifact. Both lack of PAP1 mutations in this cohort and our experience with the paralogous variant raise the possibility that PAP1 is not, in fact, the RP9-causing gene.
RPGR
Two families predicted to have adRP actually have X-linked RP (1%). Mutations in ORF 15 of RPGR were found in each family. Both mutations were small deletions, causing a frameshift and premature termination. One family had at least five known, affected females. Both families have more than four generations of affected individuals, but no male-to-male transmission.
Discussion
Prevalence of Mutations in adRP Genes
The mutations found in this survey are summarized in Table 4A. The findings are applicable largely to Americans of Western European origin. We identified definitely pathogenic or probably pathogenic changes in 107 families, that is, in 53.5% of the cohort (95% CI = 50.0%-57.0%). Of these, rhodopsin mutations are the most common cause of disease, representing 26.5% (23.4%-29.6%) of the total. RDS mutations account for the second largest group, 9% (7%-11%), and PRPF31 accounts for the third largest group, 5.5% (3.9%-7.1%). Among the other genes, IMPDH1, PRPF8, and RP1 account for 1% to 5% each, and CRX, PRPF3, and RPGR account for roughly 1% each. Digenic inheritance of RDS-ROM1 mutations was observed in one family. No mutations were found in CA4, FSCN2, NRL, or ROM1 (other than digenic). These latter genes are, most likely, rare causes of adRP in whites of Western European origin (Fig. 1).
Table 4.
Mutations in the adRP Cohort
| A. Summary | ||
|---|---|---|
| Gene | Probands (n) | % of Total |
| CA4 | 0 | 0.0 |
| CRX | 2 | 1.0 |
| FSCN2 | 0 | 0.0 |
| IMPDH1 | 5 | 2.5 |
| NRL | 0 | 0.0 |
| PRPF3 (RP18) | 2 | 1.0 |
| PRPF8 (RP13) | 6 | 3.0 |
| PRPF31 (RP11) | 11 | 5.5 |
| RDS | 18 | 9.0 |
| RDS-ROM1 digenic | 1 | 0.5 |
| RHO | 53 | 26.5 |
| ROM1 | 0 | 0.0 |
| RP1 | 7 | 3.5 |
| RP9 | 0 | 0.0 |
| RPGR | 2 | 1.0 |
| Total | 107 | 53.5 |
| B. Frequent Mutations | ||
|---|---|---|
| Gene | Mutation | % of Total |
| IMPDH1 | Asp226Asn | 2.5 |
| PRPF3 (RP18) | Thr494Met | 1.0* |
| PRPF8 (RP13) | Glu2331fs/ter 2358 | 1.0 |
| RDS | IVS2+3 A→T Pro216Leu Gly266Asp |
2.0 1.0 1.0 |
| RHO | Pro23His Gly106Trp Arg135Leu Arg135Trp Asp190Asn Pro347Leu |
10.0 1.0 1.5 2.0 1.0 1.0 |
| RP1 | Arg677ter Leu762fs/ter 777 |
1.5 1.5 |
| Total | 28.0 | |
| C. Frequent Exons or Regions | ||
|---|---|---|
| Gene | Region | % of Total |
| IMPDH1 (RP10) | Exon 7 | 2.5 |
| PRPF8 (RP13) | Exon 42 | 1.0 |
| RDS | Exon 2 | 7.0 |
| RHO | Exon 1 Exon 2 Exon 5 |
14.5 5.5 4.0 |
| RP1 | Nt 1948-2338 | 3.5 |
| Total | 38.0 | |
One per 200 in this study but 1% in Chakarova et al.15
Figure 1.
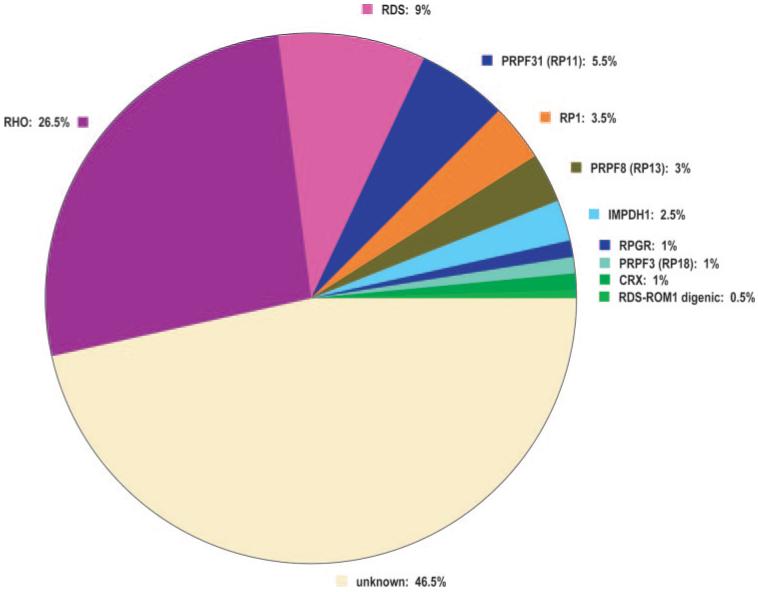
Fraction of dominant retinitis pigmentosa caused by mutations in known genes and in the remaining unknown genes.
Do Mutations in PAP1 Cause the RP9 Form of adRP?
We also failed to find mutations in PAP1, which has been proposed as the RP9 gene.27 Other groups have also failed to find PAP1 mutations in adRP families.31 One of the PAP1 “mutations” we detected in the cohort turned out to be a paralogous variant, that is, the result of PCR amplification of two nearly identical PAP1 gene copies. This variant is the same as one of the purported disease-causing mutations.27 These findings cast doubt on whether PAP1 is, in fact, the RP9 gene.
Common Mutations and Screening Strategies
Several “common” mutations account for a substantial fraction of the total in this population. Fourteen mutations (Table 4B) were each found in 1% or more of families. In aggregate, they account for 28% of the total, that is, for at least 5% of all RP cases, if the percent of adRP among all cases is 20% (a conservative estimate). Further, the common mutations and many of the remainder are clustered within just seven exons of IMPDH1, PRPF8, RDS, RHO, and RP1, accounting for 38% of the total (Table 4C). Thus, sequencing seven gene regions, spanned by 14 PCR primer sets (listed in Supplementary Tables, http://www.iovs.org/cgi/content/full/47/7/3052/DC1), will detect a large fraction of the total, making this a useful initial screening strategy for diagnostic testing. Note, though, that this conclusion applies to this ethnicgeographic group only, and that screening the remaining gene regions will potentially detect an additional 16% of mutations, most rare or unique.
The fraction of adRP cases caused by the genes tested in this cohort differs considerably from observations in other ethnic-geographic populations, as summarized in Table 1. One reason is that some of the common mutations are frequent as a result of founder effect and, thus, likely to be rare or absent from other populations. For example, the RDS IVS2+3 and RHO Pro23His mutations each descend from a common white ancestor and have not been reported in other ethnic groups19 (Shankar SP, et al. IOVS 2004;45:ARVO E-Abstract 3719). In contrast, several common mutations arise from independent, recurrent events, among them the IMPDH1 Asp226Asn, RHO Arg135Leu, RHO Arg135Trp, and RP1 Arg677ter mutations9,71 (and this publication). Whether these will be recurrent mutations in other populations has not been established.
Does Evaluation of Pedigrees and Clinical Findings Help to Prioritize Screening?
Before testing, we categorized the 200 adRP families by seveal criteria (Table 1 and Supplementary Tables, http://www.iovs.org/cgi/content/full/47/7/3052/DC1). Further, we have clinical data on affected members of most families, so we can compare phenotypes with genotypes. Is either the pedigree structure or the clinical picture a useful guide to the underlying gene and mutation?
Uncommon Retinal Findings in RP Families
Secondary retinal findings may be useful in prioritizing gene testing. For example, families with both dominant RP and complex macular disease are much more likely to have RDS mutations than predicted by prevalence alone.3,72,73 Thus, RDS screening should be a high priority in families with macular findings in some (not necessarily all) affected individuals. Like-wise, CRX mutations are more likely in families with cone-rod dystrophy, but because CRX mutations are rare among adRP families, this is not particularly helpful in setting priorities.
PRPF31 in adRP Families with Skipped Generations
Families with multiple skipped generations, but otherwise typical adRP, are more likely to have mutations in PRPF31 (RP11).74,75 Of the 11 families in the cohort with PRPF31 mutations, 5 (45%) have pedigree evidence of one or more skipped generations, which is significantly different from the overall fraction, 24%. Although other adRP genes, such as RP1,24 may show nonpenetrance, it is a more common phenomenon with PRPF31, making this a high-priority candidate in such cases.
X-Linkage in Autosomal Dominant Families
Families without male-to-male transmission but with affected females may have mutations in ORF 15 of RPGR.28,33,34 Thus, some large, multigeneration families with affected females may haveX-linked RP masquerading as autosomal dominant RP. For example, the two families with ORF 15 mutations in the cohort have five and seven affected females, respectively, and both were calculated to have odds favoring autosomal dominant inheritance of 103 or greater. However, in all cases, females are more mildly affected than males, which should be a strong indicator of X-linked inheritance.
Detectable Mutations in Families with a High Probability of Autosomal Dominant Inheritance
Finally, among the cohort of 200 families, 121 had a calculated odds ratio of 104 or greater favoring autosomal dominant over X-linked inheritance. Of these, 76 (63%) had detectable mutations. Conversely, 24 families had a calculated odds ratio of 1 or less—that is, either both modes are equally likely, or X-linkage is favored. Of the second set of families, 5 (21%) have detectable mutations, a highly significant difference. Thus, the probability calculations confirm the intuitive expectation: The more certain it is that a family has autosomal dominant inheritance, the more likely a mutation will be found.
Strengths and Limitations of Computational Analysis of Missense and Splice-Site Mutations
Establishing pathogenicity of novel, rare variants, particularly missense mutations and intronic variants, is challenging. We used a staged, systematic approach that incorporates segregation information (including prior reports), cross-species comparisons, and several computational tools (Table 3). However, it is possible that a few variants labeled “pathogenic” or “probably pathogenic” are not pathogenic; and that an occasional predicted “benign” variant is not benign. This problem has been addressed by others.35 Several mutations listed in Table 3 as “probably pathogenic,” based on all available information, are reported in pubic databases as definitely pathogenic. Thus, as a caution, where the evidence for pathogenicity is limited or ambiguous, these doubts should be incorporated into counseling of patients and families.
Supplementary Material
Acknowledgments
The authors thank the many families and patients who participated in this study, and the many clinicians who contributed patient and family samples to the project, in addition to the authors. They also thank Sarah Barton, Diane Bierke-Nelson, Lauri Black, Bernie Chodiricer, Patti Furman, James Garbern, Janet Glover, Brian Horsman, Henry Kaplan, Vivian Kim, Rachel LaFramboise, Sylvie Langlois, John Lehr, Janine Lewis, Jan Liebelt, Gabrielle Mettler, Kristy Morian, Jill Oversier, Mary Kay Pelias, Anjana Pettigrew, John Phillips, William Rameseur, David Saperstein, John Saunders, Jacqueline Siegel-Bartelt, Kent Small, Cathy Streinhorse, and Patricia Wheeler for access to patients and other assistance.
Supported by grants from the Foundation Fighting Blindness, the William Stamps Farish Fund, and the Hermann Eye Fund; and by National Eye Institute Grants EY07142 and EY05235. RAL is a Senior Scientific Investigator of Research to Prevent Blindness, which also provided unrestricted funds to the Cullen Eye Institute.
Footnotes
Disclosure: L.S. Sullivan, None; S.J. Bowne, None; D.G. Birch, None; D. Hughbanks-Wheaton, None; J.R. Heckenlively, None; R.A. Lewis, None; C.A. Garcia, None; R.S. Ruiz, None; S.H. Blanton, None; H. Northrup, None; A.I. Gire, None; R. Seaman, None; H. Duzkale, None; C.J. Spellicy, None; J. Zhu, None; S.P. Shankar, None; S.P. Daiger, None
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
References
- 1.Haim M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand Suppl. 2002;233:1–34. doi: 10.1046/j.1395-3907.2002.00001.x. [DOI] [PubMed] [Google Scholar]
- 2.Rebello G, Ramesar R, Vorster A, et al. Apoptosis-inducing signal sequence mutation in carbonic anhydrase IV identified in patients with the RP17 form of retinitis pigmentosa. Proc Natl Acad Sci USA. 2004;101:6617–6622. doi: 10.1073/pnas.0401529101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sohocki MM, Daiger SP, Bowne SJ, et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat. 2001;17:42–51. doi: 10.1002/1098-1004(2001)17:1<42::AID-HUMU5>3.0.CO;2-K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Milla E, Maseras M, Martinez-Gimeno M, et al. Genetic and molecular characterization of 148 patients with autosomal dominant retinitis pigmentosa (ADRP) Arch Soc Esp Oftalmol. 2002;77:481–484. [PubMed] [Google Scholar]
- 5.Wada Y, Abe T, Takeshita T, Sato H, Yanashima K, Tamai M. Mutation of human retinal fascin gene (FSCN2) causes autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2001;42:2395–2400. [PubMed] [Google Scholar]
- 6.Wada Y, Tamai M. Molecular genetic analysis for Japanese patients with autosomal dominant retinitis pigmentosa. Nippon Ganka Gakkai Zasshi. 2003;107:687–694. [PubMed] [Google Scholar]
- 7.Payne AM, Downes SM, Bessant DA, et al. Genetic analysis of the guanylate cyclase activator 1B (GUCA1B) gene in patients with autosomal dominant retinal dystrophies. J Med Genet. 1999;36:691–693. [PMC free article] [PubMed] [Google Scholar]
- 8.Sato M, Nakazawa M, Usui T, Tanimoto N, Abe H, Ohguro H. Mutations in the gene coding for guanylate cyclase-activating protein 2 (GUCA1B gene) in patients with autosomal dominant retinal dystrophies. Graefes Arch Clin Exp Ophthalmol. 2005;243:235–242. doi: 10.1007/s00417-004-1015-7. [DOI] [PubMed] [Google Scholar]
- 9.Bowne SJ, Sullivan LS, Blanton SH, et al. Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:559–568. doi: 10.1093/hmg/11.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kennan A, Aherne A, Palfi A, et al. Identification of an IMPDH1 mutation in autosomal dominant retinitis pigmentosa (RP10) revealed following comparative microarray analysis of transcripts derived from retinas of wild-type and Rho(-/-) mice. Hum Mol Genet. 2002;11:547–557. doi: 10.1093/hmg/11.5.547. [DOI] [PubMed] [Google Scholar]
- 11.Wada Y, Sandberg MA, McGee TL, Stillberger MA, Berson EL, Dryja TP. Screen of the IMPDH1 gene among patients with dominant retinitis pigmentosa and clinical features associated with the most common mutation, Asp226Asn. Invest Ophthalmol Vis Sci. 2005;46:1735–1741. doi: 10.1167/iovs.04-1197. [DOI] [PubMed] [Google Scholar]
- 12.Bessant DA, Payne AM, Mitton KP, et al. A mutation in NRL is associated with autosomal dominant retinitis pigmentosa. Nat Genet. 1999;21:355–356. doi: 10.1038/7678. [DOI] [PubMed] [Google Scholar]
- 13.Bessant DA, Payne AM, Plant C, Bird AC, Swaroop A, Bhattacharya SS. NRL S50T mutation and the importance of ‘founder effects’ in inherited retinal dystrophies. Eur J Hum Genet. 2000;8:783–787. doi: 10.1038/sj.ejhg.5200538. [DOI] [PubMed] [Google Scholar]
- 14.DeAngelis MM, Grimsby JL, Sandberg MA, Berson EL, Dryja TP. Novel mutations in the NRL gene and associated clinical findings in patients with dominant retinitis pigmentosa. Arch Ophthalmol. 2002;120:369–375. doi: 10.1001/archopht.120.3.369. [DOI] [PubMed] [Google Scholar]
- 15.Chakarova CF, Hims MM, Bolz H, et al. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:87–92. doi: 10.1093/hmg/11.1.87. [DOI] [PubMed] [Google Scholar]
- 16.Martinez-Gimeno M, Gamundi MJ, Hernan I, et al. Mutations in the pre-mRNA splicing-factor genes PRPF3, PRPF8, and PRPF31 in Spanish families with autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2003;44:2171–2177. doi: 10.1167/iovs.02-0871. [DOI] [PubMed] [Google Scholar]
- 17.McKie AB, McHale JC, Keen TJ, et al. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13) Hum Mol Genet. 2001;10:1555–1562. doi: 10.1093/hmg/10.15.1555. [DOI] [PubMed] [Google Scholar]
- 18.Kajiwara K, Hahn LB, Mukai S, Travis GH, Berson EL, Dryja TP. Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature. 1991;354:480–483. doi: 10.1038/354480a0. [DOI] [PubMed] [Google Scholar]
- 19.Dryja TP, McGee TL, Hahn LB, et al. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N Engl J Med. 1990;323:1302–1307. doi: 10.1056/NEJM199011083231903. [DOI] [PubMed] [Google Scholar]
- 20.Dryja TP, McEvoy JA, McGee TL, Berson EL. Novel rhodopsin mutations Gly114Val and Gln184Pro in dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2000;41:3124–3127. [PubMed] [Google Scholar]
- 21.Bascom RA, Liu L, Heckenlively JR, Stone EM, McInnes RR. Mutation analysis of the ROM1 gene in retinitis pigmentosa. Hum Mol Genet. 1995;4:1895–1902. doi: 10.1093/hmg/4.10.1895. [DOI] [PubMed] [Google Scholar]
- 22.Dryja TP, Hahn LB, Kajiwara K, Berson EL. Dominant and digenic mutations in the peripherin/RDS and ROM1 genes in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1997;38:1972–1982. [PubMed] [Google Scholar]
- 23.Sakuma H, Inana G, Murakami A, et al. A heterozygous putative null mutation in ROM1 without a mutation in peripherin/RDS in a family with retinitis pigmentosa. Genomics. 1995;27:384–386. doi: 10.1006/geno.1995.1066. [DOI] [PubMed] [Google Scholar]
- 24.Sullivan LS, Heckenlively JR, Bowne SJ, et al. Mutations in a novel retina-specific gene cause autosomal dominant retinitis pigmentosa. Nat Genet. 1999;22:255–259. doi: 10.1038/10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Payne AM, Vithana E, Khaliq S, et al. RP1 protein truncating mutations predominate at the RP1 adRP locus. Invest Ophthalmol Vis Sci. 2000;41:4069–4073. [PubMed] [Google Scholar]
- 26.Pierce EA, Quinn T, Meehan T, McGee TL, Berson EL, Dryja TP. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat Genet. 1999;22:248–254. doi: 10.1038/10305. [DOI] [PubMed] [Google Scholar]
- 27.Keen TJ, Hims MM, McKie AB, et al. Mutations in a protein target of the Pim-1 kinase associated with the RP9 form of autosomal dominant retinitis pigmentosa. Eur J Hum Genet. 2002;10:245–249. doi: 10.1038/sj.ejhg.5200797. [DOI] [PubMed] [Google Scholar]
- 28.Rozet JM, Perrault I, Gigarel N, et al. Dominant X linked retinitis pigmentosa is frequently accounted for by truncating mutations in exon ORF15 of the RPGR gene. J Med Genet. 2002;39:284–285. doi: 10.1136/jmg.39.4.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roepman R, Bauer D, Rosenberg T, et al. Identification of a gene disrupted by a microdeletion in a patient with X-linked retinitis pigmentosa (XLRP) Hum Mol Genet. 1996;5:827–833. doi: 10.1093/hmg/5.6.827. [DOI] [PubMed] [Google Scholar]
- 30.Ramesar RS, Roberts L, Rebello G, et al. Retinal degenerative disorders in Southern Africa: a molecular genetic approach. Adv Exp Med Biol. 2003;533:35–40. doi: 10.1007/978-1-4615-0067-4_5. [DOI] [PubMed] [Google Scholar]
- 31.Ziviello C, Simonelli F, Testa F, et al. Molecular genetics of autosomal dominant retinitis pigmentosa (ADRP): a comprehensive study of 43 Italian families. J Med Genet. 2005;42:e47. doi: 10.1136/jmg.2005.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bowne SJ, Daiger SP, Hims MM, et al. Mutations in the RP1 gene causing autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1999;8:2121–2128. doi: 10.1093/hmg/8.11.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mears AJ, Hiriyanna S, Vervoort R, et al. Remapping of the RP15 locus for X-linked cone-rod degeneration to Xp11.4-p21.1, and identification of a de novo insertion in the RPGR exon ORF15. Am J Hum Genet. 2000;67:1000–1003. doi: 10.1086/303091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vervoort R, Lennon A, Bird AC, et al. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet. 2000;25:462–466. doi: 10.1038/78182. [DOI] [PubMed] [Google Scholar]
- 35.Stone EM. Finding and interpreting genetic variations that are important to ophthalmologists. Trans Am Ophthalmol Soc. 2003;101:437–484. [PMC free article] [PubMed] [Google Scholar]
- 36.Lathrop GM, Lalouel JM, Julier C, Ott J. Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA. 1984;81:3443–3446. doi: 10.1073/pnas.81.11.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dausset J, Cann H, Cohen D, Lathrop M, Lalouel JM, White R. Centre d’etude du polymorphisme humain (CEPH): collaborative genetic mapping of the human genome. Genomics. 1990;6:575–577. doi: 10.1016/0888-7543(90)90491-c. [DOI] [PubMed] [Google Scholar]
- 38.Sohocki MM, Sullivan LS, Mintz-Hittner HA, et al. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am J Hum Genet. 1998;63:1307–1315. doi: 10.1086/302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowne SJ, Sullivan LS, Mortimer SE, et al. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2006;47:34–42. doi: 10.1167/iovs.05-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 41.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 44.Swain PK, Chen S, Wang QL, et al. Mutations in the cone-rod homeobox gene are associated with the cone-rod dystrophy photoreceptor degeneration. Neuron. 1997;19:1329–1336. doi: 10.1016/s0896-6273(00)80423-7. [DOI] [PubMed] [Google Scholar]
- 45.Gamundi MJ, Hernan I, Maseras M, et al. Sequence variations in the retinal fascin FSCN2 gene in a Spanish population with autosomal dominant retinitis pigmentosa or macular degeneration. Mol Vis. 2005;11:922–928. [PubMed] [Google Scholar]
- 46.Meins M, Gruning G, Blankenagel A, et al. Heterozygous ‘null allele’ mutation in the human peripherin/RDS gene. Hum Mol Genet. 1993;2:2181–2812. doi: 10.1093/hmg/2.12.2181. [DOI] [PubMed] [Google Scholar]
- 47.Wells J, Wroblewski J, Keen J, et al. Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nat Genet. 1993;3:213–218. doi: 10.1038/ng0393-213. [DOI] [PubMed] [Google Scholar]
- 48.Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science. 1994;264:1604–1608. doi: 10.1126/science.8202715. [DOI] [PubMed] [Google Scholar]
- 49.Feist RM, White MF, Jr, Skalka H, Stone EM. Choroidal neovascularization in a patient with adult foveomacular dystrophy and a mutation in the retinal degeneration slow gene (Pro 210 Arg) Am J Ophthalmol. 1994;118:259–260. doi: 10.1016/s0002-9394(14)72913-7. [DOI] [PubMed] [Google Scholar]
- 50.Budu Hayasaka S, Matsumoto M, Yamada T, Zhang XY, Hayasaka Y. Peripherin/RDS gene mutation (Pro210Leu) and polymorphisms in Japanese patients with retinal dystrophies. Jpn J Ophthalmol. 2001;45:355–358. doi: 10.1016/s0021-5155(01)00334-3. [DOI] [PubMed] [Google Scholar]
- 51.Fishman GA, Stone E, Gilbert LD, Vandenburgh K, Sheffield VC, Heckenlively JR. Clinical features of a previously undescribed codon 216 (proline to serine) mutation in the peripherin/retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Ophthalmology. 1994;101:1409–1421. doi: 10.1016/s0161-6420(94)31156-0. [DOI] [PubMed] [Google Scholar]
- 52.Sheffield VC, Fishman GA, Beck JS, Kimura AE, Stone EM. Identification of novel rhodopsin mutations associated with retinitis pigmentosa by GC-clamped denaturing gradient gel electrophoresis. Am J Hum Genet. 1991;49:699–706. [PMC free article] [PubMed] [Google Scholar]
- 53.Dryja TP, McGee TL, Reichel E, et al. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343:364–366. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- 54.Inglehearn CF, Keen TJ, Bashir R, et al. A completed screen for mutations of the rhodopsin gene in a panel of patients with autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1992;1:41–45. doi: 10.1093/hmg/1.1.41. [DOI] [PubMed] [Google Scholar]
- 55.Dryja TP, Hahn LB, Cowley GS, McGee TL, Berson EL. Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88:9370–9374. doi: 10.1073/pnas.88.20.9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sung CH, Davenport CM, Hennessey JC, et al. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88:6481–6485. doi: 10.1073/pnas.88.15.6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fuchs S, Kranich H, Denton MJ, et al. Three novel rhodopsin mutations (C110F, L131P, A164V) in patients with autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1994;3:1203. doi: 10.1093/hmg/3.7.1203. [DOI] [PubMed] [Google Scholar]
- 58.Vaithinathan R, Berson EL, Dryja TP. Further screening of the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. Genomics. 1994;21:461–463. doi: 10.1006/geno.1994.1301. [DOI] [PubMed] [Google Scholar]
- 59.Antinolo G, Sanchez B, Borrego S, Rueda T, Chaparro P, Cabeza JC. Identification of a new mutation at codon 171 of rhodopsin gene causing autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1994;3:1421. doi: 10.1093/hmg/3.8.1421. [DOI] [PubMed] [Google Scholar]
- 60.Scott K, Sieving PA, Bingham E, et al. Rhodopsin mutations associated with autosomal dominant retinitis pigmentosa. Am J Hum Genet. 1993;53:147. [Google Scholar]
- 61.Keen TJ, Inglehearn CF, Lester DH, et al. Autosomal dominant retinitis pigmentosa: four new mutations in rhodopsin, one of them in the retinal attachment site. Genomics. 1991;11:199–205. doi: 10.1016/0888-7543(91)90119-y. [DOI] [PubMed] [Google Scholar]
- 62.Macke JP, Davenport CM, Jacobson SG, et al. Identification of novel rhodopsin mutations responsible for retinitis pigmentosa: implications for the structure and function of rhodopsin. Am J Hum Genet. 1993;53:80–89. [PMC free article] [PubMed] [Google Scholar]
- 63.Bell C, Converse CA, Hammer HM, Osborne A, Haites NE. Rhodopsin mutations in a Scottish retinitis pigmentosa population, including a novel splice site mutation in intron four. Br J Ophthalmol. 1994;78:933–938. doi: 10.1136/bjo.78.12.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bascom RA, Liu L, Humphries P, Fishman GA, Murray JC, McInnes RR. Polymorphisms and rare sequence variants at the ROM1 locus. Hum Mol Genet. 1993;2:1975–1977. doi: 10.1093/hmg/2.11.1975. [DOI] [PubMed] [Google Scholar]
- 65.Sharon D, Sandberg MA, Rabe VW, Stillberger M, Dryja TP, Berson EL. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am J Hum Genet. 2003;73:1131–1146. doi: 10.1086/379379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miller MP, Kumar S. Understanding human disease mutations through the use of interspecific genetic variation. Hum Mol Genet. 2001;10:2319–2328. doi: 10.1093/hmg/10.21.2319. [DOI] [PubMed] [Google Scholar]
- 67.Briscoe AD, Gaur C, Kumar S. The spectrum of human rhodopsin disease mutations through the lens of interspecific variation. Gene. 2004;332:107–118. doi: 10.1016/j.gene.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 68.Freund CL, Gregory-Evans CY, Furukawa T, et al. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell. 1997;91:543–553. doi: 10.1016/s0092-8674(00)80440-7. [DOI] [PubMed] [Google Scholar]
- 69.Bessant DA, Khaliq S, Hameed A, et al. Severe autosomal dominant retinitis pigmentosa caused by a novel rhodopsin mutation (Ter349Glu). Mutations in brief no. 208. Online Hum Mutat. 1999;13:83. doi: 10.1002/(SICI)1098-1004(1999)13:1<83::AID-HUMU12>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 70.Berson EL, Grimsby JL, Adams SM, et al. Clinical features and mutations in patients with dominant retinitis pigmentosa-1 (RP1) Invest Ophthalmol Vis Sci. 2001;42:2217–2224. [PubMed] [Google Scholar]
- 71.Schwartz SB, Aleman TS, Cideciyan AV, Swaroop A, Jacobson SG, Stone EM. De novo mutation in the RP1 gene (Arg677ter) associated with retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2003;44:3593–3597. doi: 10.1167/iovs.03-0155. [DOI] [PubMed] [Google Scholar]
- 72.Felbor U, Schilling H, Weber BH. Adult vitelliform macular dystrophy is frequently associated with mutations in the peripherin/RDS gene. Hum Mutat. 1997;10:301–309. doi: 10.1002/(SICI)1098-1004(1997)10:4<301::AID-HUMU6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 73.Kajiwara K, Sandberg MA, Berson EL, Dryja TP. A null mutation in the human peripherin/RDS gene in a family with autosomal dominant retinitis punctata albescens. Nat Genet. 1993;3:208–212. doi: 10.1038/ng0393-208. [DOI] [PubMed] [Google Scholar]
- 74.McGee TL, Devoto M, Ott J, Berson EL, Dryja TP. Evidence that the penetrance of mutations at the RP11 locus causing dominant retinitis pigmentosa is influenced by a gene linked to the homologous RP11 allele. Am J Hum Genet. 1997;61:1059–1066. doi: 10.1086/301614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vithana EN, Abu-Safieh L, Pelosini L, et al. Expression of PRPF31 mRNA in patients with autosomal dominant retinitis pigmentosa: a molecular clue for incomplete penetrance? Invest Ophthalmol Vis Sci. 2003;44:4204–4209. doi: 10.1167/iovs.03-0253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.