

Abstract
We are developing a multielectrode silicon “neuroprobe” for maintaining a long-term, specific, two-way electrical interface with nervous tissue. Our approach involves trapping a neuron (from an embryonic rat hippocampus) in a small well with a stimulation/recording electrode at its base. The well is covered with a grillwork through which the neuron's processes are allowed to grow, making synaptic contact with the host tissue, in our case a cultured slice from a rat hippocampus. Each neuroprobe can accommodate 15 neurons, one per well.
As a first step in studying neurite outgrowth from the neuroprobe, it was necessary to develop new staining techniques so that neurites from the probe neurons can be distinguished from those belonging to the host, without interference from non-specific background staining. We virtually eliminated background staining through a number of innovations involving dye solubility, cell washing, and debris removal. We also reduced photobleaching and phototoxicity, and enhanced imaging depth by using a 2-photon laser-scanning microscope. We focused on using the popular membrane dye, DiI, however a number of other membrane dyes were shown to provide clear images of neural processes using pulsed illumination at 900 nm. These techniques will be useful to others wishing to follow the growth of transplanted neurons over time, in a non-destructive way.
Keywords: Neural prosthesis, cell suspension staining, cellular debris, phototoxicity, non-destructive imaging, rat hippocampus, carbocyanine dye, grafting, two-photon fluorescence, neuroprobe
Introduction
One of the enduring problems associated with the study of neural transplants is discerning the transplanted cells from the host cells, to assess their survival and integration with the host tissue. In cases where the transplanted cells are immunologically distinguishable from the host, many immunohistochemical methods exist for visualizing them post-mortem. The increasing popularity of organotypic culture of neural tissue (Gahwiler, 1988; Stoppini et al., 1991) and the use of embryos capable of growing on a microscope stage (O'Rourke and Fraser, 1990) have created the possibility of studying transplant integration while the tissue is still living, in order to understand the dynamic changes transplanted cells undergo. Attempts at vital labeling and imaging of neural transplants have met with limited success (e.g., see monumental efforts by: (Pyapali et al., 1992; Onifer et al., 1993)) due to the difficulty of finding a labeling method that (1) is persistent for days or weeks, (2) is specific to the transplanted cells, (3) labels fine neuronal processes, and (4) is compatible with non-destructive imaging.
As a tool for basic research into the relationship between synaptic plasticity and neuronal morphology, and eventually as an aid to handicapped people, we are developing a long-term, specific, two-way electrical connection to neural tissue called the neuroprobe (Fig. 1). Fabricated from silicon using technology developed for microelectronics (Tatic-Lucic, 1994; Tatic-Lucic and Tai, 1994), the neuroprobe has cell-sized wells with electrodes in them, and grillwork on top designed to keep the neuron's cell body in the well, next to the electrode, while allowing the neuron's processes to grow out and make synaptic contact with the host tissue. We chose the embryonic rat hippocampus as a source of donor cells, and used cultured hippocampal slices from newborn rats. The well-studied physiology of cultured rat hippocampal slices (Stoppini et al., 1991; Buchs et al., 1993; Muller et al., 1993; Stoppini et al., 1993), the role of the hippocampus in learning and memory, and its deterioration in pathological states such as Alzheimer's disease (Etcheberrigaray et al., 1994), all make this system a fertile and relevant testing ground.
Fig. 1. The neuroprobe.
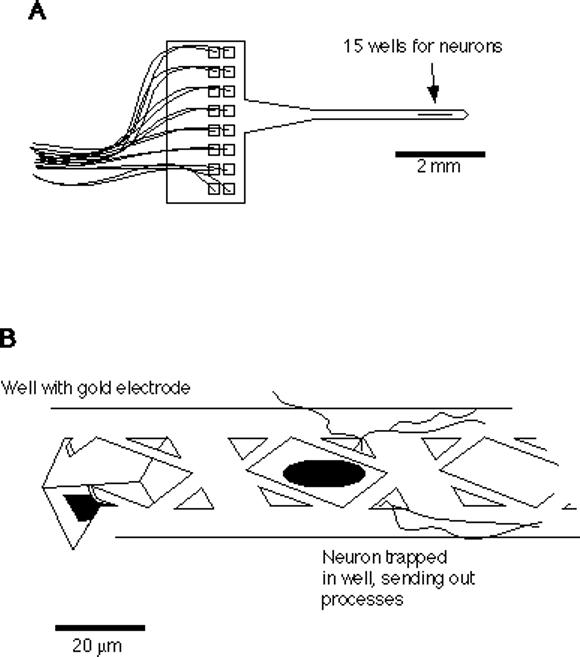
A. A schematic drawing of the neuroprobe is shown, with 16 leads (15 well electrodes plus ground) connected to bonding pads on the handle. B. Close-up schematic of the neuroprobe wells, showing grillwork that allows a immature neuron to be dropped through the center hole with a microsyringe, and smaller corner holes that permit the outgrowth of neurites.
Our collaborators Gyuri Buzsaki and Anatol Bragin at Rutgers University in New Jersey make use of the present labeling techniques with their studies of neuroprobes inplanted into living rats. Although our neuroprobe neurons are not conventional transplants as such, we share the same concerns as most of the transplant community regarding the clear observation of the integration of donor cells with the host. At present, membrane dyes, such as the carbocyanine dye DiI (1,1′-dioctadecyl-3−3−3′-3′ tetramethylindocarbocyanine, reviewed in (Honig and Hume, 1989)), give the most hope for satisfying the above constraints. DiI brightly labels fine neuronal processes, can persist in neurons for weeks, and is relatively harmless to the labeled cells (Honig and Hume, 1986; Vidal-Sanz et al., 1988). The greatest difficulty with the use of membrane dyes is to ensure that the label does not spread to host neurons. We refined techniques for the staining of donor cells in suspension in a way that minimizes artifactual staining of host cells and maximizes cell viability and brightness.
With confocal and wide-field fluorescence microscopy, the non-destructive observation of stained, living transplants is extremely difficult, if not impossible, due to the problems of photobleaching of the dye and dye-induced phototoxicity. To greatly reduce these difficulties, we constructed a 2-photon laser-scanning microscope (TPLSM). Two-photon microscopy, pioneered by Webb and co-workers (reviewed in (Williams et al., 1994)) takes advantage of the 2-photon absorption effect. High-energy pulses of infrared (IR) light are used to excite fluorophores normally excited by visible light of approximately half the wavelength. Two IR photons excite the fluorophore to emit a single visible photon. Because of the squared dependence of 2-photon emission on excitation intensity, only at the focus of the laser beam within the specimen is the photon flux great enough to induce fluorescence. Thus, the tissue above and below the plane of focus is spared from phototoxicity and photobleaching. This is especially helpful if numerous images are taken, e.g., for time-series analysis and three-dimensional reconstructions. Because there is no source of out-of-focus fluorescence, no confocal aperture is necessary, allowing more sensitive detection of weak labeling than with confocal microscopy. An additional advantage is provided by the greater penetration of IR light into the specimen, which allows imaging of deeper structures than hitherto possible, including DiI-stained neurons transplanted to cultured hippocampal slices.
Materials and Methods
Preparation of cell suspensions
Unless otherwise indicated, all donor cells were obtained from E18 Wistar rat embryo hippocampi. Pregnant rats were killed by inhalation of carbon dioxide (CO2), and embryos were immediately removed by cesarean section. Hippocampi were rapidly removed using a stereomicroscope under sterile conditions, cut into 1 mm pieces, and digested with 0.25% trypsin, 0.02 mg/mL DNase (Sigma), in Hanks balanced salt solution, no calcium or magnesium (Gibco), at 37deg.C for 15 min. Pieces were gently washed twice in plating medium (Neurobasal with B27 supplement (Gibco), with 500 uM glutamine, 25 uM glutamate), and gently triturated in 1 mL of plating medium with 5 passes through the 0.78 mm opening of a blue tip of a P-1000 Pipetman. Suspended cells were decanted and the remaining pieces were triturated once more. Cell suspensions were gravity-filtered through a 70-um nylon mesh (Falcon) to remove large debris, and centrifuged for 2 min at 160 × g through a layer of 5% bovine serum albumin in phosphate-buffered saline (BSA/PBS), to remove small debris. Pellets were resuspended by gentle trituration (3 passes) as above.
For debris removal studies, we used whole cerebral hemispheres (including the hippocampus) of newborn rats (postnatal day 1) with meninges and choroid plexus removed. 2−4 hemispheres were minced, digested, triturated, and filtered through a 70 um mesh as above, producing 20−50 million cells. These were diluted to approximately one million cells/mL in plating medium, and kept on ice in a 50-mL centrifuge tube that was stirred by inverting every 15 min, to prevent cells from settling during the experiments. Cell suspension (1 mL) was added to the indicated volume of plating medium in a 15-mL centrifuge tube and mixed by inverting. This was underlayed with 0.5 mL of BSA/PBS using a 1 mL syringe with a 16-gauge needle. All spins were conducted in triplicate (volume study) or duplicate (time study) at 160 × g (calculated at 1 cm from the end of the tube) in a swinging-bucket rotor at room temperature. Spin-up time (30 sec) was included in spin times, and spin-down (10 sec) was not. After spinning, all but 30 uL of the supernatant was aspirated, transferred to a separate tube and mixed by inverting a few times. Approximately 0.5 mL of this was plated under a 25 mm diam. coverslip raised 1.0 mm off the base of a petri dish using pieces of glass capillary tubing. Pellets were immediately resuspended in 1 mL of plating medium with 3 passes of a P-1000 Pipetman, and 0.5 mL of this was plated into the coverslip dishes, as above. Cells and debris were allowed to settle for 2 h in the incubator, and then video images were taken, one from a random spot on each dish, with phase-contrast optics using a 20x/0.4 NA objective on a Nikon Diaphot inverted microscope. The coverslips allowed excellent homogeneity of plating density across the dish (less than 7% variance from field to field), since there was no meniscus effect. The video images were analyzed using NIH Image on a Macintosh Power PC computer. Cells in each 492 × 375 um field were counted manually, and debris and cells were quantified together using the “analyze particles” function. Cell counts were subtracted to produce final debris counts. Platings of the uncentrifuged cell suspension were used to set recovery maxima.
Staining of neurons with DiI
A 40 mg/mL stock solution of DiI-C12 (Molecular Probes, Eugene, Oregon) was prepared by dissolving the tar-like dye in dimethylformamide containing 2.5% (w/v) of Pluronic F127 (BASF). This was stored at −20deg.C and dissolved easily upon warming to room temperature. Staining solutions were prepared by adding 2 uL of dye stock to 2 mL of plating medium, producing a final concentration of 40 ug/mL DiI, 0.0025% F127, and 0.1% dimethylformamide. Approximately 0.5 mL of cell suspension was added with gentle mixing to the staining solution after it was warmed to 37deg.C. Cells were stained for 15 min at 37deg.C. BSA/PBS (0.5 mL) was layered under the cells in staining solution, using a long Pasteur pipet, and the cells were pelleted with a 6 min spin at 160 × g. The staining solution and most of the BSA/PBS was carefully decanted, and the stained cells were resuspended by trituration (3 passes) in 1 mL of plating medium. Another centrifugation through BSA/PBS and resuspension, as above, was performed to remove any remaining dissolved dye.
Phototoxicity testing
DiI-stained cells, or control cells “stained” in vehicle lacking the dye, were plated at a density of approximately 300 cells/mm2 onto custom-made numbered-grid petri dishes coated with polylysine and laminin (Banker and Goslin, 1991). After 3 days in culture, the stained cultures and unstained control cultures (viabilities of 69+/−10% and 73+/−10%, respectively) were exposed for varying periods (10 sec to 6 min) to the green line of a 200 W mercury arc lamp on the Nikon Diaphot epifluorescence microscope with a 20x/0.75 NA objective. Illuminating with the 50 W tungsten lamp, video images of the exposed regions, and unexposed control regions in the same dish, were recorded using phase contrast optics (10x/0.25 NA objective) just before and 1 day after arc lamp exposure.
Preparation of neuroprobes
Neuroprobes were fabricated at Caltech in the laboratory of Yu-Chong Tai by Svetlana Tatic-Lucic and John Wright (Tatic-Lucic, 1994). They were glued to plastic petri dishes with a tiny drop of silicone rubber, with the shank resting on a piece of silicon wafer for support. They were sterilized by soaking in 95% ethanol overnight, washed with sterile water, and coated with polylysine and laminin. Neuroprobes were submerged in a drop of plating medium after rinsing off the laminin. Stained cells were plated in a 50 uL drop onto a silicone rubber-coated 10 mm diam. coverslip which was placed in the petri dish next to the neuroprobe. The rubber discouraged the cells from adhering to the coverslip before they could be transferred to the neuroprobe. Stained cells were transferred from the coverslip to the neuroprobe wells using a microsyringe consisting of a 1 mm capillary pulled (using a Brown-Flaming pipet puller), cut (using a diamond knife), and polished (using a hot tungsten filament) to have a smooth opening of approximately 100 um diam. The microsyringe was connected with Intramedic tubing to a 250 uL micrometer syringe (Gilmont), used to pick up and aspirate cells by varying the air pressure in the tubing. The tubing was also connected to a 60 cc syringe, to adjust the magnitude of the pressure difference created by the micrometer syringe that picked up and expelled the cells. The microsyringe was moved using a Leitz micromanipulator, while observing using Nomarski optics on an Olympus upright BHMJ metallurgical microscope with a 20x/0.4 NA ultra-long working distance objective. Tungsten lamp epi-illumination was filtered to remove green light using a Hoya 25A red photographic filter. Routine inspections and Nomarski photography were conducted with unfiltered tungsten light. After filling the neuroprobe with cells (which took 20−40 min/neuroprobe), the coverslip was removed from the dish, and the dish was flooded with plating medium. Cells in control dishes or neuroprobes were incubated at 37deg.C in a 5% CO2 atmosphere. Great care was taken to assure that the air in the incubator remained moist and of the proper CO2 level, by limiting the frequency and duration of door-openings.
Preparation of cultured slices
Hippocampal slices (400 um) were prepared from postnatal day 7−11 Wistar rat pups, according to the method developed by Stoppini and co-workers (Stoppini et al., 1991). Membranes inserts upon which the slices were grown (Millipore CM) were cut down to allow them to fit into 35 mm petri dishes, and to allow imaging from above with a large objective. Slice culture medium was Gibco MEM (Hank's salts, no glutamine) with 25% horse serum (Hyclone), supplemented with 5 mM sodium bicarbonate, 30 mM HEPES, 30 mM glucose, 3 mM glutamine, 2.5 mM magnesium sulfate, 2 mM calcium chloride, and 1 mg/L insulin (Sigma), 10 mL/L penicillin/streptomycin (Sigma P0781), pH 7.2 at 37deg.C. Slices were maintained at 37deg.C in a 5% CO2 atmosphere and fed twice weekly by replacing half of the medium. They remained healthy for at least 6 weeks. DiI-stained cells were diluted in slice medium to a density of 20,000−50,000 cells/mL, and a 5 uL drop of them was placed on each slice (at least one day after preparing the slice culture) which gave approximately 20−50 neurons/slice, depending on how far the drop spread before soaking through the membrane.
2-photon imaging
The TPLSM was built from a Molecular Dynamics Sarastro 2000 upright confocal laser-scanning microscope, using a Titanium:sapphire mode-locked IR laser (Coherent Mira 900) as the excitation source. With the use of three different mirror sets, the laser is continuously tunable across a range from 710 to 970 nm (10 nm bandwidth), emitting 200 fs pulses with a peak amplitude of 50 kW, a repetition rate of 76 MHz, and a mean power of a few tens of milliwatts at the sample, which did not cause detectable sample heating. The Mira 900 is pumped by an 8 W argon-ion laser (Coherent Innova 310) operating in multiline visible mode. Visible and IR laser beams were covered with metal tubing to allow safe operation by biologists. One of the mirrors in the Sarastro 2000 was replaced with a 680 nm long-pass dichroic mirror, which allowed us to use either the Ti:sapphire or the Sarastro's on-board 25 mW argon-ion laser (if standard confocal operation is desired). The scanning mirrors and one stationary mirror in the Sarastro were replaced with broad-band mirrors (Newport) to enhance the IR throughput. We used no confocal aperture, a 680 nm short-pass primary beamsplitter and a 680 nm short-pass IR-blocking filter (Chroma) when operating in TPLSM mode. The Sarastro 2000 has a Nikon Optiphot-2 upright microscope with an epifluorescence attachment. The stage height is adjusted by a computer-controlled stepper motor to 0.1 um accuracy. Microscope control, data collection and image analysis were carried out on an Indigo workstation (Silicon Graphics) using ImageSpace software (Molecular Dynamics).
Neuroprobes were transferred to Hibernate (air-buffered Neurobasal) medium (Gibco) before imaging, since the pH drift of Neurobasal in air harmed cultures left out for more than 30 min. Because the focus of the IR beam is different than that of the visible light used for positioning the specimen (10−40 um), and because the optical sectioning of the 2-photon effect is so extreme, it was easiest to find the neural processes by first executing a vertical (y-z) scan through the neuroprobe surface, and setting the stage height to bring the IR focus to the level of the neuroprobe grillwork. For neuroprobe imaging, we used pulsed excitation at 900 nm and a 40x/0.65 NA water-immersion objective. 512 × 512 pixel images were taken, with a pixel size of 0.5 um. For z-series scans, 10 z-steps of 1−2 um were used. Two scans were averaged per frame, and raw data were projected along the z-axis using the “lookthrough” function.
For slice imaging, 20x/0.75 NA air objective and a 63x/1.2 NA water immersion objectives were used. When we tuned the laser from 900 to 960 nm, we reduced autofluorescence from slice cells (presumably due to endogenous flavins), increasing our signal-to-noise ratio substantially. For z-series scans, 30 z-steps of 2 um were used, with 5 scans averaged/frame. Three-dimensional data were processed with a 3×3×3-pixel median filter and projected along the z-axis using the “maximum intensity” function.
Results
1. Preparation of stained cells
Any method for producing cell suspensions by mechanically dissociating neural tissue is bound to produce debris, consisting of pieces of broken cells, neurites from the surviving dissociated cells, and clumps of undissociated cells. If this debris is stained and transplanted along with the cells of interest, unacceptable background staining of host tissue is inevitable (Onifer et al., 1993). DiI has generally not been observed to be transferred from labeled to unlabeled neurons in culture (St. John, 1991), however, using the TPLSM, we observed non-specific labeling of host neurons in rats injected with DiI-labeled cells and debris. It is important, therefore, to prepare a cell suspension that is as free of debris as possible. We found that suspensions prepared from embryonic day 18 rats (E18, 21 day gestation) had far less debris than suspensions prepared from newborn (postnatal day 1−3) rats. The embryonic hippocampal preparation also has the advantage of fewer non-neuronal cell types. We routinely observed that 85−90% of viable cells in dissociated E18 cultures had neuronlike morphology.
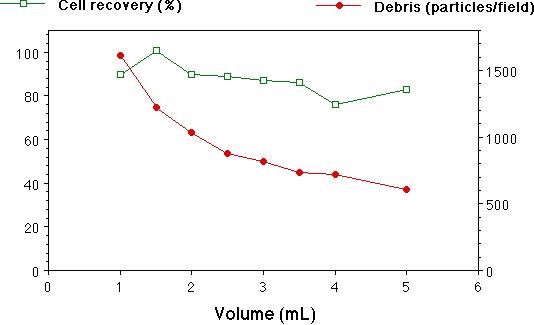
Preparing a clean suspension of cells is unfortunately more of an art than a science, requiring a customized approach for each new tissue, animal age, etc. We found trituration of brain tissue to be especially variable in cell yield and viability. To reduce some of this variability, we triturated trypsin-digested hippocampal pieces using a P-1000 Pipetman, so that the tip opening (0.78 mm) was well-defined, unlike that of the ubiquitous fire-polished Pasteur pipet. This also made it easy to control the volume of each trituration pass. After trituration, debris larger than the cells (10−20 um diam) was removed by gravity filtration of the suspension through a 70 um nylon mesh. The small debris was more troublesome to eliminate. It is traditional to centrifuge cell suspensions to remove small debris, but a plethora of different protocols exist, so we quantified debris removal and cell recovery under a variety of different conditions. At 160 × g, we observed that essentially all of the cells were pelleted from a suspension of 2.5 mL in 6 min (Fig. 2, A), however, a substantial proportion of the debris was also pelleted. Thus, one must sacrifice some of the healthy cells by spinning for a shorter time, in order to leave most of the debris behind in the supernatant fraction. Diluting the suspension beforehand reduced the debris in the pellet, while having little effect on cell recovery (Fig. 2, B). We routinely centrifuged our suspensions before staining for 2 min at 160 × g in a volume of 2−3 mL. Repeated centrifuging adversely affected cell recovery and viability, although this effect was not quantified.
Fig. 2. Centrifugation of cell suspensions.
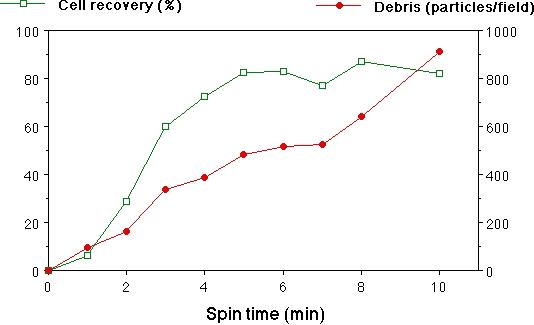
A. Spin-time vs. cell recovery and debris counts. Cells and debris from whole cerebral hemispheres of newborn rats, in 2 mL of suspension, were centrifuged through 0.5 mL of a 5% BSA solution, and cells and debris were quantified as described in Materials and Methods. Although recovery of cells at 2 min was only about 30%, we found unacceptable amounts of debris in samples spun for longer. The majority of the debris was still in the supernatant fraction after 10 min, hence the approximately linear increase in debris with spin time. Means of duplicate spins are plotted, with error bars indicating the range.
B. Volume of suspension vs. cell recovery and debris counts. All samples were spun for 6 min. Means+/-SEM of triplicate spins are plotted.
DiI is most commonly used as a neural tracer (in living and fixed tissue) by placing a small crystal or suspension of crystals in contact with the cells to be labeled. It is also dissolved in oil which is then injected in small droplets into the tissue. This has allowed numerous excellent studies in which living processes of endogenous neurons were traced (for example, see: (Hosokawa et al., 1992; Dailey et al., 1994; Hosokawa et al., 1994; O'Rourke et al., 1994)). Crystals or oil solutions of DiI are used because the most common form of the dye (DiI-C18) is only slightly soluble in aqueous media. This presents some problems for staining of cells in suspension. The worst is that DiI crystals are difficult if not impossible to wash from the cells by centrifugation. If any crystals of DiI are transferred to the host along with the stained cells, then background staining of host cells will result. Furthermore, we and others (Paramore et al., 1992) observed the lethal clumping of cells around DiI crystals. We deemed unacceptable the filtration of DiI-C18 to remove crystals, due to clogging of filters, uncertainty of the concentration of dye in the final “solution,” and subsequent crystal formation during staining.
We completely eliminated the problem of DiI insolubility by (1) using the more soluble form of DiI with a shorter alkyl tail, DiI-C12 instead of DiI-C18, and (2) using the surfactant Pluronic F127 in the staining solutions. We observed substantially brighter staining with DiI-C12, compared to an equal concentration of DiI-C18, presumably because all of the dye was dissolved and available to stain the cells. Pluronic F127, a non-ionic macromolecular polyol, serves to “catalyze” the transfer of dye molecules to the cell membrane (Lojewska and Loew, 1986; Lojewska and Loew, 1987), and has been used by researchers to harmlessly stain neurons with voltage-sensitive membrane dyes at ten times the concentration we use (Davila et al., 1973). The DiI-C12/F127 solution passed freely through a 0.2 um filter, with no diminution in its light absorption, indicating it was completely dissolved in cell culture medium at the highest concentration used, 40 ug/mL.
Unlike crystals, dissolved DiI can be removed from the suspension of stained cells by centrifugation, although this is not trivial. Even with three washings with 1.5 mL of medium, we observed unacceptable background staining of cell culture dishes, and of cultured slices onto which stained cells were plated. In order to almost completely remove dissolved DiI from the cell suspension in two washings, we centrifuged (in a swinging-bucket rotor) the stained cells through a 0.5 mL layer of 5% bovine serum albumin (BSA) in phosphate-buffered saline. The dye solution remained above the dense protein solution, and was easily decanted after spinning without disturbing the pellet of stained cells. An additional, unexpected advantage of using a BSA layer was that the cells were easier to resuspend. Cells pelleted in culture medium often clump, and one must re-filter the suspension through a nylon mesh or triturate more vigorously, killing cells and generating more small debris. Cells pelleted in BSA were easily dispersed with one or two passes of the P-1000 Pipetman, with few if any clumps remaining. The two centrifugations to remove dissolved DiI also serve to further reduce the level of small debris in the final suspension.
In a study of DiI-stained rat motor and sensory neurons (St. John, 1991), the longer chain C18 form of DiI was found to be toxic, killing all labeled neurons by 10 days in culture, while the DiI-C12-stained neurons remained healthy. Unlike St. John (who did not use F127), we did not find DiI-C12 to be completely harmless. We observed toxicity of DiI-C12 in cultured hippocampal neurons if they were stained for longer than 30 min in a 40 ug/mL solution. No adverse effects from a 15 min stain were observed, compared to unstained control cultures. Although much of the dye was concentrated in intracellular vesicles after one week in culture, all of the neurons’ fine processes were still clearly visible, including growth cones. Thus, it is crucial to adjust staining conditions such that control cultures of stained donor cells show good viability and staining.
Cells brightly stained with DiI by the above procedures remained healthy for at least two weeks in culture, unless we attempted to photograph them using the 545 nm line of a 200 W mercury arc lamp as the excitation source (a common arrangement for wide-field fluorescence microscopy of rhodamine-like dyes). Significant fading of the dye was observed during 30-sec exposures, and the photographed cells usually died by the next day. This was surprising, since DiI has a reputation for being stable and non-phototoxic(Honig and Hume, 1989). To quantify the effect, we grew stained hippocampal neurons on numbered-grid petri dishes and exposed a 500 um-diameter region to green light from the arc lamp for varying amounts of time. Upon returning to the same regions the next day, we found that exposures as short as 10 sec (using a 20x/0.75 NA objective) adversely affected cell morphology, and 30 sec exposures were lethal to all exposed cells (Fig. 3). Exposures as long as 6 min had no effect on unstained control cultures, and unexposed regions in the stained cultures remained healthy.
Fig. 3. Phototoxicity of DiI.

Stained embryonic rat hippocampal cells were cultured for 3 days on numbered-grid dishes, and exposed to green light from the mercury arc lamp of the Nikon Diaphot microscope for varying amounts of time from 10 sec to 6 min. Shown here are video images of a culture before, during, and 1 day after a 30 sec exposure. This was lethal to all the cells in the illuminated area, indicated by condensation of cell nuclei, swelling of the soma, and disintegration of the processes. Cells in unexposed areas remained healthy, as did unstained cultures exposed for as long as 6 min. Each square on the grid is 100 um.
2. TPLSM imaging
To avoid the above problems with photobleaching and phototoxicity, we used a custom-made TPLSM to non-destructively image stained cells (see Materials and Methods). Stained cells were placed into the wells of neuroprobes and checked for outgrowth after one day in culture, using Nomarski optics on an upright microscope with a tungsten lamp. Even the light from the tungsten lamp caused phototoxicity in cells that were exposed for over 30 min (during the filling of the wells), so we used a green-blocking filter (Hoya 25A) whenever possible. A representative Nomarski photograph of outgrowth one day after filling the wells is shown in Fig. 4. The same neuroprobe was subsequently imaged using the TPLSM, with 900 nm excitation, shown in the lower panel of Fig. 4. The grillwork, made from silicon nitride, is transparent, but slightly autofluorescent. Repeated scanning (at least one hour) had no adverse effect on the neurons, which were still healthy and growing the next day. Little, if any photobleaching was observed, except when vertical scans were made in which several lines were scanned in rapid succession (to enhance the signal-to-noise ratio) before advancing the stage. This produced a thin line of bleaching visible for several minutes in subsequent horizontal scans. When all of the debris removal and dye rinsing steps were rigorously followed, there was virtually no background staining on the surface of the probes or control dishes.
Fig. 4. Outgrowth of neurites from neuroprobe.
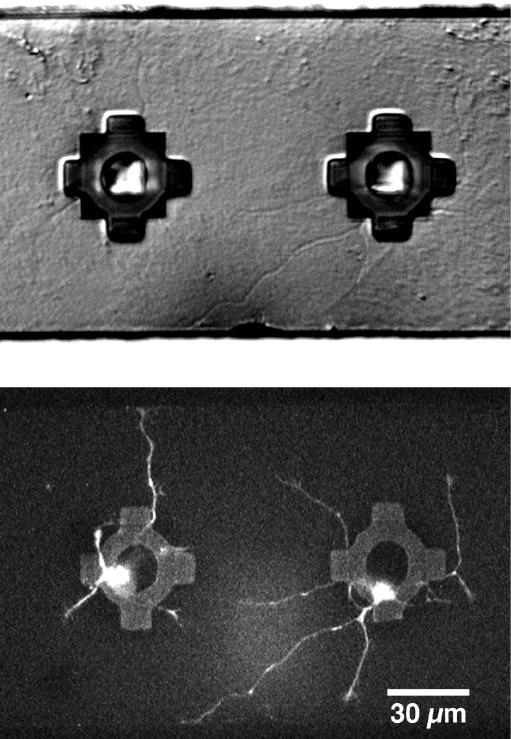
Stained embryonic rat hippocampal cells were implanted into neuroprobes and grown in culture for one day, as described in Materials and Methods. The top panel shows a Nomarski optics photograph of a pair of wells from which living neurons are sending their processes. The same neuroprobe was imaged using the TPLSM, shown in the bottom panel. This image is a lookthrough projection of unfiltered data, along the z-axis of 10 slices taken at 1 um intervals, clearly showing the cell bodies trapped under the transparent grillwork, as well as the extending processes and their growth cones. The neuroprobe was returned to the incubator, and the neurons were still healthy and growing the next day.
Eventually, we intend to use the TPLSM to image the outgrowth of neurites from neuroprobes after they have been placed on cultured hippocampal slices. In order to optimize imaging parameters, we plated stained cells directly onto the slices, at low density (20−50 cells/slice). These grew and extended processes, some reaching across the entire slice (>2 mm) by one week after plating. The TPLSM allowed us to obtain clear images of stained processes deeper than 100 um below the surface of the slice, more than twice as deep as possible using visible excitation in confocal mode. Many of the stained cells attained pyramidal cell morphology, with apical and basal dendrites, by 6 days post-transplant (Fig. 5). Fine processes were still easily visible at 17 days, the longest time after plating that 2-photon imaging was carried out.
Fig. 5. DiI-stained neurons transplanted to a cultured slice.
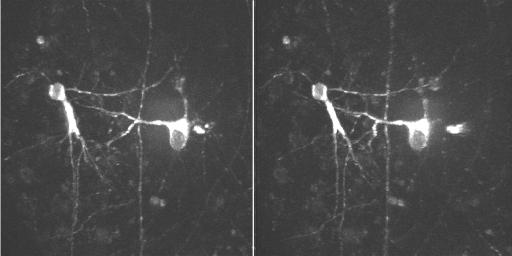
Stained embryonic hippocampal neurons were plated onto cultured hippocampal slices as described in Materials and Methods. This image was made 6 days after plating, using the TPLSM at 960 nm, from a z-series of 30 scans, 2 um apart. No photobleaching or phototoxicity was detectable during or after scanning.
Discussion
We developed a procedure for producing hippocampal cells stained brightly with DiI, with a minimum of stained debris, that will be useful to anyone wishing to study prelabeled neural transplants. Although most of the methods described have been used by others in the past, such as the use of DiI-C12, Pluronic F127, centrifugation through BSA solutions, and imaging with pulsed IR light, they seem to be greatly underappreciated. We combined them for the first time and applied them to the study of transplanted neural cells while they were still alive and growing.
We showed that DiI can have substantial phototoxicity and photobleaching in cultured hippocampal neurons, using light doses commonly used for standard 35 mm fluorescence photomicroscopy. [benefit over confocal] It was demonstrated for the first time that 2-photon laser-scanning microscopy of DiI-stained neurons is possible using pulsed IR light at 900 nm. Repeated scanning of stained neurons with the TPLSM resulted in no significant photobleaching or phototoxicity. The TPLSM allowed the visualization of processes of labeled neurons transplanted to cultured hippocampal slices to a depth of over 100 um.
With the combination of careful DiI staining methods and 2-photon imaging, we accomplished persistent labeling and non-destructive imaging of fine processes of transplanted living neurons without significant background staining of the host tissue. We are now facing the serious task of increasing the viability and outgrowth of neurons from our neuroprobe. Since each neuroprobe holds only 15 cells, we want to maximize the chance that the implanted cells are neurons (not glia, which are not electrically excitable) and that they are healthy. We consider ourselves lucky when half of the wells show neurite outgrowth after a couple of days in culture, and have not yet seen outgrowth after transfer of probes to cultured slices. Others routinely observe 5% survival of transplanted neurons (Demierre et al., 1990; Ruiz-Flandes et al., 1993), which means that the other 95% break up and may cause troublesome background staining. To address the issue of cell type and health, we are making time-lapse movies of cells immediately following plating in petri dishes, in order to observe the fate of each cell, and follow it back to time-zero to see if the healthy neurons have any distinguishing features immediately after dissociation and staining. This may help us to select only those healthy neurons for implantation into the neuroprobes.
We focused our efforts on DiI because it is the most popular membrane dye for tracing neuronal processes. DiI has a one-photon absorption maximum of 550 nm. Thus, we were hitting the short-wavelength tail of its excitation curve with 2-photon excitation at 900 nm. Although the Titanium:sapphire laser we used is continuously tunable from 710 to 970 nm, the decrease in output of the laser as it was tuned to wavelengths longer than 900 nm outweighed the increase in absorption by DiI (however, see note in last paragraph of Materials and Methods). Thus, we chose 900 nm in most cases because it gave us the maximum signal, not because it is best suited for DiI. In preliminary studies using dissociated cultures of rat hippocampal neurons, we observed that other membrane dyes normally excited by blue light provided an even brighter signal than DiI when excited by pulsed IR light at 900 nm. The dyes tested include BODIPY-ceramide (503 nm ex. max.), DiO (3−3′ dioctadecyloxacarbocyanine perchlorate, 484 nm ex. max.), and DiA (4-(4-(dihexadecylamino)styryl)-N-methylpyridinium iodide, 491 nm ex. max.) (all available from Molecular Probes). With 2-photon excitation, there is no difficulty separating excitation light from emitted light, since they are hundreds of nm apart. By using a 680 nm short-pass beamsplitter, we can detect virtually all of the emitted light from any visible-emitting fluorophore. In contrast, with normal fluorescence microscopy, one must select filters carefully suited to each fluorophore used, accepting the loss of some emitted light due to the overlap between one-photon excitation and emission spectra.
2-photon microscopy has also been proven successful for the imaging of fruit fly neurons expressing Green Fluorescent Protein (GFP, in press), a naturally fluorescent protein from jellyfish (Chalfie et al., 1994) normally excited by blue light (480 nm ex. max.). We are currently working on getting rat hippocampal neurons to express GFP. Onifer and co-workers (Onifer et al., 1993) used a retrovirus to transfer the bacterial LacZ gene to a dividing neuronally-derived cell line (RN33B cells). Transplanted cells expressing [[beta]]-galactosidase, the LacZ gene product, were observed post-mortem, and no [[beta]]-galactosidase reaction was observed in tissue from animals that received injections of transfected but lysed cells. This gives hope that GFP may provide a background-free, long-lasting way to label transplanted neurons.
Acknowledgments
We thank Sheri McKinney for her expert technical assistance; Pete Vanderklish for teaching us hippocampal slice preparation; Yu-Chong Tai, Svetlana Tatic-Lucic and John Wright for neuroprobe design and fabrication; Gyuri Buzsaki and Anatol Bragin for their continued collaboration on the Neural Prosthesis project; The NINDS at the NIH for funding the Neural Prosthesis project; and Molecular Dynamics, a Silvio Conte Center award from the NIMH, and the Beckman Institute for support of the 2-photon imaging facility.
Footnotes
[Presented at the XIV Pfefferkorn Conference: The Science of Biological Specimen Preparation for Microscopy, Aug, 1995, Belleville, IL. Proceedings to appear in Scanning Microscopy International]
References
- Banker G, Goslin K. Culturing Nerve Cells. MIT Press; Cambridge, Mass.: 1991. [Google Scholar]
- Buchs PA, Stoppini L, Muller D. Structural modifications associated with synaptic development in area CA1 of rat hippocampal organotypic cultures. Dev. Brain Res. 1993;71:81–91. doi: 10.1016/0165-3806(93)90108-m. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene-expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Dailey ME, Buchanan J, Bergles DE, Smith SJ. Mossy fiber growth and synaptogenesis in rat hippocampal slices in vitro. J. Neurosci. 1994;14:1060–1078. doi: 10.1523/JNEUROSCI.14-03-01060.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila HV, Salzberg BM, Cohen LB, Waggoner AS. A large change in axon fluorescence that provides a promising method for measuring membrane potential. Nature. 1973;241:159–160. doi: 10.1038/newbio241159a0. [DOI] [PubMed] [Google Scholar]
- Demierre B, Martinou JC, Kato AC. Embryonic motoneurons grafted into the adult CNS can differentiate and migrate. Brain Res. 1990;510:355–359. doi: 10.1016/0006-8993(90)91391-s. [DOI] [PubMed] [Google Scholar]
- Etcheberrigaray R, Gibson GE, Alkon DL. Molecular mechanisms of memory and the pathophysiology of Alzheimers-disease. Ann. N.Y. Acad. Sci. 1994;747:245–255. doi: 10.1111/j.1749-6632.1994.tb44413.x. [DOI] [PubMed] [Google Scholar]
- Gahwiler BH. Organotypic cultures of neural tissue. Trends. Neurosci. 1988;11:484–489. doi: 10.1016/0166-2236(88)90007-0. [DOI] [PubMed] [Google Scholar]
- Honig MG, Hume RI. Fluorescent carbocyanine dyes allow living neurons of identified origin to be studied in long-term cultures. J. Cell Biol. 1986;103:171–187. doi: 10.1083/jcb.103.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig MG, Hume RI. DiI and DiO - versatile fluorescent dyes for neuronal labeling and pathway tracing. TINS. 1989;12:333–341. [PubMed] [Google Scholar]
- Hosokawa T, Bliss TVP, Fine A. Persistence of individual dendritic spines in living brain slices. Neuroreport. 1992;3:477–480. doi: 10.1097/00001756-199206000-00005. [DOI] [PubMed] [Google Scholar]
- Hosokawa T, Bliss TVP, Fine A. Quantitative 3-dimensional confocal microscopy of synaptic structures in living brain-tissue. Microscopy Research and Technique. 1994;29:290–296. doi: 10.1002/jemt.1070290405. [DOI] [PubMed] [Google Scholar]
- Lojewska Z, Loew LM. Pluronic F127: An effective and benign vehicle for the insertion of hydrophobic molecules into membranes. Biophys. J. 1986;49:521a. [Google Scholar]
- Lojewska Z, Loew LM. Insertion of amphiphilic molecules into membranes is catalyzed by a high molecular weight non-ionic surfactant. Biochim. Biophys. Acta. 1987;899:104–112. doi: 10.1016/0005-2736(87)90244-6. [DOI] [PubMed] [Google Scholar]
- Muller D, Buchs PA, Stoppini L. Time course of synaptic development in hippocampal organotypic cultures. Dev. Brain Res. 1993;71:93–100. doi: 10.1016/0165-3806(93)90109-n. [DOI] [PubMed] [Google Scholar]
- O'Rourke NA, Cline HT, Fraser SE. Rapid remodeling of retinal arbors in the tectum with and without blockade of synaptic transmission. Neuron. 1994;12:921–934. doi: 10.1016/0896-6273(94)90343-3. [DOI] [PubMed] [Google Scholar]
- O'Rourke NA, Fraser SE. Dynamic changes in optic fiber terminal arbors lead to retinotopic map formation - an in vivo confocal microscopic study. Neuron. 1990;5:159–171. doi: 10.1016/0896-6273(90)90306-z. [DOI] [PubMed] [Google Scholar]
- Onifer SM, White LA, Whittemore SR, Holets VR. In vitro labeling strategies for identifying primary neural tissue and a neuronal cell-line after transplantation in the CNS. Cell Transplantation. 1993;2:131–149. doi: 10.1177/096368979300200207. [DOI] [PubMed] [Google Scholar]
- Paramore CG, Turner DA, Madison RD. Fluorescent labeling of dissociated fetal cells for tissue culture. J. Neurosci. Meth. 1992;44:7–17. doi: 10.1016/0165-0270(92)90108-p. [DOI] [PubMed] [Google Scholar]
- Potter SM, Wang C-M, Garrity PA, Fraser SE. Intravital imaging of green fluorescent protein using two-photon laser-scanning microscopy. Gene. 1996 doi: 10.1016/0378-1119(95)00681-8. (in press) [DOI] [PubMed] [Google Scholar]
- Pyapali GK, Turner DA, Madison RD. Anatomical and physiological localization of prelabeled grafts in rat hippocampus. Experimental Neurology. 1992;116:133–144. doi: 10.1016/0014-4886(92)90161-i. [DOI] [PubMed] [Google Scholar]
- Ruiz-Flandes P, Demierre B, Mattenberger L, Kato AC. Migration of purified embryonic motoneurons grafted into adult-mouse CNS. Int. J. Dev. Neurosci. 1993;11:525–533. doi: 10.1016/0736-5748(93)90042-c. [DOI] [PubMed] [Google Scholar]
- St. John PA. Toxicity of ”DiI” for embryonic rat motoneurons and sensory neurons in vitro. Life Sciences. 1991;49:2013–2021. doi: 10.1016/0024-3205(91)90644-q. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Meth. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. Lesion-induced neurite sprouting and synapse formation in hippocampal organotypic cultures. Neuroscience. 1993;57:985–994. doi: 10.1016/0306-4522(93)90043-f. [DOI] [PubMed] [Google Scholar]
- Tatic-Lucic S. PhD Dissertation. California Institute of Technology; 1994. Silicon Micromachined Devices for In Vitro and In Vivo Studies of Neural Networks. [Google Scholar]
- Tatic-Lucic S, Tai YC. Novel extra-accurate method for 2-sided alignment on silicon-wafers. Sensors and Actuators a Physical. 1994;42:573–577. [Google Scholar]
- Vidal-Sanz M, Villegas-Perez MP, Bray GM, Aguayo AJ. Persistent retrograde labeling of adult-rat retinal ganglion-cells with the carbocyanine dye DiI. Experimental Neurology. 1988;102:92–101. doi: 10.1016/0014-4886(88)90081-7. [DOI] [PubMed] [Google Scholar]
- Williams RM, Piston DW, Webb WW. 2-photon molecular-excitation provides intrinsic 3-dimensional resolution for laser-based microscopy and microphotochemistry. FASEB J. 1994;8:804–813. doi: 10.1096/fasebj.8.11.8070629. [DOI] [PubMed] [Google Scholar]