

Abstract
Inhibition of PARP activity results in extreme sensitization to MMS-induced cell killing in cultured mouse fibroblasts. In these MMS-treated cells, PARP inhibition is accompanied by an accumulation of S-phase cells that requires signaling by the checkpoint kinase ATR (Horton et al. (2005) J. Biol. Chem., 280, 15773-15785). Here, we examined mouse fibroblast extracts for formation of a complex that may reflect association between the damage responsive proteins PARP-1 and ATR. Co-immunoprecipitation of PARP-1 and ATR was observed in extracts prepared from MMS-treated cells, but not under conditions of PARP inhibition. Further, our experiments demonstrated PAR-adduction of ATR in extracts from control and MMS-treated cells. An interaction between purified ATR and PARP-1 was similarly demonstrated, suggesting that the observed co-immunoprecipitation of ATR and PARP-1 from cell extracts may be due to a direct interaction between the two enzymes. In addition, purified recombinant ATR is a substrate for poly(ADP-ribosyl)ation by PARP-1, and poly(ADP-ribose) (PAR) adduction of PARP-1 and ATR resulted in an increase in PARP-1 and ATR co-immunoprecipitation.
Keywords: ATR, PARP-1, PARP inhibitor, methylation damage
1. Introduction
Methylating agents of the SN2 type, such as methyl methanesulfonate (MMS), show a preference for methylation at nucleophilic nitrogens of DNA purine bases [1]. The base excision repair (BER) pathway responds to these modifications by excision of methylated bases with a damage-specific DNA glycosylase and replacing them with normal undamaged bases [2,3]. The mechanism of BER results in formation of various cytotoxic DNA intermediates that are passed from one step to the next during repair without triggering a cytotoxic DNA damage response [4,5]. However, if BER intermediates persist when cells enter S-phase, these intermediates may trigger signaling for a replication checkpoint [6]. It is known that such checkpoint signaling is not active in wild-type repair proficient mouse fibroblasts that have been exposed to a minimal sub-lethal concentration of MMS [7].
Poly(ADP-ribose) polymerase-1 (PARP-1) is an abundant and constitutively expressed enzyme that detects and binds to DNA nicks and strand breaks, including those formed during BER [8,9]. Once bound to DNA, PARP-1 is strongly activated for poly(ADP-ribose) (PAR) synthesis, adducting itself and a number of other nuclear proteins [10,11]. Although PARP-1 is only one of many PARP isoforms, it is thought to contribute the majority (∼90%) of the PAR adducting activity in the cell [12]. As a consequence of PAR automodification, PARP-1 is released from its binding site on DNA allowing repair to continue. Inhibition of PARP activity in MMS-treated mouse fibroblasts by 4-amino-1,8-naphthalimide (4-AN) results in dramatic sensitization to MMS and cell death by apoptosis [13]. The results suggest that PARP inhibition prevents the normally efficient repair of MMS-induced strand breaks [14,15].
Mechanisms of ATM (ataxia telangiectasia-mutated)- and ATR (ATM and Rad3-related)-dependent checkpoint signaling after DNA damage by ionizing or UV irradiation have been investigated extensively [16,17]. Considerable information is also available regarding DNA damage checkpoints resulting from alkylating agent-induced DNA strand breaks [18-20]. For example, it is known that ATR signals replication fork stalling following formation of single-stranded DNA (ssDNA) during BER of alkylated base lesions [21]. In mouse fibroblasts treated with a low dose of MMS combined with the PARP inhibitor 4-AN, there is a rapid block in DNA synthesis followed by a prolonged S-phase arrest accompanied by phosphorylation of Chk1 [7]. Caffeine, an inhibitor of ATR and ATM kinase activities [22], abrogated the S-phase arrest and also blocked phosphorylation of Chk1 [7], an essential downstream kinase primarily regulated by ATR [23]. Since both ATR and Chk1 are known to be involved in signaling a replication checkpoint for stalled replication forks [23], we proposed that DNA bound, activity-inhibited, PARP-1 triggered a replication block, and that the persistence of the S-phase arrest required ATR/Chk1-mediated signaling [7]. Signaling is activated in these MMS-treated cells only when PARP-1 is inhibited [7], and inhibition of PARP activity is known to result in increased levels of MMS-induced DNA strand breaks [24]. Activation of PARP-1, release of the protein from DNA, as well as the associated enhanced repair of DNA damage may prevent checkpoint signaling under these conditions.
An interaction between ATM and PAR-adducted PARP-1 has already been demonstrated, and in addition PARP-1 activity modulates the activation of ATM kinase [25,26]. Given that both ATR and PARP-1 are associated with the cellular response to MMS, we now examine the possibility that ATR and PARP-1 may also exist as a complex in mouse cells. We show that these proteins can be co-immunoprecipitated from extract of untreated control cells, but that the interaction intensifies when cells are treated with MMS. Interestingly, this co-immunoprecipitation was blocked by co-treatment with a PARP inhibitor. An interaction between purified ATR and PARP-1 was similarly demonstrated, suggesting that the observed co-immunoprecipitation of ATR and PARP-1 from cell extracts may be due to a direct interaction between the two enzymes.
2. Materials and methods
2.1. Materials
Dulbecco’s modified Eagle’s medium (DMEM), GlutaMAX-1 and L-glutamine were from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was from HyClone (Logan, UT), and hygromycin B from Roche Molecular Biochemicals (Indianapolis, IN). MMS and the PARP inhibitors 4-AN and PJ-34 were obtained from Sigma-Aldrich (St. Louis, MO).
Anti-ATR (sc-1887) affinity purified goat polyclonal antibody was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), anti-ATR mouse monoclonal antibody to human ATR (MS-ATR11-PX1) was from GeneTex, Inc. (San Antonio, TX) and anti-PARP-1 rabbit polyclonal antibody (ALX-210-222) from Alexis Biochemicals (San Diego, CA). Mouse anti-human PARP-1 monoclonal antibody (556494) was from BD Biosciences Pharmingen (San Jose, CA) and anti-PAR mouse monoclonal antibody (4335-MC-100) was from Trevigen (Gaithersburg, MD). Normal goat (for ATR) IgG-AC (sc-2346) and normal rabbit (for PARP-1) IgG-AC (sc-2345) were obtained from Santa Cruz and used for immunoprecipitation controls. Normal mouse IgG-HRP (sc2748) was obtained from Santa Cruz and used as a negative control for immunoblotting.
Anti-Rad9 mouse monoclonal antibody (IMG-164) was from Imgenex (San Diego, CA) and anti-replication protein A (RPA) mouse monoclonal antibody (RPA34-20) was from Oncogene (Cambridge, MA). Anti-Chk1 (sc-8408) mouse monoclonal antibody was purchased from Santa Cruz Biotechnology, Inc., anti-phospho-Chk1 (serine345) polyclonal antibody (2348S) from Cell Signaling (Beverly, MA), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from Alpha Diagnostic International, Inc. San Antonio, TX.
Restore Western Blot Stripping Buffer and Super Signal West Pico Chemiluminescent substrate were from Pierce Biotechnology, Inc. (Rockford, IL). Western Lightning Chemiluminescence Reagent Plus was from Perkin Elmer Life Sciences, Inc. (Boston, MA). The mouse IgG secondary antibody was goat anti-mouse IgG (H+L) binding grade affinity purified horseradish peroxidase (HRP) conjugate, and the rabbit IgG secondary antibody was goat anti-rabbit IgG (H+L)-HRP conjugate, both from Bio-Rad Laboratories (Hercules, CA). Protein A-sepharose CL-4B was from GE Healthcare Life Sciences (Piscataway, NJ). Protein G-agarose and protease inhibitor complete (EDTA-free) were from Roche Diagnostics Corporation (Indianapolis, IN). Leupeptin, aprotinin, and phenylmethylsulfonyl fluoride (PMSF) were from Calbiochem (La Jolla, CA).
2.2. Cell lines
The cell line used was a clone of the wild-type mouse fibroblast cell line M16tsA and has been described previously [5]. Cells were routinely grown at 34 °C in a 10% CO2 incubator in DMEM supplemented with GlutaMAX-1, 10% FBS, and hygromycin (80 μg/ml). PARP-1 +/+ and PARP-1 -/- spontaneously immortalized mouse embryonic fibroblasts [27] were obtained from Dr. Josianne Ménissier-de Murcia (CNRS, Illkirch-Graffenstaden, France). These cells were cultured at 37 °C in a 10% CO2 incubator in DMEM containing L-glutamine and 10% FBS. All cells were routinely tested and found to be free of mycoplasma contamination.
2.3. Extract preparation, co-immunoprecipitation and immunoblotting of extracts
Cells (either treated as described, or control, untreated) were washed in phosphate buffered saline (PBS) and collected by scraping, suspended in two volumes of lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 25 mM NaF, 0.1 mM sodium orthovanadate, 0.2% Triton X-100, and 0.3% NP-40) containing protease inhibitors, 0.1 mM PMSF, 1 μg/ml aprotinin, and 5 μg/ml leupeptin [28] and incubated on ice for 30 min. After agitating the tubes briefly, the lysates were centrifuged at 20,800 × g for 30 min at 4 °C, and the supernatant fraction (extract) was divided into aliquots and stored at −80 °C. The protein concentrations of the extracts (typically approximately 6 mg/ml) were determined using the Bio-Rad protein assay with bovine serum albumin as standard.
For co-immunoprecipitations, an equal amount (1 mg protein) of each cell extract was mixed with 1-2 μg of affinity purified anti-ATR goat polyclonal antibody. The mixture was incubated with rotation for 4 h at 4 °C. The immunocomplexes were adsorbed onto protein A-sepharose and protein G-agarose beads (1:1 mixture) by incubating the mixture for 16 h at 4 °C. The beads were then washed four times with lysis buffer containing protease inhibitors. Finally, the beads were resuspended in SDS sample buffer, heated at 95 °C for 5 min, and briefly centrifuged. The soluble proteins were separated by electrophoresis on 6% SDS-polyacrylamide gels in Tris-glycine buffer as described previously [7]. The proteins were then transferred to a nitrocellulose membrane (overnight at 25V).
The membrane was blocked with 5% nonfat dry milk in Tris-buffered saline (TBS) containing 0.1% (v/v) Tween-20 (TBS-T) and first probed with anti-PARP-1 monoclonal antibody (1:1,000 dilution). Goat anti-mouse IgG conjugated to HRP (1:10,000 dilution) was used as secondary antibody, and immobilized HRP activity was detected by enhanced chemiluminescence (ECL). The blot was stripped by incubating with buffer containing 62.5 mM Tris-HCl, pH 6.8, 100 mM β-mercaptoethanol, and 1% SDS for 30 min at 50 °C, then washed twice for 30 min with room temperature TBS-T, or with Restore Western Blot Stripping Buffer as suggested by the manufacturer (Pierce Biotechnology, Inc.). The presence of ATR was confirmed by probing with mouse anti-ATR monoclonal antibody (1:1,000). Similarly, the cell extract was immunoprecipitated with anti-PARP-1 rabbit polyclonal antibody as described above, and the blot was probed first with anti-ATR mouse monoclonal antibody. After stripping the blot, the presence of PARP-1 was confirmed using anti-PARP-1 mouse monoclonal antibody. In some control experiments, the immunoprecipitating antibody was substituted with normal goat or rabbit agarose-conjugated IgG. As a loading control, we compared the presence of goat or rabbit heavy chain IgG in the immunoprecipitate samples. Cell extract (20 μg) without immunoprecipitation was also used as a source of marker proteins (Input) and was submitted directly to SDS-PAGE and immunoblotting as presented in the figures. Where indicated, the intensity of the immunoblotted bands was measured using ImageQuant TL software (GE Healthcare).
In other experiments, cell extract was immunoprecipitated with anti-ATR goat polyclonal antibody and the blot was probed with anti-PAR monoclonal antibody. Similarly, cell extracts were immunoprecipitated with anti-PAR mouse monoclonal antibody and the blot was probed with anti-ATR goat polyclonal antibody. Other experiments involved immunoprecipitation with anti-ATR antibody and probing of the blot with antibodies to Rad9 or RPA.
Cell lysates (40 μg) prepared from control and treated cells were also directly separated by 4-12% SDS-PAGE. After transfer and blocking as described above, the blots were first probed with anti-phospho-Chk1 polyclonal antibody (1:1,000 dilution), and then, after stripping, with anti-Chk1 monoclonal antibody (1:1,000 dilution). Finally, as a loading control, blots were probed for GAPDH. In each case, proteins were detected by ECL following incubation with secondary antibody conjugated to HRP.
2.4. Expression and purification of recombinant proteins
Human kidney HEK293T cells were grown in DMEM supplemented with 10% FBS and 100 U/ml of penicillin and streptomycin. Cells (8 × 106/225-cm2 flask) were transfected with 25 μg of pcDNA3-Flag-ATR plasmid, a gift from Dr. Aziz Sancar [29]. Briefly, 48 h after transfection, cells were washed with PBS and lysed in 5 ml of lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM β-glycerophosphate, 10% glycerol, 1% Tween 20, 0.1% NP-40, 1 mM sodium orthovanadate, 1 mM NaF, and protease inhibitors) for 30 min on ice. The supernatant fraction obtained after centrifugation at 32,000 x g for 30 min was incubated at 4 °C overnight with 40 μl of anti-Flag antibody-affinity gel beads previously washed in TBS. The Flag-ATR-bound beads were washed three times with 1.5 ml of TBS, then once with 1.5 ml TBS plus 1 M NaCl, and then once with 1.5 ml TBS. ATR bound to the beads was eluted with 200 μl elution buffer containing 200 μg/ml Flag peptide, 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% glycerol, and 1 mM dithiothreitol. The eluted fraction was collected following centrifugation, and the purified ATR protein sample was divided into aliquots and stored at -80 °C. Recombinant human wild-type PARP-1 was expressed and purified as described previously by an adaptation of published methodology [9,30,31]. The catalytically inactive Lys893 mutant of PARP-1 has been described previously [32] and was a generous gift from Dr. Eric Ackerman (Pacific Northwest National Laboratory, Richland, WA).
2.5. Poly(ADP-ribosyl)ation of ATR in vitro by purified recombinant PARP-1
The poly(ADP-ribosyl)ation reaction was performed as described previously [33]. Briefly, wild-type or inactive mutant PARP-1 (15 nM) or ATR (20 nM) alone, or a mixture of proteins, was incubated in a reaction mixture (final volume 50 μl) containing 50 mM Tris-HCl, pH 7.8, 25 mM MgCl2, 1 mM DTT, 4 μg nicked calf thymus DNA, and 100 μM NAD+. The incubation was carried out at 37 °C for 30 min and the reaction terminated by addition of SDS sample buffer. The solution was heated for 5 min at 95 °C, and proteins were separated by 6% SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was incubated with 5% nonfat dry milk in TBS-T and probed with anti-PAR monoclonal antibody (1:1,000 dilution). Goat anti-mouse IgG conjugated to HRP (1:8,000 dilution) was used as secondary antibody, and immobilized HRP activity was detected by ECL. The blot was stripped and the presence of ATR was confirmed with mouse anti-ATR monoclonal antibody. The filter was stripped a second time and re-probed with anti-PARP-1 monoclonal antibody.
2.6. Co-immunoprecipitation of purified ATR and PARP-1 proteins
A pre-incubation of purified recombinant ATR and PARP-1 proteins for 30 min at 37 °C with or without NAD+ was conducted to determine the effect of poly(ADP-ribosyl)ation on the co-immunoprecipitation. Then co-immunoprecipitation was performed in the presence of binding buffer (25 mM Tris, pH 8.0, 10% glycerol, 100 mM NaCl, 0.01% NP40) containing protease inhibitors, 0.1 mM PMSF, 1 μg/ml aprotinin and 5 μg/ml leupeptin. In other experiments, NaCl concentrations of 200 and 300 mM were also used. To a mixture of 1.5 μM PARP-1 and 1.5 μM ATR in a final volume of 50 μl, either anti-PARP-1 rabbit polyclonal antibody or anti-ATR goat polyclonal antibody was added, and the mixture was incubated with rotation for 4 h at 4 °C. The protein complexes then were adsorbed onto protein A-sepharose and protein G-agarose beads by incubating the mixture overnight at 4 °C in a final volume of 500 μl of binding buffer. The beads were collected by centrifugation and washed four times with binding buffer containing protease inhibitors. The beads were then suspended in SDS-sample buffer and heated for 5 min at 95 °C. The soluble proteins were separated by 6% SDS-PAGE and the transferred blot was probed with anti-PARP-1 monoclonal antibody or anti-ATR monoclonal antibody as described above. The blots were then stripped and the presence of ATR or PARP-1 was confirmed by incubating the membrane with the respective primary mouse monoclonal antibodies followed by incubation with secondary anti-mouse antibody. The poly (ADP-ribosyl)ation reaction was monitored by probing the blots with anti-PAR antibody.
3. Results
3.1. Co-immunoprecipitation of ATR and PARP-1 from cell extracts
The PAR synthesis activity of PARP-1 is triggered following its binding to DNA strand breaks, including those generated during BER [34]. Since ATR is an apical checkpoint protein kinase responding to MMS-induced DNA damage that includes strand breaks [18], we investigated the possibility of complex formation between the damage responsive proteins ATR and PARP-1. Experiments were conducted with the same wild-type mouse fibroblast cell line that had been characterized earlier for cell cycle arrest as a function of MMS exposure in the presence and absence of the PARP inhibitor 4-AN [7].
Whole cell extracts were prepared at 2 h from logarithmically growing cells treated with MMS (0.25 mM for 1 h followed by 1 h incubation in drug-free medium) or medium alone as a control. In the first experiments, the extracts were immunoprecipitated with an antibody against ATR, or with normal goat agarose-conjugated IgG as a control, and the immunoprecipitated fractions were subjected to SDS-PAGE and immunoblotting with anti-PARP-1 or anti-ATR antibodies, or with normal IgG as a negative control. PARP-1 was found to co-immunoprecipitate with ATR from both cell extracts, however, the level of PARP-1 immunoprecipitated from the MMS-treated cell extract was significantly higher than that from the control cell extract (Fig. 1A, compare lane 1, top panel, of left and right panels). A plot of the intensities of the ATR immunoprecipitated and PARP-1 immunoblotted bands in control extracts compared with extracts from MMS-treated cells is presented in Figure 1B. These data confirm that the interaction between ATR and PARP-1 intensifies following treatment with MMS. The observed interaction was direct and not mediated by DNA since it was still observed in the presence of ethidium bromide (data not shown).
Fig. 1.

Interaction of ATR and PARP-1 as revealed by co-immunoprecipitation. Mouse fibroblast cell extracts were subjected to co-immunoprecipitation using anti-ATR antibody as described in “Materials and methods.” (A) Cell extracts from untreated wild-type mouse cells, (left panel, Control Cells) or cells 2 h after treatment for 1 h with 0.25 mM MMS (right panel, MMS-treated Cells) were immunoprecipitated (IP) with anti-ATR antibody or normal goat IgG as a negative control, and proteins were separated by SDS-PAGE and then blotted onto a membrane. The blot was probed (IB) for PARP-1 (top panel) and then, after stripping, for ATR (middle panel) or normal IgG in the same size range as PARP-1 as a negative control (bottom panel). “Input” represents 20 μg of cell extract mixed directly with SDS sample buffer. (B) Plot of the intensities (in arbitrary units, A.U.) of the ATR immunoprecipitated and PARP-1 immunoblotted bands from extracts of control and MMS-treated cells. Data represents results averaged from 3 experiments. (C) Untreated, control wild-type cell extracts (lanes 1 and 2) or cells treated with 0.25 mM MMS for 1 h (lane 3, MMS), or 20 J/m2 UV (lane 4, UV), in each case harvesting cells at 2 h, were subjected to immunoprecipitation with normal goat IgG (lane 1) or with anti-ATR antibody (lanes 2-4). The blot was probed (IB) for PARP-1 (top panel), for ATR (middle panel) and for normal IgG (bottom panel), as described above. (D) Plot of the intensities (in arbitrary units, A.U.) of the ATR immunoprecipitated and PARP-1 immunoblotted bands from extracts of control, MMS-treated and UV-exposed cells, and represents results averaged from 3 experiments.
We next evaluated co-immunoprecipitation of ATR and PARP-1 after treatment of the same cells with UV, another agent known to induce an ATR-mediated response. Extracts from control, or MMS- or UV-treated cells were again immunoprecipitated with an antibody against ATR, or with goat IgG, used as a control. Precipitated fractions were then separated by SDS-PAGE, and immunoblotted with anti-PARP-1 and anti-ATR antibodies, or normal IgG as a negative control (Fig. 1C). With extract derived from UV-treated cells (20 J/m2), an enhanced immunoprecipitation of PARP-1 by anti-ATR antibody was observed, a result similar to that obtained following MMS treatment (Fig. 1C, top panel, compare lanes 3 and 4 with lane 2). A plot of the intensities of the ATR immunoprecipitated and PARP-1 immunoblotted bands in control extracts compared with extracts from MMS- and UV-treated cells is presented in Figure 1D.
We next asked whether inhibition of PARP activity by 4-AN would affect the increased co-immunoprecipitation of PARP-1 and ATR observed following treatment with MMS alone. Immunoprecipitation with either anti-ATR antibody (Fig. 2A) or anti-PARP-1 antibody (Fig. 2B) was used. PARP-1 and ATR co-immunoprecipitation was not observed in extract from cells treated with 4-AN (10 μM) alone (i.e., under conditions where PAR synthesis activity of PARP-1 is blocked; Fig. 2A and B, top panels, lane 2), and negligible PARP-1 co-immunoprecipitation was observed with the extract from cells treated with the combination of MMS and 4-AN (Fig. 2A and B, top panels, lane 3). Control experiments indicated this was not due to a failure of immunoprecipitation of PARP-1 and ATR, respectively, or to a lack of expression of the enzymes in these cells. Such controls included immunoprecipitation with normal goat or rabbit agarose-conjugated IgG or immunoblotting with normal IgG as negative controls, comparison of the levels of heavy chain IgG in the immunoprecipitate samples as loading controls, immunoblotting with either anti-ATR or anti-PARP-1 to verify immunoprecipitation with the primary antibody, and finally assessment of the PARP-1 and ATR levels in the extracts without immunoprecipitation (Fig. 2A and B, bottom panels).
Fig. 2.

Effect of 4-AN on reciprocal co-immunoprecipitation of ATR and PARP-1 after MMS treatment. (A and B) Mouse fibroblast cells were treated with 0.25 mM MMS for 1 h (lane 1, MMS), with 4-AN (10 μM) for 2 h (lane 2, 4-AN), or with MMS plus 4-AN (lane 3, MMS+4-AN), as described in “Materials and methods.” Extracts were prepared after 2 h and subjected to immunoprecipitation with (A) anti-ATR, or (B) anti-PARP-1 antibodies or goat (for ATR) or rabbit (for PARP-1) normal IgG as negative controls. The blots were probed (IB) with antibodies to PARP-1, ATR, or with normal IgG at the size range of the co-immunoprecipitated protein (PARP-1 in A, and ATR in B). The level of heavy chain IgG in the immunoprecipitated samples was used as an indicator of loading. Extract without immunoprecipitation was probed with (A) anti-PARP-1 or (B) anti-ATR antibody (bottom panels). “Input” (lane 4 in A and B) represents 20 μg of MMS-treated cell extract analyzed directly, without immunoprecipitation. (C) Immunoprecipitation (IP) of the same extracts with normal goat IgG (lane 1) as a negative control or anti-ATR antibody (lanes 2-5), and immunoblotting (IB) with anti-Rad9, anti-RPA, anti-ATR antibodies or normal IgG as indicated. “Input” (lane 6) represents 20 μg of MMS-treated cell extract analyzed directly, without immunoprecipitation.
Next, we carried out co-immunoprecipitation experiments with other known DNA damage sensor proteins, RPA and Rad9. Following stalling of replication forks, as well as during normal replication, extensive regions of ssDNA are exposed, and this ssDNA becomes coated with the ssDNA-binding protein RPA, which is critical for ATR checkpoint activation [35]. Rad9, part of the 9-1-1 complex, also binds to the RPA/ssDNA complex and recruits signaling proteins such as ATR [35]. We found that anti-ATR antibody was able to immunoprecipitate both Rad9 and RPA from control as well as extracts from treated mouse fibroblasts. However, the amount of each protein immunoprecipitated did not change as a function of the cell treatments (MMS, 4-AN, MMS+4-AN) used here (Fig. 2C).
Additional control experiments were conducted to verify that co-immunoprecipitation of PARP-1 and ATR could only be observed from extracts of PARP-1-expressing mouse fibroblast cells (Fig. 3). As expected, co-immunoprecipitation of PARP-1 and ATR was not observed when similar experiments were conducted with MMS-treated PARP-1 -/- cells, whereas the isogenic PARP-1 +/+ cells used were positive for PARP-1 and ATR co-immunoprecipitation (Fig. 3A, top panel). Among the various conditions and cell extracts tested in this experiment and presented in Figure 3, panels B and C, the only extract showing co-immunoprecipitation of PARP-1 and ATR was prepared from PARP-1 +/+ cells treated with MMS alone (Fig. 3B and C, top panels, lane 2). As expected, ATR could be immunoprecipitated from all of the cell extracts (Fig. 3B, middle panel), and conversely, PARP-1 could be immunoprecipitated from the PARP-1 +/+ cell extracts, but not from the PARP-1 -/- cell extracts (Fig. 3C, middle panel).
Fig. 3.

Effect of PARP-1 gene deletion on co-immunoprecipitation of ATR and PARP-1 from mouse fibroblast cell extracts. Cell extracts were from PARP-1 +/+ cells or isogenic PARP-1 -/- gene-deleted cells, and cell treatments were as described in Figure 2 and in “Materials and methods.” Immunoprecipitation (IP) of (A) control cell extracts or (B) extracts from treated cells, as indicated, with anti-ATR antibody, and immunoblotting (IB) with anti-PARP-1, anti-ATR, or normal IgG at the size range of PARP-1 as indicated. “Input” (panel A, lane 3) represents 20 μg of untreated PARP-1 +/+ cell extract analyzed directly, without immunoprecipitation. (C) Immunoprecipitation (IP) with anti-PARP-1 antibody, and immunoblotting (IB) with anti-ATR, anti-PARP-1, or normal IgG at the size range of ATR, as indicated. As a negative control, results from immunoprecipitations of extract from MMS-treated PARP-1 +/+ cells using normal goat or rabbit IgG are shown in lanes 1 of panels B and C (MMS: IgG). “Input” (panels B and C, lane 8) represents 20 μg of MMS-treated PARP-1 +/+ cell extract analyzed directly, without immunoprecipitation.
We next determined whether PJ-34, a more recently described potent water-soluble PARP inhibitor [36,37], would be as effective as 4-AN in preventing the interaction between ATR and PARP-1 observed in extracts from MMS-treated cells. The combination of MMS (0.25 mM) and PJ-34 (5 μM) used in these immunoprecipitation experiments resulted in a significant accumulation of cells in S-phase of the cell cycle (data not shown). A similar treatment protocol was used as described above for experiments with 4-AN. As shown in Figure 4A, and as discussed above, an interaction between ATR and PARP-1 was observed in ATR-immunoprecipitated extracts prepared from MMS-treated cells (Fig. 4A, top panel, compare lane 5 with control extracts in lane 3). The level of interacting PARP-1 protein was considerably decreased when using extracts from cells additionally treated with 4-AN (lane 4) or PJ-34 (5 μM) (lane 6), and was undetectable following treatment with 4-AN or PJ-34 alone (lanes 2 and 7). Thus inhibition of PARP activity with either 4-AN or PJ-34 was able to significantly block the interaction between ATR and PARP-1 that results following exposure of cells to a low concentration of MMS. Possibly PAR-adduction of ATR and/or PARP-1 facilitates the interaction of these two proteins.
Fig. 4.
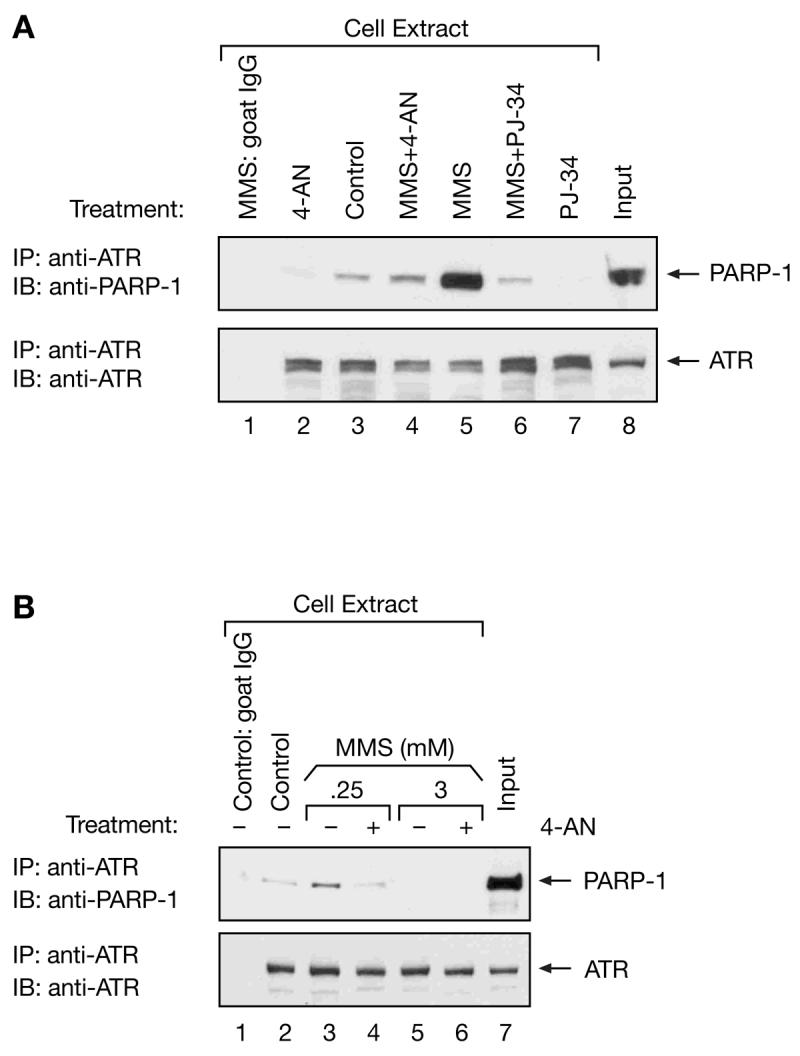
Effect of PJ-34 on co-immunoprecipitation of PARP-1 and ATR, and MMS dose dependence of the co-immunoprecipitation. (A) Extracts were prepared at 2 h from control, untreated mouse fibroblast cells (lanes 1 and 3), or cells treated with 0.25 mM MMS for 1 h (lane 5, MMS), with 4-AN (10 μM) for 2 h (lane 2, 4-AN), with PJ-34 (5 μM) for 2 h (lane 7, PJ-34) or with MMS plus 4-AN (lane 4, MMS+4-AN), or MMS plus PJ-34 (lane 6, MMS+PJ-34) as described in “Materials and methods.” Extracts were subjected to immunoprecipitation (IP) with anti-ATR antibody (lanes 2-7) or goat normal IgG (lane 1) as a negative control, and blotted (IB) with anti-PARP-1 and anti-ATR antibodies. “Input” (lane 8) represents 20 μg of MMS-treated cell extract analyzed directly, without immunoprecipitation. (B) Extracts from control, untreated mouse fibroblast cells (lanes 1 and 2), or cells treated for 1 h with 0.25 mM (lanes 3 and 4) or 3 mM MMS (lanes 5 and 6) in the absence (-) or presence (+) of 4-AN were immunoprecipitated with anti-ATR antibody (lanes 2-6) or with normal goat IgG (lane 1) as a negative control. Blots were probed with anti-PARP-1 or anti-ATR antibody as indicated. “Input” (lane 7) represents 20 μg of 0.25 mM MMS-treated cell extract analyzed directly, without immunoprecipitation.
Next we compared the interaction of ATR and PARP-1 in cells treated with low dose MMS (0.25 mM), as utilized in all the experiments described above, or with a highly toxic concentration (3 mM) of MMS. Whereas an association of ATR and PARP-1 was seen only in the absence of 4-AN for low dose MMS (Fig. 4B, top panel, compare lanes 3 and 4), there was no detectable association between ATR and PARP-1 following treatment with 3 mM MMS either in the presence or absence of the PARP inhibitor (Fig. 4B, top panel, lanes 5 and 6). It appears that the interaction of ATR and PARP-1 is prevented in cells receiving highly cytotoxic levels of MMS-induced DNA damage, whether PARP activity is inhibited or not.
3.2. Dose-dependent activation of Chk1 after exposure to MMS
We have shown that the S-phase arrest observed in mouse fibroblasts treated with a low dose of MMS combined with the PARP inhibitor 4-AN, is accompanied by caffeine-sensitive phosphorylation of Chk1 [7]. Follow-up experiments in human cells demonstrated that the Chk1 activation was ATR-dependent [15]. Here, we looked for appearance of phosphorylated Chk1 protein (phospho-Chk1) in cells treated with a range of concentrations of MMS in the presence (+) or absence (-) of 4-AN (Fig. 5). As observed previously with the lowest concentrations (0.25 - 1 mM) of MMS [7], phosphorylated Chk1 was detected in cells also treated with 4-AN, but not (or at a significantly reduced level) in its absence (Fig. 5, top panel, lanes 3 - 8). However, at higher doses of MMS (2 and 3 mM), phosphorylated Chk1 was observed both in the presence and absence of 4-AN (Fig. 5, top panel, lanes 9 -12). The level of non-phosphorylated Chk1 protein remained constant in control, untreated cells as well as following all of the cell treatments (Fig. 5, middle panel). Probing the blot for GAPDH demonstrated equal loading of samples (Fig. 5, bottom panel). It appears that a highly cytotoxic concentration of MMS alone (3 mM) is sufficient to activate an ATR/Chk1-mediated damage response pathway even in the absence of PARP inhibitor.
Fig. 5.

Immunoblot analysis of phosphorylated Chk1 in MMS-treated cell extracts. Extracts were prepared from control, untreated cells (lane 1), cells treated for 2 h with 4-AN alone (lane 2) or 2 h after a 1 h treatment with a range of concentrations of MMS in the absence (-) or presence of 4-AN (+) (lanes 3-12) as described in “Materials and methods,” and equal amounts of protein (40 μg) were resolved by 4-12% SDS-PAGE. Blots were probed for phosphorylated Chk1 (phospho-Chk1, top panel), non-phosphorylated Chk1 (middle panel) and GAPDH (bottom panel) as a loading control.
Our results demonstrate that an ATR-mediated pathway is activated in our system following exposure to high dose MMS (3 mM) either in the presence or absence of 4-AN (Fig. 5, top panel, lanes 11 and 12), and under treatment conditions where ATR and PARP-1 do not associate (Fig. 4B, top panel, lanes 5 and 6). The ATR pathway is similarly activated following exposure to a minimal concentration of MMS, but only in the presence of 4-AN (Fig. 5, top panel, compare lanes 3 and 4). It is not activated by the same low sub-lethal concentration of MMS alone (Fig. 5, top panel, lane 3) under conditions where a strong interaction of ATR and PARP-1 is also observed (Fig. 4B, top panel, lane 3). Taken together, our results suggest that the observed association of PARP and ATR prevents activation of ATR-mediated damage responses.
3.3. Poly(ADP-ribose) adduction of ATR in MMS-treated cells
The observed interaction between PARP-1 and ATR raised the possibility that PARP-1 may be able to PAR adduct ATR in MMS-treated cells. In order to evaluate this suggestion, we conducted co-immunoprecipitation experiments in cell extracts with anti-ATR and anti-PAR antibodies (Fig. 6). In the absence of PARP inhibitor and using extracts from control or MMS-treated cells, the anti-ATR antibody precipitated PAR-containing material recognized by anti-PAR antibody in the size range of ATR (300 kDa), along with material in larger size ranges consistent with PAR-adducted ATR (Fig. 6A, top panel, lanes 1 and 2). Similarly, the anti-PAR antibody immunoprecipitated ATR of the native protein size, and also precipitated ATR of larger mass, probably due to PAR-adduction (Fig. 6B, top panel). More PAR-adducted protein was observed in immunoprecipitations from the extract prepared from MMS-treated cells than from the control cell extract (Fig. 6, top panel, compare lanes 1 and 2). Thus, our experiments demonstrated PAR-adduction of ATR in extracts from control and MMS-treated cells. As expected, with extracts from cells treated with 4-AN alone or with the combination of MMS and 4-AN, no PAR-adducted material was observed in the immunoprecipitates (Fig. 6A and B, top panel, lanes 3 and 4). In addition, less PAR-adducted protein was detected in cell extracts directly immunoblotted with PAR antibody (Fig. 6B, bottom panel). In both ATR- and PAR-immunoprecipitated extracts, normal IgG used as a negative control failed to detect ATR- or PAR-containing material, and comparison of heavy chain IgG in the immunoprecipitate samples was used as an indicator of equal loading as outlined previously (Fig. 6A and B).
Fig. 6.

Effect of cell treatment on poly(ADP-ribosyl)ation of immunoprecipitated ATR in cell extracts. Mouse fibroblasts were either untreated (lane 1, Control) or treated with MMS for 1 h (lane 2, MMS), with 4-AN for 2 h (lane 3, 4-AN), or with MMS plus 4-AN (lane 4, MMS+4-AN), as described in Figure 2 and “Materials and methods.” Extracts were prepared after 2 h and subjected to immunoprecipitation (IP) with (A) anti-ATR or (B) anti-PAR antibody. Immunoblotting (IB) was with anti-ATR, anti-PAR, or normal IgG at the size range of ATR as indicated. The level of heavy chain IgG in the immunoprecipitated samples was used as an indicator of loading. Extract without immunoprecipitation was probed with (A) anti-ATR or (B) anti-PAR antibody (bottom panels of A and B). Migration of purified ATR in the SDS-PAGE system corresponded to 300 kDa.
3.4. Poly(ADP-ribose) adduction of purified PARP-1 and ATR
Next, we ascertained the ability of purified PARP-1 to poly(ADP-ribosyl)ate purified ATR in an in vitro reaction. Self-poly(ADP-ribosyl)ation was apparent in our sample of purified wild-type PARP-1 protein incubated with calf thymus DNA and NAD+ (Fig. 7, lane 1 of top panel). However, we were able to find reaction conditions and relative protein concentrations where PARP-1 self-adduction was less than PAR-adduction of ATR in reactions containing both proteins (Fig. 7, top panel, compare lanes 1 and 2). Purified recombinant ATR incubated alone under the same experimental conditions required for achieving poly(ADP-ribosyl)ation, had no PAR-adduction detectable by immunoblotting with anti-PAR antibody (Fig. 7, lane 3 of top panel). Incubation of ATR with a catalytically inactive Lys893 mutant PARP-1 protein also failed to result in PAR-adduction (Fig. 7, top panel, lane 4). The mobilites of unmodified ATR and PARP-1 proteins (300 and 116 kDa, respectively) are indicated on this top panel of Figure 7, and immunoblotting with antibodies to ATR and PARP-1 are presented in the lower panels. These results confirm that ATR is a substrate for poly(ADP-ribosyl)ation by PARP-1.
Fig. 7.

Poly(ADP-ribosyl)ation of purified ATR by purified PARP-1. Ribosylation reaction mixtures containing 15 nM PARP-1 (lane 1), 15 nM PARP-1 plus 20 nM ATR (lane 2), 20 nM ATR (lane 3) or 15 nM catalytically inactive Lys893 mutant of PARP-1 plus ATR (lane 4) were incubated as described under “Materials and methods,” and proteins were analyzed by 6% SDS-PAGE. Immunoblotting (IB) in the top panel was with anti-PAR antibody. The migration positions in the gel of unmodified ATR and PARP-1 are indicated and correspond to 300 kDa and 116 kDa, respectively. Immunoblotting (IB) of the reaction mixture with antibodies to ATR and PARP-1 are presented in the lower panels.
3.5. Co-immunoprecipitation of purified ATR and PARP-1 proteins
In the next experiments, we asked whether purified ATR and PARP-1 could be co-immunoprecipitated, and whether poly(ADP-ribosyl)ation might influence this interaction. Purified human enzymes were used in co-immunoprecipitation experiments that included a pre-incubation with or without NAD+ (Fig. 8). ATR and PARP-1 were found to co-immunoprecipitate after a control pre-incubation without NAD+ (Fig. 8A and B, top panels, lane 1). Inclusion of NAD+ in the pre-incubation allowing for poly(ADP-ribosyl)ation of PARP-1 and ATR resulted in an increase in PARP-1 and ATR co-immunoprecipitation (Fig. 8A and B, top panels, compare lanes 1 and 2). These results suggested that the interaction between ATR and PARP-1 was stronger under conditions where both proteins can become poly(ADP-ribosyl)ated, yet note that the the immunoprecipitates contain unmodified as well as PAR-adducted protein.
Fig. 8.

Effect of poly(ADP-ribosyl)ation on co-immunoprecipitation of purified ATR and PARP-1. Purified ATR and PARP-1 proteins were pre-incubated for 30 min in a ribosylation reaction mixture, either without NAD+ (- NAD+) or with NAD+ (+ NAD+) as described under “Materials and methods.” After the pre-incubation, reaction mixtures were subjected to the usual co-immunoprecipitation protocol in buffer containing 100 mM NaCl. (A) Immunoprecipitation (IP) with anti-PARP-1 antibody and (B) with anti-ATR antibody, and immunoblotting (IB) with anti-ATR, anti-PARP-1 or anti-PAR, as indicated. The migration of unmodified PARP-1 and ATR proteins in the SDS-PAGE system are indicated in the bottom panels of (A) and (B), and correspond to 116 and 300 kDa, respectively. (C) Following pre-incubation of purified ATR and PARP-1 in a reaction without (-) or with (+) NAD+, immunoprecipitation with anti-ATR antibody was carried out in buffer containing 100 mM NaCl as previously, or with 200 or 300 mM NaCl. The samples were immunoblotted (IB) with anti-PARP-1 or anti-ATR, as shown. Purified ATR or PARP-1 proteins, as indicated, were analyzed directly by 6% SDS-PAGE.
To confirm poly(ADP-ribosyl)ation of PARP-1 and ATR in the NAD+-containing incubations, the blots were stripped and probed with antibody against PAR (Fig. 8A and B, bottom panels, lane 2). PAR-adducted protein migrating slower than unmodified PARP-1 (116 kDa) was observed for the immunoprecipitations with anti-PARP-1 antibody (Fig. 8A, bottom panel, lane 2). With the anti-ATR immunoprecipitations (Fig. 8B), PAR-adduction of immunoprecipitated protein migrating slower than unmodified ATR was strong (Fig. 8B, bottom panel, lane 2).
The immunoprecipitation experiments presented in panels A and B of Figure 8 were conducted in buffer containing 100 mM NaCl. When using buffer containing higher salt concentrations (200 or 300 mM NaCl), co-immunoprecipitation of PARP-1 by ATR was less than in reactions containing 100 mM NaCl, but was still significant after the poly(ADP-ribosyl)ation reaction (Fig. 8C, compare lanes 4 and 6 with lane 2). At all salt concentrations, the co-immunoprecipitation was less apparent without the NAD+-containing pre-incubation (Fig. 8C, lanes 1, 3 and 5). The results suggest that the observed co-immunoprecipitation of purified ATR and PARP-1 was stronger when the proteins were PAR-adducted, and also that the co-immunoprecipitation from cell extracts may be due to a direct interaction between the two enzymes.
4. Discussion
The ATR and PARP-1 co-immunoprecipitation results presented here were obtained with a mouse fibroblast cell system that has been characterized extensively with regard to cell growth, cell cycle arrest, and apoptotic responses following treatment with MMS combined with a PARP inhibitor [7,13]. The S-phase arrest initially observed following exposure to MMS and 4-AN was caffeine-sensitive pointing to involvement of ATM or ATR. An ATR-mediated pathway was a good candidate for this effect since it was shown that the ATR substrate Chk1 was activated, and the appearance of phosphorylated activated Chk1 was suppressed by caffeine [7]. In addition, later experiments in human cells showed that ATR inhibition through expression of kinase-dead ATR (ATRkd) blocked the MMS plus 4-AN-induced delay in progression through S-phase [15].
Since these earlier experiments suggested sensitization to MMS by a PARP inhibitor involved ATR-mediated signaling, we now examine the possibility of an interaction between ATR and PARP-1. We were able to observe co-immunoprecipitation of these enzymes from extracts of control and, to a greater extent, MMS-treated mouse fibroblasts (Fig. 1A). A similar increase in co-immunoprecipitation was observed following UV exposure (Fig. 1C). Anti-ATR antibody was also able to immunoprecipitate both Rad9 and RPA, however the interaction of these proteins with ATR did not correlate with PARP activity in the cells (Fig. 2C).
Since the MMS-induced co-immunoprecipitation was considerably weakened by PARP inhibition (Fig. 2 and 4), we proposed that PAR-adduction of one or both enzymes was required to facilitate the ATR/PARP-1 interaction. Our experiments demonstrated PAR-adduction of ATR in extracts from control and MMS-treated cells, and that this modification was blocked by treatment with a PARP inhibitor (Fig. 6). Studies in vitro with recombinant proteins confirmed that ATR is a substrate for poly(ADP-ribosyl)ation by PARP-1 (Fig. 7). Additionally, when using purified proteins, poly(ADP-ribosyl)ation of PARP-1 and ATR resulted in an increase in PARP-1 and ATR co-immunoprecipitation confirming that PAR-adduction did enhance formation of the ATR/PARP-1 complex (Fig. 8). This enhanced interaction following PAR formation became especially apparent as the salt concentration in the binding buffer was increased (Fig. 8C). Co-immunoprecipitation of PARP-1 and ATM has been demonstrated [25], and it was proposed that PAR mediates the interaction between ATM and PARP-1 via specific PAR-binding domains in ATM [26].
Exposure of wild-type mouse fibroblast cells to a low concentration of MMS in the absence of a PARP inhibitor does not result in an ATR-mediated S-phase checkpoint [7]. Thus under conditions of normal poly(ADP-ribosyl)ation, and when MMS-induced damage can be efficiently repaired, an interaction of ATR and PARP-1 is observed by co-immunoprecipitation (Fig. 4B) and ATR activation does not occur (Fig. 5). The results suggest that ATR-mediated signaling is prevented by PARP activation, PAR-adduction of ATR, and the interaction of ATR and PARP-1. Conversely, in the presence of a PARP inhibitor and in the absence of PAR synthesis, the ATR/PARP-1 interaction is prevented (Fig. 4B) and ATR-mediated signaling is initiated (Fig. 5). Also of interest is the observed activation of Chk1 in the absence of an interaction between ATR and PARP-1 occurring after a highly toxic dose of MMS without PARP inhibitor (Fig. 4B and Fig. 5). Again, activation of the damage signaling pathway is observed in the absence of complex formation. These results are in contrast to the situation with ATM, where optimal activation of a response to DNA damage requires PAR synthesis and ATM/PARP-1 complex formation, and is delayed in cells treated with a PARP inhibitor [25,26]. It should be noted that the S-phase checkpoint was not observed in PARP-1 -/- cells following exposure to a similar concentration of MMS plus 4-AN [7], suggesting that activity-inhibited PARP-1 protein is required for triggering ATR-mediated signaling. The relationship between the ATR and PARP-1 interaction and the mechanism(s) accounting for ATR signaling remain to be elucidated.
Acknowledgments
We thank Dr. William A. Beard for help with figure preparation and Jennifer Myers for editorial assistance. We thank Dr. Michael J. Carrozza for critical reading of the manuscript. We also thank Dr. Robert Petrovich for assistance with expression of Flag-tagged human ATR in 293 cells. This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. Dr. Kedar’s contribution was funded in whole with federal funds from NIH/NIEHS, under delivery order HHSN273200700046U to Constella/SRA, LLC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Beranek DT. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat. Res. 1990;231:11–30. doi: 10.1016/0027-5107(90)90173-2. [DOI] [PubMed] [Google Scholar]
- [2].Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Requirement of mammalian DNA polymerase-βin base-excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- [3].Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu. Rev. Genet. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- [4].Engelward BP, Weeda G, Wyatt MD, Broekhof JL, de Wit J, Donker I, Allan JM, Gold B, Hoeijmakers JH, Samson LD. Base excision repair deficient mice lacking the Aag alkyladenine DNA glycosylase. Proc. Natl. Acad. Sci. USA. 1997;94:13087–13092. doi: 10.1073/pnas.94.24.13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Horton JK, Joyce-Gray DF, Pachkowski BF, Swenberg JA, Wilson SH. Hypersensitivity of DNA polymerase β null mouse fibroblasts reflects accumulation of cytotoxic repair intermediates from site-specific alkyl DNA lesions. DNA Repair (Amst) 2003;2:27–48. doi: 10.1016/s1568-7864(02)00184-2. [DOI] [PubMed] [Google Scholar]
- [6].Engelward BP, Allan JM, Dreslin AJ, Kelly JD, Wu MM, Gold B, Samson LD. A chemical and genetic approach together define the biological consequences of 3-methyladenine lesions in the mammalian genome. J. Biol. Chem. 1998;273:5412–5418. doi: 10.1074/jbc.273.9.5412. [DOI] [PubMed] [Google Scholar]
- [7].Horton JK, Stefanick DF, Naron JM, Kedar PS, Wilson SH. Poly(ADP-ribose) polymerase activity prevents signaling pathways for cell cycle arrest following DNA methylating agent exposure. J. Biol. Chem. 2005;280:15773–15785. doi: 10.1074/jbc.M413841200. [DOI] [PubMed] [Google Scholar]
- [8].Lindahl T, Satoh MS, Poirier GG, Klungland A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci. 1995;20:405–411. doi: 10.1016/s0968-0004(00)89089-1. [DOI] [PubMed] [Google Scholar]
- [9].Lavrik OI, Prasad R, Sobol RW, Horton JK, Ackerman EJ, Wilson SH. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J. Biol. Chem. 2001;276:25541–25548. doi: 10.1074/jbc.M102125200. [DOI] [PubMed] [Google Scholar]
- [10].D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- [11].Amé JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- [12].Burkle A. Poly(ADP-ribose). The most elaborate metabolite of NAD+ FEBS J. 2005;272:4576–4589. doi: 10.1111/j.1742-4658.2005.04864.x. [DOI] [PubMed] [Google Scholar]
- [13].Horton JK, Stefanick DF, Wilson SH. Involvement of poly(ADP-ribose) polymerase activity in regulating Chk1-dependent apoptotic cell death. DNA Repair (Amst) 2005;4:1111–1120. doi: 10.1016/j.dnarep.2005.05.011. [DOI] [PubMed] [Google Scholar]
- [14].Caldecott KW. Mammalian single-strand break repair: mechanisms and links with chromatin. DNA Repair (Amst) 2007;6:443–453. doi: 10.1016/j.dnarep.2006.10.006. [DOI] [PubMed] [Google Scholar]
- [15].Horton JK, Stefanick DF, Kedar PS, Wilson SH. ATR signaling mediates an S-phase checkpoint after inhibition of poly(ADP-ribose) polymerase activity. DNA Repair (Amst) 2007;6:742–750. doi: 10.1016/j.dnarep.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- [17].Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- [18].Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998;17:159–169. doi: 10.1093/emboj/17.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Caporali S, Falcinelli S, Starace G, Russo MT, Bonmassar E, Jiricny J, D’Atri S. DNA damage induced by temozolomide signals to both ATM and ATR: role of the mismatch repair system. Mol. Pharmacol. 2004;66:478–491. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- [20].Stojic L, Mojas N, Cejka P, Di Pietro M, Ferrari S, Marra G, Jiricny J. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 2004;18:1331–1344. doi: 10.1101/gad.294404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Beardsley DI, Kim WJ, Brown KD. N-methyl-N’-nitro-N-nitrosoguanidine activates cell-cycle arrest through distinct mechanisms activated in a dose-dependent manner. Mol. Pharmacol. 2005;68:1049–1060. doi: 10.1124/mol.105.013888. [DOI] [PubMed] [Google Scholar]
- [22].Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- [23].Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–14459. [PMC free article] [PubMed] [Google Scholar]
- [24].Horton JK, Watson M, Stefanick DF, Shaughnessy DT, Taylor JA, Wilson SH. XRCC1 and DNA polymerase β in cellular protection against cytotoxic DNA single-strand breaks. Cell Res. 2008;18:48–63. doi: 10.1038/cr.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Aguilar-Quesada R, Muñoz-Gámez JA, Martín-Oliva D, Peralta A, Valenzuela MT, Matínez-Romero R, Quiles-Pérez R, Menissier-de Murcia J, de Murcia G, de Almodóvar MR, Oliver FJ. Interaction between ATM and PARP-1 in response to DNA damage and sensitization of ATM deficient cells through PARP inhibition. BMC Mol. Biol. 2007;8:29. doi: 10.1186/1471-2199-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Haince J-F, Kozlov S, Dawson VL, Dawson TM, Hendzel MJ, Lavin MF, Poirier GG. ATM signaling network is modulated by a novel PAR-dependent pathway in the early response to DNA damaging agents. J. Biol. Chem. 2007:16441–16453. doi: 10.1074/jbc.M608406200. [DOI] [PubMed] [Google Scholar]
- [27].Dantzer F, de la Rubia G, Ménissier-de Murcia J, Hostomsky Z, de Murcia G, Schreiber V. Base excision repair is impaired in mammalian cells lacking poly(ADP-ribose) polymerase-1. Biochemistry. 2000;39:7559–7569. doi: 10.1021/bi0003442. [DOI] [PubMed] [Google Scholar]
- [28].Kedar PS, Kim SJ, Robertson A, Hou E, Prasad R, Horton JK, Wilson SH. Direct interaction between mammalian DNA polymerase β and proliferating cell nuclear antigen. J. Biol. Chem. 2002;277:31115–31123. doi: 10.1074/jbc.M201497200. [DOI] [PubMed] [Google Scholar]
- [29].Ünsal-Kaçmaz K, Makhov AM, Griffith JD, Sancar A. Preferential binding of ATR protein to UV-damaged DNA. Proc. Natl. Acad. Sci. USA. 2002;99:6673–6678. doi: 10.1073/pnas.102167799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ikejima M, Noguchi S, Yamashita R, Suzuki H, Sugimura T, Miwa M. Expression of human poly(ADP-ribose) polymerase with DNA-dependent enzymatic activity in Escherichia coli. Biochem. Biophys. Res. Commun. 1989;163:739–745. doi: 10.1016/0006-291x(89)92285-7. [DOI] [PubMed] [Google Scholar]
- [31].Marsischky GT, Wilson BA, Collier RJ. Role of glutamic acid 988 of human poly-ADP-ribose polymerase in polymer formation. Evidence for active site similarities to the ADP-ribosylating toxins. J. Biol. Chem. 1995;270:3247–3254. doi: 10.1074/jbc.270.7.3247. [DOI] [PubMed] [Google Scholar]
- [32].Simbulan-Rosenthal CM, Rosenthal DS, Luo RB, Samara R, Jung M, Dritschilo A, Spoonde A, Smulson ME. Poly(ADP-ribosyl)ation of p53 in vitro and in vivo modulates binding to its DNA consensus sequence. Neoplasia. 2001;3:179–188. doi: 10.1038/sj.neo.7900155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Simbulan-Rosenthal CM, Rosenthal DS, Luo R, Samara R, Espinoza LA, Hassa PO, Hottiger MO, Smulson ME. PARP-1 binds E2F-1 independently of its DNA binding and catalytic domains, and acts as a novel coactivator of E2F-1-mediated transcription during re-entry of quiescent cells into S phase. Oncogene. 2003;22:8460–8471. doi: 10.1038/sj.onc.1206897. [DOI] [PubMed] [Google Scholar]
- [34].Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 2005;19:1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- [35].Parrilla-Castellar ER, Arlander SJH, Karnitz L. Dial 9-1-1 for DNA damage: the Rad9-Hus1-Rad1 (9-1-1) clamp complex. DNA Repair (Amst) 2004;3:1009–1014. doi: 10.1016/j.dnarep.2004.03.032. [DOI] [PubMed] [Google Scholar]
- [36].Altmann SM, Muryshev A, Fossale E, Maxwell MM, Norflus FN, Fox J, Hersch SM, Young AB, MacDonald ME, Abagyan R, Kazantsev AG. Discovery of bioactive small-molecule inhibitor of poly ADP-ribose polymerase: implications for energy-deficient cells. Chem. Biol. 2006;13:765–770. doi: 10.1016/j.chembiol.2006.05.012. [DOI] [PubMed] [Google Scholar]
- [37].Soriano F. Garcia, Virág L, Jagtap P, Szabó É, Mabley JG, Liaudet L, Marton A, Hoyt DG, Murthy KG, Salzman AL, Southan GJ, Szabó C. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat. Med. 2001;7:108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]