

Abstract
Deep brain stimulation (DBS) of the globus pallidus pars interna (GPi) is an effective therapy option for controlling the motor symptoms of medication-refractory Parkinson's disease and dystonia. Despite the clinical successes of GPi DBS, the precise therapeutic mechanisms are unclear and questions remain on the optimal electrode placement and stimulation parameter selection strategies. In this study, we developed a three-dimensional computational model of GPi-DBS in nonhuman primates to investigate how membrane channel dynamics, synaptic inputs, and axonal collateralization contribute to the neural responses generated during stimulation. We focused our analysis on three general neural elements that surround GPi-DBS electrodes: GPi somatodendritic segments, GPi efferent axons, and globus pallidus pars externa (GPe) fibers of passage. During high-frequency electrical stimulation (136 Hz), somatic activity in the GPi showed interpulse excitatory phases at 1–3 and 4–5.5 ms. When including stimulation-induced GABAA and AMPA receptor dynamics into the model, the somatic firing patterns continued to be entrained to the stimulation, but the overall firing rate was reduced (78.7 to 25.0 Hz, P < 0.001). In contrast, axonal output from GPi neurons remained largely time-locked to each pulse of the stimulation train. Similar entrainment was also observed in GPe efferents, a majority of which have been shown to project through GPi en route to the subthalamic nucleus. The models suggest that pallidal DBS may have broader network effects than previously realized and the modes of therapy may depend on the relative proportion of GPi and/or GPe efferents that are directly affected by the stimulation.
INTRODUCTION
Multiple movement disorders, including Parkinson's disease and dystonia, are characterized by abnormal neuronal activity in the globus pallidus pars interna (GPi) (Hutchison et al. 1994, 1997; Vitek et al. 1999). In 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated monkeys, for instance, parkinsonian symptoms are present with altered GPi neuronal firing rates (Filion and Tremblay 1991), firing patterns (Wichmann and Soares 2006), oscillations (Nini et al. 1995), and receptive fields (Filion et al. 1988). Since the GPi is a primary output nucleus of the basal ganglia sending GABAergic projections to the ventral tier of thalamus, the centromedian-parafascicular nucleus of thalamus, and the pedunculopontine tegmental nucleus (PPN) (Baron et al. 2001; Kuo and Carpenter 1973; Parent et al. 2001; Shink et al. 1997; Sidibe et al. 1997), pathological activity within the GPi can have widely distributed network effects (Mitchell et al. 1989).
Surgical ablation of the posteroventral GPi is one approach to control the motor signs of Parkinson's disease (Bakay et al. 1992) and primary generalized dystonia (Lozano et al. 1997). Chronic high-frequency electrical stimulation (HFS) of the same region is another approach to achieve equally therapeutic effects (Krauss et al. 2004; Volkmann 2004). Unlike the immutable effects of GPi lesioning, however, GPi deep brain stimulation (DBS) enables the clinician to tailor stimulation parameter settings postoperatively to maximize efficacy and minimize the induction of undesirable side effects (Kumar 2002; Volkmann et al. 2006). The juxtaposition of therapeutic outcomes generated by DBS and lesioning led to the hypothesis that HFS effectively silences the stimulated nucleus and thereby mitigates excessive GABAergic input into the motor thalamus and brain stem (Toth and Tomka 1968). Several modeling studies have argued that although HFS may inhibit local somatic activity, action potentials can be elicited in the axonal processes of the neuron, leading paradoxically to stimulus-driven output activity (McIntyre et al. 2004; Miocinovic et al. 2006). Most experimental studies investigating GPi-HFS support these predictions showing that GPi-HFS reduces somatic firing rates in the GPi (Boraud et al. 1996; Dostrovsky et al. 2000; Wu et al. 2001) as well as in one of its downstream targets, the ventrolateral nucleus of thalamus, with inhibitory latencies in the latter case consistent with monosynaptic GABAergic signaling (Anderson et al. 2003). GPi-HFS has also been shown to induce more regular firing patterns in downstream thalamic nuclei (Montgomery 2006; Pralong et al. 2003). Moreover, similar changes in somatic firing patterns have been observed in the GPi during GPi-HFS (Bar-Gad et al. 2004) with modulatory effects propagating to regions of the GPi that exceed predictions for the spread of suprathreshold current (Wu et al. 2001).
In this study, we developed an anatomically and physiologically realistic computational model of GPi-DBS to investigate how membrane dynamics, synaptic input, and axonal collateralization contribute to these observed changes. The model simulates DBS with scaled clinical electrodes implanted in the caudal (sensorimotor) GPi of a nonhuman primate. Three-dimensional (3-D) morphologies of individual pallidal neurons were positioned within the context of a reconstructed volume of the basal ganglia. We used the model system to first investigate what membrane channel and synaptic properties contribute to the experimentally observed somatic responses to HFS. We then evaluated the GPi axonal output induced by clinically relevant stimulation parameters. Finally, we incorporated globus pallidus pars externa (GPe) projection neurons, of which a majority course through and at times collateralize within GPi en route to the subthalamic nucleus (STN) and substantia nigra pars reticulata (SNr) (Parent and Parent 2002; Sato et al. 2000a), and tested the hypothesis that GPi-DBS can also excite these fibers of passage. The model provides a tool to examine how various stimulation paradigms affect the neural output of the pallidum and to make predictions in terms of stimulation-induced network effects within the basal ganglia. Preliminary results from this work have been presented in abstract form (Johnson et al. 2007).
METHODS
Neuron morphologies
Our model system used a 3-D reconstruction of the basal ganglia from a Cynomolgus monkey (Macaca fascicularis) (Martin and Bowden 2000) to define the boundaries of individual nuclei of interest (i.e., GPi and GPe) (Miocinovic et al. 2007). Biotinylated dextran amine (BDA) labeled GPi and GPe neurons from Cynomolgus monkeys were reconstructed in 3-D using Neurolucida (MicroBrightField, Williston, VT) as shown in Fig. 4 of Parent et al. (2001) and Fig. 5 of Sato et al. (2000a). Somatodendritic processes of GPi neurons were verified by counterstaining with cytochrome oxidase and the BDA labeling was sufficient to easily follow the axonal processes through each sagittal section. These GPi neurons consisted of ovoid, spindle-shaped, or triangular cell bodies with two to five long, but poorly branched, primary dendrites with dendritic fields typically spanning ≤1 mm along the rostrocaudal and/or dorsoventral planes but <300 μm along the mediolateral plane. Their neuronal morphologies (Fig. 1A) were distributed within the atlas-based reconstruction according to anatomically realistic orientations and axonal trajectories that followed dorsally along the lenticular fasciculus (LF) on leaving the GPi (Baron et al. 2001; Parent and Parent 2004). GPe neuron morphologies (Fig. 1B) were similarly distributed within the basal ganglia framework and oriented in 3-D such that their axonal processes extended through and collateralized within GPi and subsequently targeted STN (Sato et al. 2000a; Smith et al. 1994). Since it is unknown whether these axons preferentially follow the LF or the more ventral ansa lenticularis (AL) fiber tract on exiting the GPi, GPe axonal trajectories were given a random dorsoventral distribution (Fig. 1C). An iterative algorithm developed in MATLAB (The MathWorks, Natick, MA) converted truncated versions of these morphology files into segments with length and thickness parameters defined individually for the soma, dendrites, and axons in the case of GPi cells (Fig. 1C) and only the axons and associated collaterals in the case of GPe cells. Multicompartment cable models of GPi neurons and GPe axons were simulated within NEURON v6.1 (Hines and Carnevale 1997). The model scripts are available on the ModelDB website (http://senselab.med.yale.edu/modeldb).
FIG. 1.

Anatomical reconstructions of (A) GPi and (B) GPe projection neurons were generated from biotinylated dextran amine-labeled pallidal neurons as described in Parent et al. (2001) and Sato et al. (2000a). C: these neurons were oriented within a 3-D atlas-based reconstruction of monkey pallidum containing a DBS lead with electrode contacts positioned primarily in GPi. D: from the neuron reconstructions, GPi membrane model compartments were created for axonal, somatic, and dendritic segments and instantiated with physiologically relevant passive properties, ion channels, and distributions of AMPA and GABAA receptors. E: the 3-D position of each compartment was then transformed into polar coordinate space with respect to the DBS lead. Extracellular stimulus amplitudes applied to each compartment were defined according to potential distributions simulated in an axisymmetic finite element model of neural tissue, which consisted of a 250 μm layer of encapsulation tissue (0.1 S/m) between the DBS lead and the bulk tissue (0.30 S/m). 3-D, 3-dimensional; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; DBS, deep brain stimulation; GABAA, γ-aminobutyric acid type A; GPe, globus pallidus pars externa; GPi, globus pallidus pars interna.
Membrane properties
Based on in vitro electrical properties, two distinct types of neurons are known to exist within the entopeduncular nucleus (EP), which is the rat homolog of monkey GPi (Nakanishi et al. 1990, 1991). Type I neurons exhibit anomalous rectification typical of Ih currents and the capacity for producing high-frequency activity with minimal spike adaptation. Given that the majority of EP neurons (80%) display these membrane characteristics, type I cells are thought to constitute the GABAergic projection neurons that target thalamic and brain stem nuclei. In contrast, type II neurons display small anomalous rectification, robust spike adaptation, and a steady repolarization after a hyperpolarizing pulse indicative of transient A-current. For the purposes of this study, however, we modeled only type I cells.
All somatic and dendritic compartments of the GPi neuron model were assigned a series of passive membrane properties (Table 1) (Kita 2001; Nakanishi et al. 1990, 1991; Shin and Carlen 2008; Shin et al. 2007). The action potential threshold (APth) for these cells was identified as the voltage at which the derivative of the membrane voltage (dVm/dt) was 2SDs larger than its mean during the interspike interval (Atherton and Bevan 2005). Since rat EP neurons are spontaneously active, the resting membrane potential was defined as the point halfway between the action potential repolarization trough and the APth (Shin et al. 2007). Inclusion of ion channel mechanisms was based on previous reports of rat EP neurons as well as functionally related neurons in the substantia nigra pars reticulata (SNr) (Atherton and Bevan 2005). Ion channels were placed in both somatic and dendritic compartments since it has been reported previously that APs generated in the soma can backpropagate into the dendrites (Atherton and Bevan 2005; Hausser et al. 1995). In cases where ion channel activation and inactivation functions have not yet been characterized for EP or SNr neurons, channel kinetic equations were modeled from those reported in other brain regions (Table 2). Parameter values determined from these in vitro experiments were scaled with Q10 values to match in vivo conditions at 36°C (see the appendix). Channel conductances were then adjusted manually to match the model's somatic activity to specific electrophysiological behaviors of rat EP neurons (Nakanishi et al. 1990) (see Fig. 2).
TABLE 1.
General membrane model parameters
| Model Parameter | Experimental Value | Region | Source |
|---|---|---|---|
| Vm = −57.1 mV | Vm < −50 mV | EP | Nakanishi et al. 1990; Shin et al. 2007 |
| Rin = 101 MΩ | Rin = 112 ± 38 MW | EP | Nakanishi et al. 1990 |
| τm = 14.7 ms | τm = 14 ms | GP | Hanson et al. 2004; Kita and Kitai 1991 |
| ρa = 174Ω·cm | ρa = 174 Ω·cm | GP | Hanson et al. 2004 |
TABLE 2.
Active channel properties
| Current | Description | gion, S/cm2 |
|---|---|---|
| NaF | Fast-acting sodium current | 2.0e-2 |
| NaL | Sodium leakage current | 1.8e-5 (in vitro) |
| 2.8e-5 (in vivo) | ||
| HCN | Hyperpolarization-activated inward cation current | 2.0e-4 |
| KDRf | Fast rectifier potassium (Kv3.1) current | 4.0e-3 |
| KDRs | Delayed rectifier potassium (Kv2.1) current | 1.0e-3 |
| SK | Small-conductance calcium-activated potassium current | 1.0e-5 |
| CaL | L-type high-voltage–activated calcium current | 5.0e-4 |
| CaN | N-type high-voltage–activated calcium current | 2.0e-3 |
FIG. 2.

Membrane properties of the multicompartment cable models of GPi neurons were fit to data from in vitro studies of rat entopeduncular nucleus (EP) neurons (Nakanishi et al. 1990; Shin et al. 2007). A: simulations of somatic activity yielded comparable action potential and resting membrane potential characteristics to those observed experimentally. B: the firing rate depended on the conductance of the sodium leakage channels (gNaL), and this parameter was used to tune the model to produce firing rates consistent with in vitro EP neurons (18 μS/cm2) as well as in vivo GPi spike recording data (28 μS/cm2) from nonhuman primates. C: current–frequency plots demonstrated that model neurons could generate high firing rates during intracellular current injection with (D) little spike accommodation. E: in a hyperpolarized state, model neurons also responded to a depolarizing pulse with both short-duration sodium and long-duration calcium spikes (compare with Fig. 2 from Nakanishi et al. 1990). F: using a gNaL of 18 μS/cm2, simulations were able to capture hyperpolarization-activated currents that (G) were consistent with experimental recordings (compare with Fig. 1 from Nakanishi et al. 1990).
Myelinated axons used in the GPi neuron simulations consisted of a double-cable model that included representations of the nodes of Ranvier (diameter, 1.4 μm), paranodal (1.4 μm) and internodal (1.6 μm) segments of the axon, and a myelin sheath (outer diameter, 2.0 μm) (McIntyre et al. 2002, 2004; Miocinovic et al. 2006). Nodal compartments were instantiated with nonlinear fast Na+ channels, persistent sodium channels, slow K+ channels, and a leakage current. The paranodal segment included a slow K+ current, whereas the internodal and myelin segments contained only passive mechanisms (membrane capacitance of 2 μF/cm2, axoplasmic resistivity of 70 Ω·cm). These axonal properties were also used to model GPe axons that collateralized en passant through GPi. However, histological studies have suggested that axonal collaterals making synaptic boutons within the pallidum may be unmyelinated and of a smaller diameter than the primary efferent processes (Fox et al. 1975; Galvan et al. 2005; Kita and Kitai 1987; Korotchenko 1979). Given these unknowns in the GPe model, we investigated how collateral myelination and diameter affect the response of fibers of passage to deep brain stimulation. Unmyelinated axon compartments (100 μm in length) were modeled with modified Hodgkin–Huxley membrane dynamics (“hh” mechanism in NEURON) as described by Grill et al. (2008). The sodium channel conductance was increased from 0.24 to 0.35 S/cm2 to generate a stable resting membrane potential and facilitate antidromic and orthodromic action potential propagation through the axonal branches. All simulations were run at 36°C.
Synaptic properties
Anatomical tracing studies have shown that GPi cells are innervated by glutamatergic afferents arising from the subthalamic nucleus (STN) (Carpenter et al. 1981) and GABAergic afferents originating from the striatum and GPe (Percheron et al. 1984), thereby forming sites of convergent integration (Bolam and Smith 1992). More specifically, in primates, GPi neurons are richly innervated by dense baskets of GABAergic terminals around both the soma and the dendrites (Hazrati et al. 1990). γ-Aminobutyric acid (GABA)-positive boutons from the striatum account for about 32% of the axosomatic terminals and 80% of the axodendritic terminals in GPi (Shink and Smith 1995). Indirect pathway GABAergic afferents from the GPe represent an additional 48% of axosomatic boutons, whereas these processes make little to no contact with dendritic segments in GPi (Shink and Smith 1995). Glutamatergic afferents from the STN also target GPi neurons making sporadic synapses on both soma and dendrite compartments (∼10% of all terminals) (Bolam and Smith 1992; Clarke and Bolam 1998; Shink and Smith 1995). The substantially higher proportion of GABAergic compared with glutamatergic synapses is thought to facilitate strong gating effects by striatal and GPe neurons on GPi output (Kita 2001).
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and GABAA receptors were incorporated into the GPi neuron models with both sets of receptors randomly distributed along the somatic and dendritic arbors according to their relative ratios (Fig. 1D). In total, receptors were present in 7/15 somatic compartments (1 AMPA and 6 GABAA) and 38/60 dendritic compartments (4 AMPA and 34 GABAA). To simulate a synaptic event, an intracellular current pulse was generated in a virtual presynaptic terminal triggering the delivery of a fixed pulse (1 mM) of transmitter occurring over 1 ms to a postsynaptic compartment that used a minimal kinetic first-order model of either a glutamate-based AMPA receptor or GABA-based GABAA receptor (Destexhe et al. 1994). The postsynaptic current generated by these models was dependent on the synaptic conductances (gAMPA and gGABAA). Synaptic conductances were tuned to reproduce firing rate and bursting features consistent with GPi spike recordings performed in monkeys treated with MPTP. This process involved generating synaptic events based on a representative set of input spike raster scans from extracellular microelectrode recordings in the putamen, GPe, and STN in the same MPTP-treated monkeys. This tuning process was simplified by using a single combination of recordings for each simulation and applying those spike patterns to all afferent axonal compartments of the same subtype. A collection of these simulations (n = 10 different sets of recorded raster scans) was then used to tune the synaptic conductances.
Neuronal recordings in parkinsonian monkeys
Female rhesus macaque monkeys (Macaca mulatta, ∼15 yr old) were treated with intracarotid injections of MPTP and evaluated behaviorally for parkinsonian contralateral limb rigidity and bradykinesia. For more methodological details, see Hashimoto et al. (2003). Briefly, the monkeys were implanted with two cranial chambers for microdrive-guided insertions of single tungsten microelectrodes (FHC, Bowdoin, ME) or linear arrays of 12 microelectrodes with each site spaced 300 μm apart (NeuroNexus, Ann Arbor, MI). All procedures complied with the National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the Cleveland Clinic IACUC. Microelectrode tracts were made along the coronal plane and cells were recorded in the putamen (n = 35), STN (n = 13), GPe (n = 46), and GPi (n = 38). Wide-band spike recordings were sampled at 25 kHz through an Alpha-Lab system (Alpha Omega, Nazareth, Israel) and band-pass filtered between 500 and 5,000 Hz. Raw data traces were thresholded and sorted off-line with Off-line Sorter (Plexon, Dallas, TX). Raster scans from these recordings were then used as input signals for weighting synaptic conductances within the GPi model.
DBS electric field model
Within the 3-D model, a monkey-scaled version of a human DBS lead was positioned along the coronal plane at an angle of 15° and along the sagittal plane at an angle of 25° such that most of the electrode contacts were present in the caudal portion of GPi (see Fig. 1C) (Gross et al. 1997; Siegfried and Lippitz 1994). The scaled DBS lead consisted of four cylindrical electrodes, each with a diameter of 750 μm and height of 500 μm (Elder et al. 2005). These dimensions were consistent with the relative volume of the Medtronic 3387 DBS lead in the human GPi. Briefly, the human GPi (478 mm3) (Yelnik 2002) is about 9.6-fold larger than the atlas-based reconstruction of the Cynomolgus monkey GPi used in this study (50 mm3). The volume ratio consumed by one of the cylindrical DBS electrodes was a comparable 8.6 (1.90 mm3 in the human and 0.22 mm3 in the monkey), although it should be noted that the proportional surface area for the scaled DBS electrode was slightly higher than that of a Medtronic 3387 DBS electrode (6.0 mm2 in the human and 2.4 mm2 in the monkey). Model neurons with segments that overlapped with any part of the DBS lead were removed from the analysis. This modeling criterion resulted in a remaining population of 63 GPi and 83 GPe neurons.
An axisymmetric finite-element model (FEM) of neural tissue was constructed around the DBS lead using COMSOL Multiphysics v3.3 (COMSOL, Burlington, MA). The FEM consisted of a mesh of tetrahedral elements within a 30 × 30-mm surface with the outside boundary set to a ground potential. Electrical properties of neural tissue were defined as homogeneous and isotropic (0.3 S/m) (Miocinovic et al. 2006), except for a 0.25-mm-thick section of encapsulation tissue at the electrode surface (0.1 S/m) (Grill and Mortimer 1994; Haberler et al. 2000; Moss et al. 2004). The stimulated metal contact boundaries were given a voltage and the potential distribution generated in the tissue was calculated using a frontal solution method of the Laplace equation (Fig. 1E). The model assumed ideal electrode behavior (i.e., no voltage drop at the electrode–tissue interface) and that the electric fields scaled linearly through the bulk tissue with increasing stimulation amplitude. Electrode impedances were calculated to be 3.4 kΩ (Butson et al. 2006).
The stimulus pulse train was based on experimentally recorded waveforms generated by a voltage-controlled implantable pulse generator (IPG; Medtronic, Minneapolis, MN). The stimulus waveform from the IPG had a pseudomonophasic profile consisting of a 90-μs cathodic phase followed by a 3-ms anodic phase separated by a 400-μs interphase delay. The waveform was then filtered according to a Fourier FEM simulation to incorporate the capacitive properties of the electrode–tissue interface (Butson and McIntyre 2005). The filtered waveform (Fig. 1E, top) was then applied to each neuron membrane compartment with peak cathodic amplitude defined by the FEM potential distribution during monopolar stimulation.
RESULTS
Characterization of membrane properties of GPi model neurons
The GPi neuron model faithfully replicated several electrophysiological features of EP neurons reported in brain slices (Nakanishi et al. 1990, 1991; Shin and Carlen 2008; Shin et al. 2007) (Fig. 2). For these simulations, synaptic currents were excluded from the somatodendritic compartments and extracellular ion concentrations for Na+, K+, and Ca2+ were consistent with those described experimentally. Action potential height (model: 88.2 mV; in vitro: 94.6 mV) depended on the fast Na+ channel (NaF) conductance, whereas the fast K+ rectifying channel (Kv3.1) conductance had a large effect on action potential duration (0.73 ms, half-height width). The model also demonstrated a spontaneous firing rate of 18.0 Hz on average, which was similar to that observed for type I EP neurons in vitro (2–30 Hz) (Nakanishi et al. 1990). A sodium leakage current (NaL) has been implicated in the generation of spontaneous, rhythmic activity of SNr neurons (Atherton and Bevan 2005). Given the functional similarity of firing properties between SNr and EP neurons, somatic (gNaL = 18 μS/cm2) and dendritic (gNaL = 0.1 μS/cm2) compartments in the GPi neuron model were instantiated with this type of leakage current. Increasing the somatic NaL conductance resulted in a linear augmentation to the baseline firing rate (Fig. 2B). For somatic conductances <15 μS/cm2, however, model GPi neurons were no longer spontaneously active.
Neurons in the entopeduncular nucleus have been reported to exhibit unique responses to depolarizing and hyperpolarizing intracellular current injections. Robust depolarizing current pulses elicited high-frequency discharge rates (>200 Hz) in model neurons (Fig. 2C) with minimal spike adaptation (Fig. 2D). This generalized membrane feature was important to replicate since some neurons recorded in the GPi of our MPTP-treated monkeys fired at rates >100 Hz with intraburst rates exceeding 300 Hz. In addition to fast spikes, type I EP neurons when continuously hyperpolarized and on switching to a depolarizing potential also evoked slow spikes (Fig. 2E; compare with Fig. 2 in Nakanishi et al. 1990). In the model, the fast spikes were dependent on the NaF current, whereas the slow spikes required the presence of calcium currents and were more pronounced with the addition of tetraethylammonium (TEA; simulated as a 100-fold decrease in K+ channel conductance). Anomalous rectification, involving a decrease in the membrane potential shift following an intracellular hyperpolarizing current pulse, was incorporated into the model as an Ih current with hyperpolarization-activated cation (HCN) channels in both somatic and dendritic compartments (Fig. 2F) (Shin and Carlen 2008). Current–voltage plots demonstrated that the final HCN channel conductance used in the model generated an anomalous rectification, especially at more hyperpolarizing current levels, which closely followed experimental data (Fig. 2G). Input resistance was calculated as 101 MΩ from the model's current–voltage plot, which was similar to that reported in brain slices (112 ± 38 MΩ) (Nakanishi et al. 1990). Moderate rebound responses at the cessation of a hyperpolarizing intracellular pulse were observed by Nakanishi et al. (1990); however, no detectable levels of transcripts of the low-threshold Ca2+ T-type channel family were reported with in situ hybridization (Talley et al. 1999) and, consequently, T-type calcium channels were not included in the model.
Characterization of synaptic properties of GPi model neurons
Raster scans of single-unit activity recorded in MPTP-treated monkeys were incorporated into the GPi neuron models as input signals for tuning synaptic conductance values of GABAA and AMPA receptors. Extracellular spike recordings were performed in the sensorimotor regions of putamen (n = 35, 12.9 ± 7.5 Hz), GPe (n = 46, 49.9 ± 21.8 Hz), and STN (n = 13, 55.3 ± 13.0 Hz) while the monkeys were awake, but quietly resting. Representative raster scans with firing rates near population mean levels from each nucleus drove the pulsatile activity of dendritic GABAA (putamen), somatic GABAA (GPe), and somatodendritic AMPA (STN) synapses (Fig. 3A). Increasing the AMPA conductance alone was not sufficient to generate baseline firing rates consistent with those recorded in the parkinsonian GPi in vivo (n = 38, 77.7 ± 38.0 Hz). Although changing NaF, KDRf, or KDRs channel conductance could increase the GPi neuron model's discharge rate, key properties of the action potential would no longer be consistent with in vitro experimental recordings. To compensate for the difference in baseline firing rates, we made the assumption that the sodium leakage current was higher in the in vivo state (i.e., gNaL increased from 18 to 28 μS/cm2). However, it should be noted that the work of Hanson et al. (2004) addressing similar issues in GPe neurons suggests that clustering of sodium channels at sites of excitatory synapses also facilitates increased pallidal firing.
FIG. 3.

Synaptic conductances of the GPi neuron models were tuned to elicit behavior consistent with in vivo spike activity recorded in the GPi of MPTP-treated monkeys. A: synaptic inputs consisted of a collection of spike raster scans recorded in vivo from the putamen, GPe, and STN. B: in this example, 10 model GPi cells (gAMPA = 0.005 μS for STN input, gGABAa = 0.0015 μS for putamen and GPe input) were simulated, each with a different set of input spike raster scans. Shown below it is a representative selection of 10 spike trains recorded from isolated neurons in the GPi of a parkinsonian monkey. C–E: synaptic conductances were adjusted to generate firing rates and patterns consistent with in vivo GPi spike recordings. Although many different combinations of gAMPA and gGABAa could generate firing rates comparable to the in vivo mean (77.7 Hz), the set of synaptic weights was much reduced when also accounting for (D) the percentage of spikes in bursts and (E) the frequency of bursts. Dotted lines represent means from the in vivo spike recordings and the shaded boxes signify their 95% confidence intervals. The large black circles indicate the final parameters values used in this study (same as B: gAMPA = 0.005 μS for STN input, gGABAa = 0.0015 μS for putamen and GPe input). MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; STN, subthalamic nucleus.
Synaptic conductances were estimated by simulating the response of GPi neurons to 10 different combinations of recorded raster scans and evaluating the model output in terms of its ability to capture overall firing rate and burstiness (Fig. 3B). Bursts within the spike train were determined according to the Poisson surprise method (Hahn et al. 2008; Legendy and Salcman 1985) and evaluated in terms of the percentage of total spikes within bursts and the frequency of bursts. Although multiple combinations of AMPA and GABAA synaptic conductances could generate firing rates comparable to those observed in our in vivo recordings in GPi (Fig. 3C), the set of parameter values was constrained when also accounting for burstiness (Fig. 3, D and E). Strengthening the conductances of both types of synapses increased both spike train burst measures, which underlied the intrinsic burst activity present in the input raster scans. Independently increasing the AMPA conductance resulted in prolonged bursts with greater than five spikes per burst, whereas increasing GABAA conductance produced longer hyperpolarized pauses between bursts. The final AMPA and GABAA conductances for each synapse used in the model were 0.005 and 0.0015 μS, respectively. The mean firing rate of the model neurons was 83.1 ± 10.9 Hz (n = 10), which was not significantly different from the firing rates recorded in vivo (P > 0.05, ANOVA) or from those observed in other studies (Boraud et al. 2002; Wichmann and Soares 2006). As shown in Fig. 3, D and E, the percentage of spikes in bursts and frequency of bursts calculated from the GPi neuron model population's spike trains (36.2 ± 14.7% and 285.8 ± 128.2 Hz, respectively) were within the 95% confidence intervals of the same measures calculated for the in vivo GPi spike trains (33.8 ± 13.9% and 280.0 ± 146.5 Hz, respectively).
Somatic firing patterns during GPi-HFS depend on membrane properties
Initially, we used a GPi neuron model lacking synaptic inputs to investigate the intrinsic response of GPi cells to high-frequency extracellular stimulation (Fig. 4A). For suprathreshold stimulus pulses, model neurons exhibited a high probability of firing an action potential in their soma before the next stimulus pulse (i.e., within the 7.4-ms interpulse window) as shown for a representative GPi cell (Fig. 4B). The overall somatic firing rate of the GPi population, which lacked synaptic input, was only slightly above the baseline rate during stimulation (from 62 to 78.7 ± 14.4 Hz, n = 63). Notably, however, the firing pattern during stimulation developed a nonuniform distribution during the interstimulus period with an excitatory crest between 1 and 3 ms (Fig. 4C). On increasing the stimulation intensity to −5 V, the initial excitatory phase became more pronounced at a latency of 1.5 ms and another excitatory phase developed between 4 and 5.5 ms (Fig. 4D).
FIG. 4.

GPi-DBS models lacking afferent synaptic input exhibited somatic spikes time-locked to the electrical stimulation train. A: a population of 63 GPi model neurons was distributed within the posterior GPi such that their axons followed the lenticular fasciculus on exiting the pallidum. B: high-frequency stimulation induced somatic action potentials following most stimulus pulses. The example somatic response is from the GPi neuron is labeled in green in A and indicated by the arrow. C: a peristimulus time histogram of the population's somatic response to monopolar stimulation at contact 1 (shown in yellow in A) at 136 Hz, 90-μs pulse width, and −2 V peak cathodic amplitude showed an overall increase in firing rate between 1 and 3 ms. D: by increasing the peak cathodic stimulation amplitude to −5 V, the population's response developed a sharper response at 1.5 ms and an additional excitatory peak at 5 ms. E–I: individually varying the membrane channel conductances of NaL, NaF, KDRf, KDRs, and SK by 33% affected the amplitude and relative ratio of excitatory peaks in the PSTH population responses. J–L: in contrast, changes in HCN, CaL, and CaN conductances resulted in little to no adaptations to the PSTH population responses. Bin widths on the PSTH plots were 0.05 ms and smoothed with a 5-point averaging window. CaL, L-type high-voltage–activated calcium current; CaN, N-type high-voltage–activated calcium current; HCN, hyperpolarization-activated cation; HFS, high-frequency stimulation; KDRf, fast deactivating potassium current; KDRs, slowly deactivating potassium current; NaF, fast-acting sodium current; NaL, sodium leakage current; PSTH, peristimulus time histogram; SK, calcium-activated potassium current.
Both of these time-locked responses depended on the conductance of specific ion channels (Fig. 4, E–L). The sodium leakage channel conductance (gNaL) played an important role in the somatic firing rate during the stimulus train. Decreasing gNaL reduced the probability of a spike occurring between stimulus pulses and, with very low gNaL levels, HFS would no longer elicit somatic spikes. The excitatory phases of the peristimulus time histogram (PSTH) were dominated by the interplay between the fast Na+ and the Kv3.1/Kv2.1 delayed rectifier K+ currents (Fig. 4, E–L). Decreasing gNaF by 33% resulted in a sharpening of the initial excitatory phase as well as the emergence of an inhibitory phase from 3.5 to 7.4 ms. Increasing either gKDRf or gKDRs generated similar augmentation to the initial excitatory phase. The second excitatory phase became more pronounced when reducing either gKDRf or gSK. Changing other ion channel currents smoothed the excitatory phases for some cells in the population, but it did not disrupt the overall ability of the neuron to become entrained to the stimulus. Whereas the average PSTH across the population exhibited a time-locked response, only neurons sufficiently close to the electrode responded with this type of behavior. For the stimulation configuration shown in Fig. 4C (−2 V), somatic entrainment was observed for the majority of somata within 700 μm of the electrode. When the stimulus amplitude was increased to −5 V (Fig. 4D), nearly all GPi neurons in our model population exhibited some degree of somatic entrainment.
Overall somatic firing rate during GPi-HFS depends on input from synaptic afferents
Integrating stimulation-evoked synaptic afferents into the GPi neuron model resulted in an overall reduction in somatic activity, but axonal output continued to be entrained to each electrical pulse of the stimulus train (Fig. 5A). A single stimulation pulse could transiently hyperpolarize the cell membrane with latencies dependent on the scaling of the synaptic conductances. The representative cell's response (Fig. 5B) had a delay of 17.5 ms following a single stimulation pulse with a peak cathodic amplitude of −2 V and with synaptic weights set according to those trained by the in vivo afferent raster scans. Decreasing both AMPA and GABAA receptor conductances by 50% resulted in a decrease in the delay to 11.6 ms. On the other hand, increasing the conductances of both receptor types by factors of 2 and 4 augmented the delay to 22.4 and 27.0 ms, respectively. In other words, increasing the overall extent of synaptic afferent input into the GPi model neurons increased the duration of the hyperpolarized response. For pulse trains using the same stimulus amplitude as in Fig. 5B, somatic spikes occurred during HFS, but did not appear after every stimulus pulse (Fig. 5C). For simulations with stronger afferent input, somatic activity during HFS was completely suppressed.
FIG. 5.

Responses of GPi model neurons to HFS depended on the extent of axonal efferent and afferent activation. A: action potentials were generated primarily in the axonal segments during DBS, which resulted in orthodromic and antidromic propagation along the axon. When stimulation voltages were suprathreshold in the axonal afferents, AMPA and GABAA receptors were both activated using synaptic conductances defined previously to match features of the in vivo GPi spike recordings. Using these parameter values in the context of the 3-D GPi-DBS model resulted in little to no somatic response following each stimulus pulse. Right: action potential time course recorded at different locations in the neuron, depicted by the lowercase letters labeled in the left panel. B: for a single stimulus pulse, increasing both AMPA and GABAA conductances prolonged the inhibitory periods. C: during HFS, similar increases in synaptic conductances led to complete somatic inhibition since the amplitude of the hyperpolarizing potential prevented the neuron from fully recovering before the next stimulus pulse.
Population responses to HFS demonstrated variability in somatic firing patterns. Similar to the models lacking stimulus-driven synaptic input, these more detailed models exhibited interpulse somatic responses with peaks centered at 1 and 4–5 ms (Fig. 6A). The addition of synaptic afferents into the model significantly reduced the population's mean somatic firing rate for both −2-V stimulation (25.0 ± 17.2 Hz, −70%, ANOVA, P < 0.001) and −5-V stimulation (45.3 ± 24.4 Hz, −45%, ANOVA, P < 0.001). Four different somatic responses to HFS were observed across the population: complete inhibition, inhibition with a sharply locked response (width <1 ms), inhibition with a broadly locked response (width >1 ms), and inhibition without locking (Fig. 6B). The population responded in approximately equal proportions within these classifications for monopolar stimulation (−2 V) through contact 1. At higher stimulus voltages, a greater number of model neurons exhibited inhibition with a broad, time-locked response (49 vs. 25%).
FIG. 6.

Models that incorporated synaptic afferent responses during DBS exhibited time-locked behavior overall, but also showed inhibition and a substantial degree of response variability across the spatially distributed population. A: averaged somatic responses to GPi-DBS from a population of model GPi neurons under baseline conditions (n = 10) and during HFS (n = 63). Somatic responses to HFS exhibited a time-locked excitatory phase at 1 and 4–5 ms and an overall decrease in firing frequency across the remainder of the interpulse interval. B: the population, however, exhibited nuanced differences in the somatic response to HFS, which depended on the potential distribution around a particular neuron. C: despite the inhibitory and time-locked behavior within the soma, HFS drove efferent activity through the axons with a prominent excitatory peak at 1 ms and a secondary peak from 5 to 6 ms. D: population responses in the axons similarly depended on stimulation amplitude. These simulations were derived from the GPi neuron population shown in Fig. 4A with stimulation applied through contact 1. Bin widths on the PSTH plots were again 0.05 ms and smoothed with a 5-point averaging window.
In terms of GPi output during HFS, the averaged PSTH showed a prominent peak at 1 ms and an attenuated secondary response from 5 to 6 ms (Fig. 6C). In most cases, the latter response derived from early somatic discharge within the interstimulus period, which in turn augmented the output firing rate (172.2 ± 42.7 Hz) beyond the stimulation frequency (136 Hz) for those axons with stimulus-driven activity. For the −2 V stimulation configuration, the overall GPi population output rate was 108.2 ± 79.0 Hz, which was not significantly different from the model's baseline rate (ANOVA, P = 0.32). However, on increasing the stimulation amplitude to −5 V, the population's overall output rate increased significantly above baseline (158.6 ± 62.1 Hz, P < 0.001). As shown in Fig. 6D, the percentage of GPi efferents entrained at or above the stimulation frequency similarly increased from −2 to −5 V (64–92%), which paralleled a decrease in the number of cells influenced by synaptic input from afferent collaterals, but located in a subthreshold potential distribution for direct axonal efferent excitation.
Activation of GPe fibers of passage during GPi-HFS
The GPi is by no means a homogeneous input–output structure; it contains numerous axonal fibers passing through from putamen, GPe, STN, as well as nigral and brain stem regions (Charara and Parent 1994; Parent and Parent 2002). To investigate the hypothesis that GPi-HFS not only activates GPi efferents but also these fibers of passage, anatomical reconstructions of GPe fibers were simulated with axonal branches collateralizing en passant within GPi (Fig. 7A). The example GPe neuron reconstruction consisted of two primary branches that projected primarily along the parasagittal plane (Sato et al. 2000a). Clinically relevant DBS settings entrained GPe axonal activity to the stimulation frequency when membrane compartments exceeded threshold. These action potentials not only developed along the primary axon, but they also could be generated in the branches (Fig. 7B). The latter occurrence suggested that DBS could lead to substantial release of neurotransmitters as assumed in our previous analysis.
FIG. 7.

DBS-excited GPe fibers of passage coursing through GPi. A: a population of 83 GPe fibers of passage were distributed within the pallidum such that their axonal collaterals were positioned randomly within the posterior GPi. B: since these fibers collateralized within GPi with multiple branches, action potentials could develop in these collaterals and propagate both antidromically and orthodromically. Arrows show the site of action potential initiation in the representative orange and red GPe fibers. C: increasing the voltage amplitude from −2 to −5 V more than doubled the number of GPe fibers activated by the stimulation regardless of whether the collaterals were myelinated or unmyelinated. At higher voltage settings, however, the percentage of activated fibers in which the action potential was initiated in the collaterals was less for unmyelinated than that for myelinated branches. Increasing the unmyelinated compartment diameter increased (D) the percentage of the GPe fiber population that was activated and (E) the percentage of fibers with action potentials first generated in their branches. F: since the site of action potential initiation varied within the population, the temporal profile of action potential innervation at the initial segment, the terminal ends of each branch, and the primary axon terminations targeting STN also varied across the GPe neuron population. These effects are shown for 2 representative GPe models, illustrated in B. Unmyelinated branch compartments are colored, whereas nodal compartments along the main axon are shown in black.
Several studies have suggested that collaterals within the pallidum are unmyelinated and of a smaller diameter than the primary fibers of passage (Fox et al. 1975; Galvan et al. 2005; Kita and Kitai 1987; Korotchenko 1979). We evaluated how collateral myelination affects the ability of electrical stimulation to influence the activity of GPe fibers of passage (Fig. 7C). The total number of fibers showing activated output (>80% entrainment to the stimulation) was similar for populations with myelinated and unmyelinated branches, which reflects the fact that most action potentials developed in the primary axon (66–84% of the population). The percentage of fibers in which the action potential first developed in one of the branches was reduced for unmyelinated collaterals at higher stimulus intensities. For fibers of passage with unmyelinated branches, the extent of activation depended on the diameter of those collaterals such that populations with larger-diameter collaterals showed higher activation ratios (Fig. 7D). Increasing the collateral diameter also augmented the percentage of activated fibers of passage that had action potentials first develop in their branches (Fig. 7E). This variability in the site of action potential initiation was found to generate a slight temporal delay (0–1.5 ms) for orthodromic and/or antidromic action potential innervation of the branch terminals (Fig. 7F). Thus depending on the spatial separation of the branches, the timing of stimulus-induced afferent input into GPi neurons may not be temporally uniform across GPi.
Whereas the previous analysis examined the responses of single GPe projection neurons during GPi-DBS using contact 1, we next asked how the population would respond if the electrode contact and stimulation voltage were varied. The likelihood of activating the output of GPe efferents was for the most part nonuniform across contact sites. For GPe fibers of passage with 0.8-μm-diameter unmyelinated collaterals, high-frequency stimulation at both −2 and −5 V intensities through contact 2 produced the largest percentage activation (34.9 and 68.7%, respectively) (Fig. 8A). On the other hand, the population responses of GPi efferents had a more spatially tuned profile at −2 V, but a more uniform activation profile when increasing the stimulation amplitude to −5 V (Fig. 8B). In the former case, stimulation through contact 1 at −2 V produced the highest percentage activation (65.1%), whereas contact 3, which was located in part outside the GPi, elicited the smallest percentage activation (15.9%). Comparing these activation percentages to those simulated from the GPe neuron population (28.9 and 20.5%, respectively) showed markedly different activation ratios that depended on the precise location of the active contact within the pallidum (GPi:GPe of 2.25 for contact 1 and 0.78 for contact 2, respectively) (Fig. 8C). On increasing the stimulus amplitude to −5 V, this spatially distinct profile of GPi to GPe activation ratios largely disappeared.
FIG. 8.

The extent of GPi and GPe efferent activation depended in different ways on the electrode contact and stimulation amplitude. A: activation of GPe fibers of passage exhibited a nonuniform spatial profile with the highest percentage activation occurring when stimulating through contact 2. B: GPi neuronal output during DBS, especially at −2 V, exhibited a distinct spatial tuning with maximum activation at contact 1. Population responses are shown for DBS voltages at −2 V (black) and −5 V (gray). Activation was defined as driving axonal output ≥80% of the stimulation frequency. C: activation ratios of GPi to GPe output were more pronounced at lower stimulation voltages such that at contact 3, a greater percentage of GPe efferents than GPi efferents were excited. On increasing the stimulus intensity to −5 V, activation ratios showed less variability and were >1 across all contacts.
DISCUSSION
High-frequency stimulation of the GPi is a promising therapy for individuals with Parkinson's disease (Volkmann 2004) and dystonia (Krauss et al. 2004). Despite its well-documented clinical successes, the therapeutic mechanisms and optimal targets remain a matter of debate (Bejjani et al. 1997; Wu et al. 2001). Our computational models of pallidal DBS provide new tools to investigate how membrane channel dynamics, synaptic inputs, and axonal collateralization contribute to neuronal responses during stimulation.
Generating parkinsonian-like activity in computational models of GPi neurons
The GPi neuron model reproduced many of the electrophysiological attributes of recordings in the rat entopeduncular nucleus, the homolog of primate GPi. Although the ion channel kinetics, conductances, and distributions are unlikely to be unique, the final parameter set did capture the signature features of EP type I neurons. These attributes included an inward rectifying current, calcium spikes in the presence of TEA, and the capacity for following high-frequency intracellular current stimulation with minimal spike adaptation. Whether these characteristics translate to neurons in the primate GPi and whether the underlying membrane channel properties differ between normal and pathological states require further investigation. Such experiments, however, are technically challenging to perform in primates and especially in primates with neurological disorders. As such, computational models are useful to test how membrane channel properties might contribute to the pathological activity observed in the GPi of human and nonhuman primates.
Pathological activity observed in GPi neurons in parkinsonian or dystonic primates may indeed be a product of changes in expression level and distribution of certain membrane channels, but could also derive from alterations in synaptic input from afferent fibers, or a combination of the two. Our simulations suggest that certain ion channels may facilitate higher firing rates (NaL, NaF, Kv3.1, Kv3.2) and burstiness (HCN, sKCa, CaL) in GPi neuronal activity. Whereas the NaL current was used to augment the baseline firing rate to match that observed in vivo, other mechanisms are possible, including an elevated leak reversal potential, a nonspecific cation conductance (Lee and Tepper 2007), or clustering of sodium channels at excitatory synapses (Hanson et al. 2004). By instantiating synaptic inputs (AMPA and GABAA) with release probabilities derived from in vivo recordings in afferent nuclei, the models demonstrated that the increased burstiness observed in the parkinsonian state could be produced entirely from pathological network activity (i.e., abnormal putamen, GPe, and STN input). Other receptor types are known to exist within the GPi, including N-methyl-d-aspartate and GABAB receptors as well as receptors for serotonin (Azmitia and Segal 1978; Bobillier et al. 1976; DeVito et al. 1980), acetylcholine (Carpenter et al. 1981; Gonya-Magee and Anderson 1983; Saper and Loewy 1982), and dopamine (Lindvall and Bjorklund 1979; Parent and Smith 1987). These inputs, although not explicitly modeled in our study, could also contribute to changes in GPi firing rates and patterns in Parkinson's disease and dystonia.
Effects of intrinsic membrane properties on somatic activity during GPi-HFS
A consistent observation with electrophysiological recordings in GPi during GPi-HFS is the regularization of somatic activity (Bar-Gad et al. 2004; Boraud et al. 1996; Dostrovsky et al. 2000; Wu et al. 2001). Bar-Gad et al. (2004) showed that the majority of pallidal neurons in nonhuman primates (70%) were time-locked during high-frequency stimulation with latencies for the first and second excitatory phases ranging from 1.5 to 3.5 and from 5.5 to 7 ms, respectively, with inhibitory phases in between. Similar temporal locking, although not specifically analyzed, has also been observed in the raw recordings of human GPi cells during 300-Hz stimulation [∼2.0–2.6 ms, Fig. 4B (Wu et al. 2001) and ∼2.4–2.7 ms, Fig. 1B (Dostrovsky et al. 2000)]. These time-locked responses were also consistent with observations from MPTP-treated parkinsonian monkeys in which GPi-HFS was shown to decrease somatic bursting in GPi (Boraud et al. 1996). Our models reproduced time-locked somatic activity with two excitatory phases and inhibitory phases in between during GPi-HFS (Fig. 6). The first excitatory phase had an interpulse latency of 1 ms, which paralleled that found in vivo. The second excitatory time-locked response (4–5 ms) emerged at higher stimulation amplitudes and appeared slightly earlier than that observed experimentally (Bar-Gad et al. 2004), which may reflect deviations in certain membrane channel properties from the actual biophysical values. Our models showed that the time-locked responses of GPi neurons during HFS depended on the direct activation of Na+ channels and an interplay between Na+ channels and Kv3.1/Kv2.1 channels. Decreasing the sodium conductance or increasing the potassium conductance sharpened the initial time-locked PSTH response. In turn, varying the conductances of either the Kv3.1 or SK channels could augment the second excitatory phase within the PSTH.
Effects of afferent input on somatic activity during GPi-HFS
The models demonstrated that afferent stimulation of both AMPA and GABAA receptors resulted in overall inhibition of GPi spike rates as predicted by (Wu et al. 2001). Our models showed a range of inhibitory periods following a stimulus pulse (12–27 ms) that depended on the intensity of synaptic input. These durations were comparable to the inhibitory periods observed following suprathreshold stimulation in humans (10–25 ms at 50% recovery) (Dostrovsky et al. 2000) and were consistent with a dependence on stimulation intensity. For example, Wu et al. (2001) found that GPi spike activity in humans resumed after 15 to 40 ms for stimulation using 50- to 100-μA levels, respectively. One explanation could be that more intense electrical stimulation induces a disproportionate increase in GABA release from striatal and pallidal afferents, which in turn augments the activation of extrasynaptic GABAA and especially GABAB receptors (Galvan et al. 2005).
However, the lower somatic firing rates observed experimentally during GPi-HFS do not necessarily indicate that inhibitory afferents from putamen or GPe or collaterals from other GPi neurons were excited more than those from STN. It is possible that the relatively higher proportion of GABAergic inputs fibers and their dense perisomal and dendritic baskets predispose GPi neurons toward inhibitory responses during HFS. For instance, Kita et al. (2005) observed that neuronal firing rates in GPi during STN stimulation were influenced more by the GPe–GPi pathway than the STN–GPi pathway. Studies have also indicated that GABAA and GABAB receptors are up-regulated in the GPi after unilateral dopaminergic denervation (Pan et al. 1985; Robertson et al. 1990). An up-regulation of GABA receptors in GPi may implicate a compensatory mechanism to decreased GABAergic transmission along the direct pathway and increased GABAergic signaling along the indirect pathway (Johnston and Duty 2003). Thus loss of dopaminergic signaling in Parkinson's disease may further “prime” GPi somata for inhibitory responses to HFS. Whether these changes, and the models' assumptions of constant distributions and conductances of AMPA and GABAA synapses, persist with long-term stimulation of the GPi will require further investigation.
The entrained firing patterns during HFS remained even after including the synaptic mechanisms in the models. Models that incorporated AMPA and GABAA receptors showed variability in firing pattern responses that depended on the potential distribution around the neuron. For instance, time-locked responses were present in 42 and 70% of the GPi population for stimulation amplitudes at −2 and −5 V, respectively. The latter of these values was comparable to that observed experimentally in nonhuman primates (70%) (Bar-Gad et al. 2004). This study also noted that a small fraction of cells demonstrated overall excitation during HFS. Our GPi-DBS models suggest that these responses could have resulted from a preferential activation of excitatory input (e.g., activation of STN afferents) or an altogether lack of afferent input, which in the nonsynaptic neuron models showed neuronal entrainment to the stimulation coupled with a slight increase in the overall firing probability.
We observed elevated GPi axonal entrainment with higher stimulation voltages, which in turn resulted in a decrease in the number of cells completely inhibited (25 to 8%). Dostrovsky et al. (2000) estimated that the currents used in their experiments would be subthreshold to activate somata or axons located >250–600 μm from the stimulating electrode. Nevertheless, they observed neuronal inhibition following HFS at recording distances >600 μm. They hypothesized that HFS could activate afferent fibers, which collateralized beyond the activation volume and thus influenced GPi activity, a phenomenon that our GPi-DBS models reproduced. In turn, GABA signaling may have important implications in the therapeutic mechanisms of DBS. Penn et al. (1998), for example, demonstrated that local injection of muscimol (a GABAA agonist) into the GPi of a patient with Parkinson's disease significantly reduced the parkinsonian motor signs.
Mechanistic implications of axonal collateralization within GPi
The models predicted that a substantial number of GPi axonal efferents were driven during HFS despite their complex and variable somatic activity. These results suggest that the pallidal receiving areas of thalamus and brain stem are inhibited during GPi-HFS. Experimental recordings in the pallidal-receiving areas of thalamus support this prediction, demonstrating reduced firing rates during GPi-HFS (Anderson et al. 2003; Montgomery 2006) with spikes time-locked to the stimulation pulse train (Pralong et al. 2003). An overall decrease in neuronal activity has also been reported in the PPN during GPi-HFS in a parkinsonian monkey (J Zhang, D Ghosh, and J Vitek, personal communication).
The GPi contains a rich set of fibers of passage, many of which form collaterals en passant (Parent and Parent 2002). The models predicted that a substantial number of GPe projection neurons were excited by direct stimulation of their primary axons and/or axon collaterals extending through GPi. Although not explicitly investigated, other fibers of passage through GPi (including efferents from the STN and putamen) may also be activated by GPi-HFS. Imaging studies support the concept of activating multiple pathways during GPi-HFS. For example, Davis et al. (1997) found a statistically significant increase in regional cerebral blood flow (rCBF) in the ipsilateral lentiform nuclei (LN, which includes the GPe and putamen) during therapeutic GPi-HFS. Similarly, Fukuda et al. (2001) observed an augmented positron emission tomographic (PET) response in the ipsilateral LN during GPi-HFS. Although orthodromic activation of STN afferents that project to pallidum could account for the larger rCBF response in GPe, the increased response in putamen is likely due to a different mechanism since STN only sparsely innervates the putamen (Parent and Parent 2002). Our models suggest that the PET results derived from antidromic activation of GPe and putamen fiber projections passing through or near the GPi and the subsequent synaptic activity induced within their collateral networks.
Activation profiles for GPe fibers of passage were nonuniform across the different electrode contacts at lower stimulation voltages. Consequently, for dorsal contacts stimulated at lower stimulation voltages, the models predicted that GPe axonal collaterals could be activated in equal or greater numbers than GPi axonal efferents. Activation of GPe fibers would be expected not only to inhibit GPi directly, but also to suppress STN, which would in turn weaken the excitatory effects of STN on both GPe and GPi. Clinical data has suggested that dorsal contacts in GPi-DBS implants were more effective than the ventral contacts at relieving bradykinesia, but ventral contacts improved levodopa-induced dyskinesias more successfully (Bejjani et al. 1997; Krack et al. 1998; Yelnik et al. 2000). According to these reports, the dorsal contacts were likely in GPe or along the medullary lamina separating GPe and GPi. Others have reported that intraoperative high-frequency stimulation through a lesioning probe putatively targeting the GPe improved bradykinesia and, at high stimulation amplitudes, produced dyskinesias (Vitek et al. 2004). Relieving certain motor symptoms may thus require balancing the activation of GPi and GPe efferents. Similar to the predicted effects of activating GPi efferents, entrainment of GPe output could also have far-reaching network effects. Its inhibitory effect on STN somatic activity could disrupt recurrent signaling of GPi, SNr, PPN, and GPe neurons not directly activated by the stimulation.
Other fibers of passage may also be activated during GPi-DBS. For example, the majority of STN axons project through GPi en route to the GPe (Sato et al. 2000b), striatofugal fibers course through GPi en route to the SNr (Parent et al. 1995), and bundles of dopaminergic fibers from the substantia nigra pars compacta, serotonergic fibers from the dorsal raphe nucleus, and cholinergic fibers from the PPN also collateralize en passant through GPi (Charara and Parent 1994). Closed-form representations of network effects resulting from activation of these fibers of passage are beyond the scope of this study. However, the results of this study demonstrate that fibers of passage activation can be an important factor to incorporate when developing network models of DBS and may be useful for determining which neural elements underlie the therapeutic benefit provided by DBS.
GRANTS
This work was supported by National Institute of Neurological Disorders and Stroke Grants F32-NS-061541 and R01-NS-047388.
Acknowledgments
We thank S. Miocinovic for assistance with the models, M. Parent and A. Parent for providing the camera lucida data for the globus pallidus neurons, and W. Xu and J. Vitek for help and support with the in vivo spike recordings.
APPENDIX
Ionic currents within both somatic and dendritic compartments of the GPi neuron model were defined according to the following summation
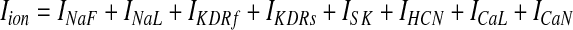 |
(1) |
Each channel's current was written as Iion = gionη(v − Eion) such that the maximum conductance gion was multiplied by one or more gating variables (η) that ranged from 0 to 1
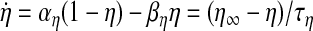 |
(2) |
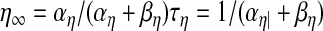 |
(3) |
Although immunohistochemical and in situ hybridization studies have established the presence of certain ion channels in the entopeduncular nucleus, the channel kinetics in many cases have not yet been established. In those cases, activation and inactivation functions were implemented from experimental data and computational models reported in the literature from other brain regions.
GPi somatodendritic sodium currents (NaF, NaL)
Fast-acting sodium currents (NaFs) were based on channel kinetics defined in Traub et al. (1991) and derived from recordings in Sah et al. (1988). Since data in these experiments were collected at 23°C, channel conductance and kinetic rates were adjusted to those present at 36°C using a Q10 value of 1.58, which led to a scaling value of 1.78 using the Arrhenius equation
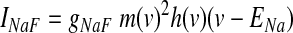 |
(4) |
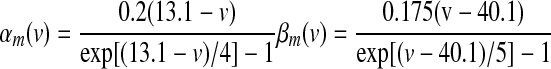 |
(5) |
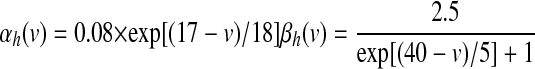 |
(6) |
Sodium leakage current (NaL) has been implicated in the generation of spontaneous, rhythmic activity with strong influence on the resting membrane potential of SNr neurons (Atherton and Bevan 2005). Given the functional similarity of firing properties between SNr and EP neurons, somatic and dendritic compartments in the GPi neuron model were instantiated with this type of leakage current, defined as
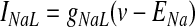 |
(7) |
GPi somatodendritic potassium delayed rectifier currents (KDRf and KDRs)
Potassium channels, which exhibit a fast deactivating component (KDRf: Kv3.1), enable a rapid recovery of Na+ channel inactivation by minimizing the hyperpolarization phase, thereby facilitating sustained high-frequency spike activity (Rudy et al. 1999). Immunohistochemistry indicates that this type of delayed rectifier channel is present in rat EP, GP, and SNr (Ozaita et al. 2002; Perney et al. 1992). Channel kinetics were derived from electrophysiological recordings in rat GP (Baranauskas et al. 1999; Hernandez-Pineda et al. 1999) and in the case of Hernandez-Pineda et al. adjusted according to a Q10 measure of 1.7
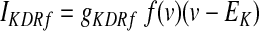 |
(8) |
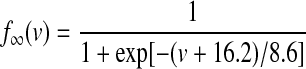 |
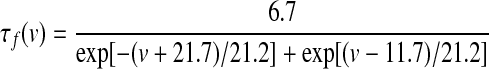 |
(9) |
Kv2.1 potassium channels, which are thought to produce the slowly deactivating potassium current (KDRs), are robustly expressed in GP and substantia nigra and appear to be present in EP as well (Vega-Saenz de Miera 2004). Kinetic rate equations were derived from Chan et al. (2007), with a Q10 of 4 and described as follows
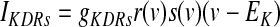 |
(10) |
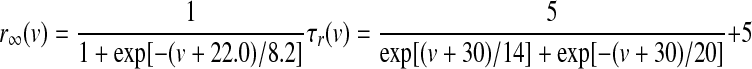 |
(11) |
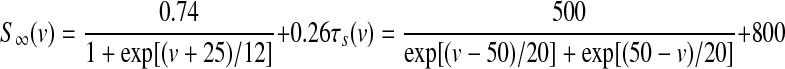 |
(12) |
GPi somatodendritic small conductance calcium-activated potassium current (SK)
Neurons in the entopeduncular nucleus (and in the SNr) express small conductance calcium-activated potassium channels (SK) with primarily SK2 subunits (Stocker and Pedarzani 2000), which when activated generate a slow afterhyperpolarization that hinders repetitive high-frequency spike activity (Vergara et al. 1998). Kinetic rate equations for the SK2 currents were based on those described by Hirschberg et al. (1998) and implemented in part by Gillies and Willshaw (2006), with a Q10 adjustment of 1.5
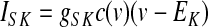 |
(13) |
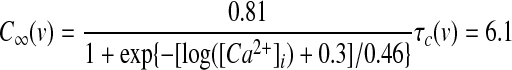 |
(14) |
GPi somatodendritic Ih current (HCN)
HCN channels, which are thought to regulate the rate and uniformity of spontaneous activity, are expressed in entopeduncular nucleus neurons (Chan et al. 2004). Kinetics for these channels were modeled according to Huguenard and McCormick (1992). No Q10 adjustment was necessary since their experiments were performed at 36°C
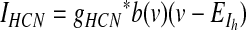 |
(15) |
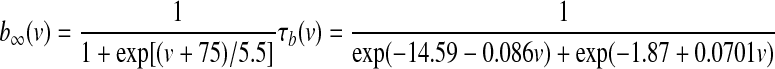 |
(16) |
GPi somatodendritic high-voltage–activated calcium currents (CaN, CaL)
High-voltage–activated (HVA) calcium channels with N- and L-type subunits are expressed in the EP (Cole et al. 2005; Ludwig et al. 1997). The kinetic rate equations for both currents were reproduced from Brown et al. (1993) and the inactivation functions for the CaN and CaL channels were from Fox et al. (1987) and Meuth et al. (2002), respectively. The model framework was implemented as described in Gillies and Willshaw (2006), with a Q10 of 1.95
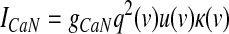 |
(17) |
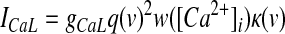 |
(18) |
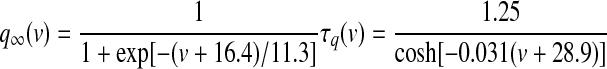 |
(19) |
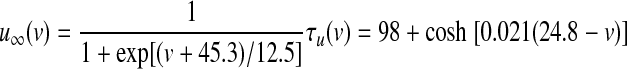 |
(20) |
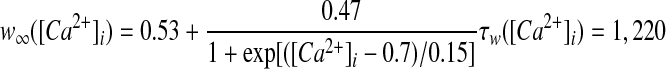 |
(21) |
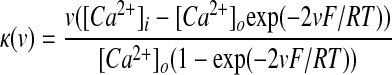 |
(22) |
where F is Faraday's constant, R is the gas constant, and T is the temperature in Kelvin. Intracellular calcium concentrations were modeled as a submembrane shell as reported by Gillies and Willshaw (2006), with buffering and diffusion mechanisms determined by
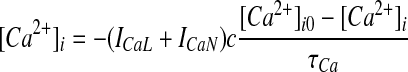 |
(23) |
where c is the conversion constant, τCa is the time constant (185.7 ms for these simulations), and [Ca2+]i0 is the basal intracellular calcium concentration.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- Anderson et al. 2003.Anderson ME, Postupna N, Ruffo M. Effects of high-frequency stimulation in the internal globus pallidus on the activity of thalamic neurons in the awake monkey. J Neurophysiol 89: 1150–1160, 2003. [DOI] [PubMed] [Google Scholar]
- Atherton and Bevan 2005.Atherton JF, Bevan MD. Ionic mechanisms underlying autonomous action potential generation in the somata and dendrites of GABAergic substantia nigra pars reticulata neurons in vitro. J Neurosci 25: 8272–8281, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azmitia and Segal 1978.Azmitia EC, Segal M. An autoradiographic analysis of the differential ascending projections of the dorsal and median raphe nuclei in the rat. J Comp Neurol 179: 641–667, 1978. [DOI] [PubMed] [Google Scholar]
- Bakay et al. 1992.Bakay RA, DeLong MR, Vitek JL. Posteroventral pallidotomy for Parkinson's disease. J Neurosurg 77: 487–488, 1992. [DOI] [PubMed] [Google Scholar]
- Baranauskas et al. 1999.Baranauskas G, Tkatch T, Surmeier DJ. Delayed rectifier currents in rat globus pallidus neurons are attributable to Kv2.1 and Kv3.1/3.2 K(+) channels. J Neurosci 19: 6394–6404, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Gad et al. 2004.Bar-Gad I, Elias S, Vaadia E, Bergman H. Complex locking rather than complete cessation of neuronal activity in the globus pallidus of a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated primate in response to pallidal microstimulation. J Neurosci 24: 7410–7419, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron et al. 2001.Baron MS, Sidibe M, DeLong MR, Smith Y. Course of motor and associative pallidothalamic projections in monkeys. J Comp Neurol 429: 490–501, 2001. [PubMed] [Google Scholar]
- Bejjani et al. 1997.Bejjani B, Damier P, Arnulf I, Bonnet AM, Vidailhet M, Dormont D, Pidoux B, Cornu P, Marsault C, Agid Y. Pallidal stimulation for Parkinson's disease. Two targets? Neurology 49: 1564–1569, 1997. [DOI] [PubMed] [Google Scholar]
- Bobillier et al. 1976.Bobillier P, Seguin S, Petitjean F, Salvert D, Touret M, Jouvet M. The raphe nuclei of the cat brain stem: a topographical atlas of their efferent projections as revealed by autoradiography. Brain Res 113: 449–486, 1976. [DOI] [PubMed] [Google Scholar]
- Bolam and Smith 1992.Bolam JP, Smith Y. The striatum and the globus pallidus send convergent synaptic inputs onto single cells in the entopeduncular nucleus of the rat: a double anterograde labelling study combined with postembedding immunocytochemistry for GABA. J Comp Neurol 321: 456–476, 1992. [DOI] [PubMed] [Google Scholar]
- Boraud et al. 1996.Boraud T, Bezard E, Bioulac B, Gross C. High frequency stimulation of the internal globus pallidus (GPi) simultaneously improves parkinsonian symptoms and reduces the firing frequency of GPi neurons in the MPTP-treated monkey. Neurosci Lett 215: 17–20, 1996. [DOI] [PubMed] [Google Scholar]
- Boraud et al. 2002.Boraud T, Bezard E, Bioulac B, Gross CE. From single extracellular unit recording in experimental and human parkinsonism to the development of a functional concept of the role played by the basal ganglia in motor control. Prog Neurobiol 66: 265–283, 2002. [DOI] [PubMed] [Google Scholar]
- Brown et al. 1993.Brown AM, Schwindt PC, Crill WE. Voltage dependence and activation kinetics of pharmacologically defined components of the high-threshold calcium current in rat neocortical neurons. J Neurophysiol 70: 1530–1543, 1993. [DOI] [PubMed] [Google Scholar]
- Butson et al. 2006.Butson CR, Maks CB, McIntyre CC. Sources and effects of electrode impedance during deep brain stimulation. Clin Neurophysiol 117: 447–454, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butson and McIntyre 2005.Butson CR, McIntyre CC. Tissue and electrode capacitance reduce neural activation volumes during deep brain stimulation. Clin Neurophysiol 116: 2490–2500, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter et al. 1981.Carpenter MB, Baton RR 3rd, Carleton SC, Keller JT. Interconnections and organization of pallidal and subthalamic nucleus neurons in the monkey. J Comp Neurol 197: 579–603, 1981. [DOI] [PubMed] [Google Scholar]
- Chan et al. 2007.Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. “Rejuvenation” protects neurons in mouse models of Parkinson's disease. Nature 447: 1081–1086, 2007. [DOI] [PubMed] [Google Scholar]
- Chan et al. 2004.Chan CS, Shigemoto R, Mercer JN, Surmeier DJ. HCN2 and HCN1 channels govern the regularity of autonomous pacemaking and synaptic resetting in globus pallidus neurons. J Neurosci 24: 9921–9932, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charara and Parent 1994.Charara A, Parent A. Brainstem dopaminergic, cholinergic and serotoninergic afferents to the pallidum in the squirrel monkey. Brain Res 640: 155–170, 1994. [DOI] [PubMed] [Google Scholar]
- Clarke and Bolam 1998.Clarke NP, Bolam JP. Distribution of glutamate receptor subunits at neurochemically characterized synapses in the entopeduncular nucleus and subthalamic nucleus of the rat. J Comp Nneurol 397: 403–420, 1998. [PubMed] [Google Scholar]
- Cole et al. 2005.Cole RL, Lechner SM, Williams ME, Prodanovich P, Bleicher L, Varney MA, Gu G. Differential distribution of voltage-gated calcium channel alpha-2 delta (alpha2delta) subunit mRNA-containing cells in the rat central nervous system and the dorsal root ganglia. J Comp Neurol 491: 246–269, 2005. [DOI] [PubMed] [Google Scholar]
- Davis et al. 1997.Davis KD, Taub E, Houle S, Lang AE, Dostrovsky JO, Tasker RR, Lozano AM. Globus pallidus stimulation activates the cortical motor system during alleviation of parkinsonian symptoms. Nat Med 3: 671–674, 1997. [DOI] [PubMed] [Google Scholar]
- Destexhe et al. 1994.Destexhe A, Mainen ZF, Sejnowski TJ. An efficient method for computing synaptic conductances based on a kinetic-model of receptor-binding. Neural Comput 6: 14–18, 1994. [Google Scholar]
- DeVito et al. 1980.DeVito JL, Anderson ME, Walsh KE. A horseradish peroxidase study of afferent connections of the globus pallidus in Macaca mulatta. Exp Brain Res 38: 65–73, 1980. [DOI] [PubMed] [Google Scholar]
- Dostrovsky et al. 2000.Dostrovsky JO, Levy R, Wu JP, Hutchison WD, Tasker RR, Lozano AM. Microstimulation-induced inhibition of neuronal firing in human globus pallidus. J Neurophysiol 84: 570–574, 2000. [DOI] [PubMed] [Google Scholar]
- Elder et al. 2005.Elder CM, Hashimoto T, Zhang J, Vitek JL. Chronic implantation of deep brain stimulation leads in animal models of neurological disorders. J Neurosci Methods 142: 11–16, 2005. [DOI] [PubMed] [Google Scholar]
- Filion and Tremblay 1991.Filion M, Tremblay L. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res 547: 142–151, 1991. [PubMed] [Google Scholar]
- Filion et al. 1988.Filion M, Tremblay L, Bedard PJ. Abnormal influences of passive limb movement on the activity of globus pallidus neurons in parkinsonian monkeys. Brain Res 444: 165–176, 1988. [DOI] [PubMed] [Google Scholar]
- Fox et al. 1987.Fox AP, Nowycky MC, Tsien RW. Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurones. J Physiol 394: 149–172, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox et al. 1975.Fox CA, Rafols JA, Cowan WM. Computer measurements of axis cylinder diameters of radial fibers and “comb” bundle fibers. J Comp Neurol 159: 201–223, 1975. [DOI] [PubMed] [Google Scholar]
- Fukuda et al. 2001.Fukuda M, Mentis MJ, Ma Y, Dhawan V, Antonini A, Lang AE, Lozano AM, Hammerstad J, Lyons K, Koller WC, Moeller JR, Eidelberg D. Networks mediating the clinical effects of pallidal brain stimulation for Parkinson's disease: a PET study of resting-state glucose metabolism. Brain 124: 1601–1609, 2001. [DOI] [PubMed] [Google Scholar]
- Galvan et al. 2005.Galvan A, Villalba RM, West SM, Maidment NT, Ackerson LC, Smith Y, Wichmann T. GABAergic modulation of the activity of globus pallidus neurons in primates: in vivo analysis of the functions of GABA receptors and GABA transporters. J Neurophysiol 94: 990–1000, 2005. [DOI] [PubMed] [Google Scholar]
- Gillies and Willshaw 2006.Gillies A, Willshaw D. Membrane channel interactions underlying rat subthalamic projection neuron rhythmic and bursting activity. J Neurophysiol 95: 2352–2365, 2006. [DOI] [PubMed] [Google Scholar]
- Gonya-Magee and Anderson 1983.Gonya-Magee T, Anderson ME. An electrophysiological characterization of projections from the pedunculopontine area to entopeduncular nucleus and globus pallidus in the cat. Exp Brain Res 49: 269–279, 1983. [DOI] [PubMed] [Google Scholar]
- Grill et al. 2008.Grill WM, Cantrell MB, Robertson MS. Antidromic propagation of action potentials in branched axons: implications for the mechanisms of action of deep brain stimulation. J Comput Neurosci 24: 81–93, 2008. [DOI] [PubMed] [Google Scholar]
- Grill and Mortimer 1994.Grill WM, Mortimer JT. Electrical properties of implant encapsulation tissue. Ann Biomed Eng 22: 23–33, 1994. [DOI] [PubMed] [Google Scholar]
- Gross et al. 1997.Gross C, Rougier A, Guehl D, Boraud T, Julien J, Bioulac B. High-frequency stimulation of the globus pallidus internalis in Parkinson's disease: a study of seven cases. J Neurosurg 87: 491–498, 1997. [DOI] [PubMed] [Google Scholar]
- Haberler et al. 2000.Haberler C, Alesch F, Mazal PR, Pilz P, Jellinger K, Pinter MM, Hainfellner JA, Budka H. No tissue damage by chronic deep brain stimulation in Parkinson's disease. Ann Neurol 48: 372–376, 2000. [PubMed] [Google Scholar]
- Hahn et al. 2008.Hahn PJ, Russo GS, Hashimoto T, Miocinovic S, Xu W, McIntyre CC, Vitek JL. Pallidal burst activity during therapeutic deep brain stimulation. Exp Neurol 211: 243–251, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson et al. 2004.Hanson JE, Smith Y, Jaeger D. Sodium channels and dendritic spike initiation at excitatory synapses in globus pallidus neurons. J Neurosci 24: 329–340, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto et al. 2003.Hashimoto T, Elder CM, Okun MS, Patrick SK, Vitek JL. Stimulation of the subthalamic nucleus changes the firing pattern of pallidal neurons. J Neurosci 23: 1916–1923, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausser et al. 1995.Hausser M, Stuart G, Racca C, Sakmann B. Axonal initiation and active dendritic propagation of action potentials in substantia nigra neurons. Neuron 15: 637–647, 1995. [DOI] [PubMed] [Google Scholar]
- Hazrati et al. 1990.Hazrati LN, Parent A, Mitchell S, Haber SN. Evidence for interconnections between the two segments of the globus pallidus in primates: a PHA-L anterograde tracing study. Brain Res 533: 171–175, 1990. [DOI] [PubMed] [Google Scholar]
- Hernandez-Pineda et al. 1999.Hernandez-Pineda R, Chow A, Amarillo Y, Moreno H, Saganich M, Vega-Saenz de Miera EC, Hernandez-Cruz A, Rudy B. Kv3.1–Kv3.2 channels underlie a high-voltage-activating component of the delayed rectifier K+ current in projecting neurons from the globus pallidus. J Neurophysiol 82: 1512–1528, 1999. [DOI] [PubMed] [Google Scholar]
- Hines and Carnevale 1997.Hines ML, Carnevale NT. The NEURON simulation environment. Neural Comput 9: 1179–1209, 1997. [DOI] [PubMed] [Google Scholar]
- Hirschberg et al. 1998.Hirschberg B, Maylie J, Adelman JP, Marrion NV. Gating of recombinant small-conductance Ca-activated K+ channels by calcium. J Gen Physiol 111: 565–581, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard and McCormick 1992.Huguenard JR, McCormick DA. Simulation of the currents involved in rhythmic oscillations in thalamic relay neurons. J Neurophysiol 68: 1373–1383, 1992. [DOI] [PubMed] [Google Scholar]
- Hutchison et al. 1994.Hutchison WD, Lozano AM, Davis KD, Saint-Cyr JA, Lang AE, Dostrovsky JO. Differential neuronal activity in segments of globus pallidus in Parkinson's disease patients. Neuroreport 5: 1533–1537, 1994. [DOI] [PubMed] [Google Scholar]
- Hutchison et al. 1997.Hutchison WD, Lozano AM, Tasker RR, Lang AE, Dostrovsky JO. Identification and characterization of neurons with tremor-frequency activity in human globus pallidus. Exp Brain Res 113: 557–563, 1997. [DOI] [PubMed] [Google Scholar]
- Johnson et al. 2007.Johnson MD, Miocinovic S, Zhang J, Xu W, Vitek JL, McIntyre CC. Model-based analysis of pallidal deep brain stimulation in parkinsonian monkeys. Soc Neurosci Abstr 693.610, 2007.
- Johnston and Duty 2003.Johnston T, Duty S. Changes in GABA(B) receptor mRNA expression in the rodent basal ganglia and thalamus following lesion of the nigrostriatal pathway. Neuroscience 120: 1027–1035, 2003. [DOI] [PubMed] [Google Scholar]
- Kita 2001.Kita H Neostriatal and globus pallidus stimulation induced inhibitory postsynaptic potentials in entopeduncular neurons in rat brain slice preparations. Neuroscience 105: 871–879, 2001. [DOI] [PubMed] [Google Scholar]
- Kita and Kitai 1987.Kita H, Kitai ST. Efferent projections of the subthalamic nucleus in the rat: light and electron microscopic analysis with the PHA-L method. J Comp Neurol 260: 435–452, 1987. [DOI] [PubMed] [Google Scholar]
- Kita et al. 2005.Kita H, Tachibana Y, Nambu A, Chiken S. Balance of monosynaptic excitatory and disynaptic inhibitory responses of the globus pallidus induced after stimulation of the subthalamic nucleus in the monkey. J Neurosci 25: 8611–8619, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotchenko 1979.Korotchenko VV Myelo- and cytoarchitectonics of the cat pallidum. Neirofiziologiia 11: 321–328, 1979. [PubMed] [Google Scholar]
- Krack et al. 1998.Krack P, Pollak P, Limousin P, Hoffmann D, Benazzouz A, Le Bas JF, Koudsie A, Benabid AL. Opposite motor effects of pallidal stimulation in Parkinson's disease. Ann Neurology 43: 180–192, 1998. [DOI] [PubMed] [Google Scholar]
- Krauss et al. 2004.Krauss JK, Yianni J, Loher TJ, Aziz TZ. Deep brain stimulation for dystonia. J Clin Neurophysiol 21: 18–30, 2004. [DOI] [PubMed] [Google Scholar]
- Kumar 2002.Kumar R Methods for programming and patient management with deep brain stimulation of the globus pallidus for the treatment of advanced Parkinson's disease and dystonia. Mov Disord 17, Suppl. 3: S198–S207, 2002. [DOI] [PubMed] [Google Scholar]
- Kuo 1973.Kuo JS, Carpenter MB. Organization of pallidothalamic projections in the rhesus monkey. J Comp Neurol 151: 201–236, 1973. [DOI] [PubMed] [Google Scholar]
- Lee and Tepper 2007.Lee CR, Tepper JM. A calcium-activated nonselective cation conductance underlies the plateau potential in rat substantia nigra GABAergic neurons. J Neurosci 27: 6531–6541, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendy and Salcman 1985.Legendy CR, Salcman M. Bursts and recurrences of bursts in the spike trains of spontaneously active striate cortex neurons. J Neurophysiol 53: 926–939, 1985. [DOI] [PubMed] [Google Scholar]
- Lindvall and Bjorklund 1979.Lindvall O, Bjorklund A. Dopaminergic innervation of the globus pallidus by collaterals from the nigrostriatal pathway. Brain Res 172: 169–173, 1979. [DOI] [PubMed] [Google Scholar]
- Lozano et al. 1997.Lozano AM, Kumar R, Gross RE, Giladi N, Hutchison WD, Dostrovsky JO, Lang AE. Globus pallidus internus pallidotomy for generalized dystonia. Mov Disord 12: 865–870, 1997. [DOI] [PubMed] [Google Scholar]
- Ludwig et al. 1997.Ludwig A, Flockerzi V, Hofmann F. Regional expression and cellular localization of the alpha1 and beta subunit of high voltage-activated calcium channels in rat brain. J Neurosci 17: 1339–1349, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin and Bowden 2000.Martin R, Bowden D. Primate Brain Maps: Structure of the Macaque Brain. Amsterdam: Elsevier Science, 2000.
- McIntyre et al. 2004.McIntyre CC, Grill WM, Sherman DL, Thakor NV. Cellular effects of deep brain stimulation: model-based analysis of activation and inhibition. J Neurophysiol 91: 1457–1469, 2004. [DOI] [PubMed] [Google Scholar]
- McIntyre et al. 2002.McIntyre CC, Richardson AG, Grill WM. Modeling the excitability of mammalian nerve fibers: influence of afterpotentials on the recovery cycle. J Neurophysiol 87: 995–1006, 2002. [DOI] [PubMed] [Google Scholar]
- Meuth et al. 2002.Meuth S, Pape HC, Budde T. Modulation of Ca2+ currents in rat thalamocortical relay neurons by activity and phosphorylation. Eur J Neurosci 15: 1603–1614, 2002. [DOI] [PubMed] [Google Scholar]
- Miocinovic et al. 2006.Miocinovic S, Parent M, Butson CR, Hahn PJ, Russo GS, Vitek JL, McIntyre CC. Computational analysis of subthalamic nucleus and lenticular fasciculus activation during therapeutic deep brain stimulation. J Neurophysiol 96: 1569–1580, 2006. [DOI] [PubMed] [Google Scholar]
- Miocinovic et al. 2007.Miocinovic S, Zhang J, Xu W, Russo GS, Vitek JL, McIntyre CC. Stereotactic neurosurgical planning, recording, and visualization for deep brain stimulation in non-human primates. J Neurosci Methods 162: 32–41, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell et al. 1989.Mitchell IJ, Clarke CE, Boyce S, Robertson RG, Peggs D, Sambrook MA, Crossman AR. Neural mechanisms underlying parkinsonian symptoms based upon regional uptake of 2-deoxyglucose in monkeys exposed to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neuroscience 32: 213–226, 1989. [DOI] [PubMed] [Google Scholar]
- Montgomery 2006.Montgomery EB Effects of GPi stimulation on human thalamic neuronal activity. Clin Neurophysiol 117: 2691–2702, 2006. [DOI] [PubMed] [Google Scholar]
- Moss et al. 2004.Moss J, Ryder T, Aziz TZ, Graeber MB, Bain PG. Electron microscopy of tissue adherent to explanted electrodes in dystonia and Parkinson's disease. Brain 127: 2755–2763, 2004. [DOI] [PubMed] [Google Scholar]
- Nakanishi et al. 1990.Nakanishi H, Kita H, Kitai ST. Intracellular study of rat entopeduncular nucleus neurons in an in vitro slice preparation: electrical membrane properties. Brain Res 527: 81–88, 1990. [DOI] [PubMed] [Google Scholar]
- Nakanishi et al. 1991.Nakanishi H, Kita H, Kitai ST. Intracellular study of rat entopeduncular nucleus neurons in an in vitro slice preparation: response to subthalamic stimulation. Brain Res 549: 285–291, 1991. [DOI] [PubMed] [Google Scholar]
- Nini et al. 1995.Nini A, Feingold A, Slovin H, Bergman H. Neurons in the globus pallidus do not show correlated activity in the normal monkey, but phase-locked oscillations appear in the MPTP model of parkinsonism. J Neurophysiol 74: 1800–1805, 1995. [DOI] [PubMed] [Google Scholar]
- Ozaita et al. 2002.Ozaita A, Martone ME, Ellisman MH, Rudy B. Differential subcellular localization of the two alternatively spliced isoforms of the Kv3.1 potassium channel subunit in brain. J Neurophysiol 88: 394–408, 2002. [DOI] [PubMed] [Google Scholar]
- Pan et al. 1985.Pan HS, Penney JB, Young AB. Gamma-aminobutyric acid and benzodiazepine receptor changes induced by unilateral 6-hydroxydopamine lesions of the medial forebrain bundle. J Neurochem 45: 1396–1404, 1985. [DOI] [PubMed] [Google Scholar]
- Parent et al. 1995.Parent A, Charara A, Pinault D. Single striatofugal axons arborizing in both pallidal segments and in the substantia nigra in primates. Brain Res 698: 280–284, 1995. [DOI] [PubMed] [Google Scholar]
- Parent and Smith 1987.Parent A, Smith Y. Differential dopaminergic innervation of the two pallidal segments in the squirrel monkey (Saimiri sciureus). Brain Res 426: 397–400, 1987. [DOI] [PubMed] [Google Scholar]
- Parent et al. 2001.Parent M, Levesque M, Parent A. Two types of projection neurons in the internal pallidum of primates: single-axon tracing and three-dimensional reconstruction. J Comp Neurol 439: 162–175, 2001. [DOI] [PubMed] [Google Scholar]
- Parent and Parent 2002.Parent M, Parent A. Axonal collateralization in primate basal ganglia and related thalamic nuclei. Thalamus Relat Syst 2: 71–86, 2002. [Google Scholar]
- Parent and Parent 2004.Parent M, Parent A. The pallidofugal motor fiber system in primates. Parkinsonism Relat Disord 10: 203–211, 2004. [DOI] [PubMed] [Google Scholar]
- Penn et al. 1998.Penn RD, Kroin JS, Reinkensmeyer A, Corcos DM. Injection of GABA-agonist into globus pallidus in patient with Parkinson's disease. Lancet 351: 340–341, 1998. [DOI] [PubMed] [Google Scholar]
- Percheron et al. 1984.Percheron G, Yelnik J, Francois C. A Golgi analysis of the primate globus pallidus. III. Spatial organization of the striato-pallidal complex. J Comp Neurol 227: 214–227, 1984. [DOI] [PubMed] [Google Scholar]
- Perney et al. 1992.Perney TM, Marshall J, Martin KA, Hockfield S, Kaczmarek LK. Expression of the mRNAs for the Kv3.1 potassium channel gene in the adult and developing rat brain. J Neurophysiol 68: 756–766, 1992. [DOI] [PubMed] [Google Scholar]
- Pralong et al. 2003.Pralong E, Debatisse D, Maeder M, Vingerhoets F, Ghika J, Villemure JG. Effect of deep brain stimulation of GPI on neuronal activity of the thalamic nucleus ventralis oralis in a dystonic patient. Clin Neurophysiol 33: 169–173, 2003. [DOI] [PubMed] [Google Scholar]
- Robertson et al. 1990.Robertson RG, Clarke CA, Boyce S, Sambrook MA, Crossman AR. The role of striatopallidal neurones utilizing gamma-aminobutyric acid in the pathophysiology of MPTP-induced parkinsonism in the primate: evidence from [3H]flunitrazepam autoradiography. Brain Res 531: 95–104, 1990. [DOI] [PubMed] [Google Scholar]
- Rudy et al. 1999.Rudy B, Chow A, Lau D, Amarillo Y, Ozaita A, Saganich M, Moreno H, Nadal MS, Hernandez-Pineda R, Hernandez-Cruz A, Erisir A, Leonard C, Vega-Saenz de Miera E. Contributions of Kv3 channels to neuronal excitability. Ann NY Acad Sci 868: 304–343, 1999. [DOI] [PubMed] [Google Scholar]
- Sah et al. 1988.Sah P, Gibb AJ, Gage PW. The sodium current underlying action potentials in guinea pig hippocampal CA1 neurons. J Gen Physiol 91: 373–398, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper and Loewy 1982.Saper CB, Loewy AD. Projections of the pedunculopontine tegmental nucleus in the rat: evidence for additional extrapyramidal circuitry. Brain Res 252: 367–372, 1982. [DOI] [PubMed] [Google Scholar]
- Sato et al. 2000a.Sato F, Lavallee P, Levesque M, Parent A. Single-axon tracing study of neurons of the external segment of the globus pallidus in primate. J Comp Neurol 417: 17–31, 2000a. [PubMed] [Google Scholar]
- Sato et al. 2000b.Sato F, Parent M, Levesque M, Parent A. Axonal branching pattern of neurons of the subthalamic nucleus in primates. J Comp Neurol 424: 142–152, 2000b. [DOI] [PubMed] [Google Scholar]
- Shin and Carlen 2008.Shin DS, Carlen PL. Enhanced Ih depresses rat entopeduncular nucleus neuronal activity from high-frequency stimulation or raised Ke+. J Neurophysiol 99: 2203–2219, 2008. [DOI] [PubMed] [Google Scholar]
- Shin et al. 2007.Shin DS, Samoilova M, Cotic M, Zhang L, Brotchie JM, Carlen PL. High frequency stimulation or elevated K+ depresses neuronal activity in the rat entopeduncular nucleus. Neuroscience 149: 68–86, 2007. [DOI] [PubMed] [Google Scholar]
- Shink et al. 1997.Shink E, Sidibe M, Smith Y. Efferent connections of the internal globus pallidus in the squirrel monkey: II. Topography and synaptic organization of pallidal efferents to the pedunculopontine nucleus. J Comp Neurol 382: 348–363, 1997. [PubMed] [Google Scholar]
- Shink and Smith 1995.Shink E, Smith Y. Differential synaptic innervation of neurons in the internal and external segments of the globus pallidus by the GABA- and glutamate-containing terminals in the squirrel monkey. J Comp Neurol 358: 119–141, 1995. [DOI] [PubMed] [Google Scholar]
- Sidibe et al. 1997.Sidibe M, Bevan MD, Bolam JP, Smith Y. Efferent connections of the internal globus pallidus in the squirrel monkey: I. Topography and synaptic organization of the pallidothalamic projection. J Comp Neurol 382: 323–347, 1997. [PubMed] [Google Scholar]
- Siegfried and Lippitz 1994.Siegfried J, Lippitz B. Bilateral chronic electrostimulation of ventroposterolateral pallidum: a new therapeutic approach for alleviating all parkinsonian symptoms. Neurosurgery 35: 1126–1130, 1994. [DOI] [PubMed] [Google Scholar]
- Smith et al. 1994.Smith Y, Wichmann T, DeLong MR. Synaptic innervation of neurones in the internal pallidal segment by the subthalamic nucleus and the external pallidum in monkeys. J Comp Neurol 343: 297–318, 1994. [DOI] [PubMed] [Google Scholar]
- Stocker and Pedarzani 2000.Stocker M, Pedarzani P. Differential distribution of three Ca(2+)-activated K(+) channel subunits, SK1, SK2, and SK3, in the adult rat central nervous system. Mol Cell Neurosci 15: 476–493, 2000. [DOI] [PubMed] [Google Scholar]
- Talley et al. 1999.Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci 19: 1895–1911, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth and Tomka 1968.Toth S, Tomka I. Responses of the human thalamus and pallidum to high frequency stimulations. Confin Neurol 30: 17–40, 1968. [DOI] [PubMed] [Google Scholar]
- Traub et al. 1991.Traub RD, Wong RK, Miles R, Michelson H. A model of a CA3 hippocampal pyramidal neuron incorporating voltage-clamp data on intrinsic conductances. J Neurophysiol 66: 635–650, 1991. [DOI] [PubMed] [Google Scholar]
- Vega-Saenz de Miera 2004.Vega-Saenz de Miera EC Modification of Kv2.1 K+ currents by the silent Kv10 subunits. Brain Res Mol Brain Res 123: 91–103, 2004. [DOI] [PubMed] [Google Scholar]
- Vergara et al. 1998.Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol 8: 321–329, 1998. [DOI] [PubMed] [Google Scholar]
- Vitek et al. 1999.Vitek JL, Chockkan V, Zhang JY, Kaneoke Y, Evatt M, DeLong MR, Triche S, Mewes K, Hashimoto T, Bakay RA. Neuronal activity in the basal ganglia in patients with generalized dystonia and hemiballismus. Ann Neurol 46: 22–35, 1999. [DOI] [PubMed] [Google Scholar]
- Vitek et al. 2004.Vitek JL, Hashimoto T, Peoples J, DeLong MR, Bakay RA. Acute stimulation in the external segment of the globus pallidus improves parkinsonian motor signs. Mov Disord 19: 907–915, 2004. [DOI] [PubMed] [Google Scholar]
- Volkmann 2004.Volkmann J Deep brain stimulation for the treatment of Parkinson's disease. J Clin Neurophysiol 21: 6–17, 2004. [DOI] [PubMed] [Google Scholar]
- Volkmann et al. 2006.Volkmann J, Moro E, Pahwa R. Basic algorithms for the programming of deep brain stimulation in Parkinson's disease. Mov Disord 21, Suppl. 14: S284–S289, 2006. [DOI] [PubMed] [Google Scholar]
- Wichmann and Soares 2006.Wichmann T, Soares J. Neuronal firing before and after burst discharges in the monkey basal ganglia is predictably patterned in the normal state and altered in parkinsonism. J Neurophysiol 95: 2120–2133, 2006. [DOI] [PubMed] [Google Scholar]
- Wu et al. 2001.Wu YR, Levy R, Ashby P, Tasker RR, Dostrovsky JO. Does stimulation of the GPi control dyskinesia by activating inhibitory axons? Mov Disord 16: 208–216, 2001. [DOI] [PubMed] [Google Scholar]
- Yelnik 2002.Yelnik J Functional anatomy of the basal ganglia. Mov Disord 17, Suppl. 3: S15–S21, 2002. [DOI] [PubMed] [Google Scholar]
- Yelnik et al. 2000.Yelnik J, Damier P, Bejjani BP, Francois C, Gervais D, Dormont D, Arnulf I, Bonnet AM, Cornu P, Pidoux B, Agid Y. Functional mapping of the human globus pallidus: contrasting effect of stimulation in the internal and external pallidum in Parkinson's disease. Neuroscience 101: 77–87, 2000. [DOI] [PubMed] [Google Scholar]