

Abstract
We studied the role of somatic mosaicism in fibrous dysplasia of bone (FD) within the context of skeletal (“mesenchymal”) stem cells by assessing the frequency of mutated colony forming unit‐fibroblasts (CFU‐Fs) from FD lesions, and in some cases, from unaffected sites, in a series of patients. There was a tight inverse correlation between the percentage mutant CFU‐F versus age, suggesting demise of mutant stem cells caused by exuberant apoptosis noted in samples from young patients. In older patients, either partially or completely normal bone/marrow histology was observed. On in vivo transplantation, FD ossicles were generated only by cell strains in which mutant CFU‐Fs were identified. Strains that lacked mutant CFU‐F (but were mutation positive) failed to regenerate an FD ossicle. These data indicate that GNAS mutations are only pathogenic when in clonogenic skeletal stem cells. From these data, we have evolved the novel concept of “normalization” of FD. As a lesion ages, mutant stem cells fail to self‐renew, and their progeny are consumed by apoptosis, whereas residual normal stem cells survive, self‐renew, and enable formation of a normal structure. This suggests that activating GNAS mutations disrupt a pathway that is required for skeletal stem cell self‐renewal.
Keywords: fibrous dysplasia, Gsα, mutation, skeletal stem cells, bone marrow stromal cells
INTRODUCTION
Fibrous dysplasia of bone (FD) is a crippling disorder occurring as either a solely skeletal disease (monostotic or polyostotic) or in conjunction with endocrinopathies and skin hyperpigmentation as part of the McCune‐Albright syndrome (MAS; OMIM 174800). Activating missense mutations of the GNAS gene, encoding the α subunit of the stimulatory G protein, Gs, underlie all forms of FD of bone1, 3, 4, 5 and are thought to occur postzygotically leading to a somatic mosaic state.5, 6 Somatic mosaicism has been invoked as the mechanism allowing for survival of mutant cells during development. The observation that the disease is never transmitted through the germ line is consistent with this view. The postzygotic de novo mutational events at R201 (primarily R201C and H, rarely S and G) lead to the development of a focal, noninherited disease phenotype. Taken together, the somatic mosaic state characterizing the abnormal genotype and the multifocal nature of the skeletal disease (as well as of the non‐obligatory extraskeletal lesions) would lend credibility to the view that each affected site represents the direct expression of local mutant cells as has been shown previously. In contrast, unaffected sites express a local mutation‐free environment generated during development by either restricted migration or negative selection of mutant cells, which has been assumed, but never formally showed.
FD lesions of bone reflect the dysfunction of cells in the osteogenic lineage, in which the relevant activating GNAS mutations have been repeatedly identified, along with distinct phenotypic anomalies. Lesions are characterized by unorganized and poorly mineralized woven bone, retraction of osteogenic cells from the bone surface and formation of Sharpey's fibers, and replacement of hematopoietic marrow with a fibrotic marrow with characteristics of osteogenic progenitors.9, 10 However, cells with both the normal and the disease‐associated genotype are found within an individual FD lesion, even at the level of individual clonogenic progenitors of bone‐forming cells (colony forming unit‐fibroblasts [CFU‐Fs]). This makes each FD lesion a somatic mosaic itself, rather than simply the “wrong” piece of a macroscopic patchwork of phenotypically abnormal tissue and normal tissue. A natural corollary, we reasoned, would be that mutant cells might also, in principle, be found in normal tissue as well. In a previous study, mutant allele was minimally detected in a biopsy of a radiograpically normal bone that was divided into two parts (one for histological analysis that proved to be normal bone and marrow and one for mutation analysis); however, the mutant cell type was not identified. If true, determinants, ranging from local mutational load to stimuli instigating initiation of a lesion, could possibly be invoked to explain not only the development but also the natural prevention of an FD lesion at different sites. To address these questions, we studied the occurrence of GNAS mutations in nonclonal and clonal adherent stromal cells, derived from histologically abnormal and normal bone of patients with polyostotic FD and the MAS. In addition, based on the fact that the stromal cell population contains a subset of clonogenic, multipotent skeletal (mesenchymal) stem cells, capable of forming different cell phenotypes associated with skeletal tissue,11, 12, 13 we assessed the frequency of mutant CFU‐Fs within the populations and the ability of the populations to recreate an FD‐like ossicle on in vivo transplantation into immunocompromised mice.
MATERIALS AND METHODS
Patients and samples
Eighteen consecutive patients with FD (ranging in age from 7 to 58 yr; average age, 25.5 yr) were enrolled in Institutional Review Board (IRB)‐approved studies of FD/MAS at the NIH and gave written informed consent (Table 1). All patients underwent 99Tc‐MDP bone scans using a standardized dosing and scanning protocol. Radiographs were obtained using standard procedures. Diagnosis of FD was confirmed based on clinical history, radiographic findings, histopathological findings, and analysis for activating mutations (R201C, R201H, R201S, R201G) in the GNAS gene. Patients with severe endocrinopathies known to affect the course and evolution of the skeletal lesions were excluded. Samples (nine biopsies and nine surgical samples) were obtained from radiographically involved sites (iliac crest, femur, phalange, or craniofacial bones) from 15/18 patients, and seven samples (five biopsies and two surgical samples) were obtained from radiographically normal sites (iliac crest, femur, mandible) from 6/18 patients. Biopsies from unaffected sites (when possible) and affected sites were routinely performed on enrollment in the study unless counter‐indicated (inaccessible site or risk of fracture). All samples were analyzed histologically and used to derive strains of bone marrow stromal cells. Because of variability in sample size (needle or core biopsy versus surgical waste), not all of the other assays described below could be applied to samples collected from all of the 18 patients. Samples from normal donors (N = 42, ranging in age from 1 to 76 yr; average age, 23.0 yr) were collected from surgical waste under approved guidelines for the use of human subjects in research.
Table Table 1.
Patient Characteristics and Summary of Mutation Analysis
|
|
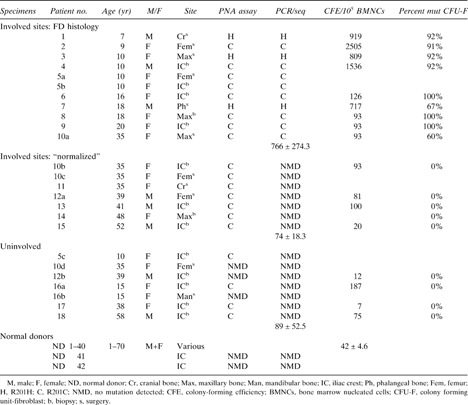
Bone marrow stromal cell cultures
Fragments of specimens were scraped gently with a steel blade into cold α‐MEM (Invitrogen, Carlsbad, CA, USA) and pipetted repeatedly. The released cells were passed consecutively through 16‐ and 20‐gauge needles and filtered through a 70‐μm pore size nylon cell strainer (Becton Dickinson, Franklin Lakes, NJ, USA) to remove cell aggregates. Single cell suspensions were plated at high density (1.0 × 107 nucleated cells) per 75‐cm flask (Becton Dickinson) to generate non‐clonal strains and at low density (1–3 × 105 nucleated cells) per 150‐mm dish (Becton Dickinson) to generate single colony‐derived strains. Growth medium consisted of α‐MEM, 2 mM l‐glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin sulfate (Invitrogen), and 20% lot‐selected FBS (Atlanta Biological, Norcross, GA, USA). After 3 h of incubation at 37°C, unattached cells were removed by aspiration, and cultures were washed thoroughly three times with α‐MEM. Cultivation was continued in growth medium at 37°C in an atmosphere of 100% humidity and 5% carbon dioxide. Non‐clonal strains were passaged at ∼10 days using trypsin/EDTA and were generally used for mutation analysis at the second or third passage. In low‐density cultures, after 13–16 days, ∼20 individual colonies from each sample were isolated using cloning cylinders and trypsin release and expanded in cell number by placing them in vessels of increasing size at consecutive passages until at least 6 million cells were available (Fig. 1).
Figure FIG. 1.
Mutational analyses of non‐clonal and clonal cultures of BMSCs derived from fibrous dysplastic lesions. Non‐clonal cultures were analyzed by selective amplification of the mutated allele using a PNA primer to block amplification of the normal allele (which detects 1:200 mutated cells) and by direct DNA sequencing (which detects 1:2 or 3 mutated cells) to approximate the mutational load within the BMSC population. Clonal cultures were established to determine the colony forming efficiency (the number of CFU‐Fs), and isolated clones were analyzed by direct DNA sequencing to determine the number of mutated clones.
Mutation analysis
Genomic DNA was isolated from cells using the DNeasy Tissue Kit according to the manufacturer's instructions (Qiagen, Valencia, CA, USA). A peptide nucleic acid (PNA)‐based assay and direct DNA sequencing of the relevant PCR‐amplified target sequence in the GNAS gene were used in these experiments to detect the GNAS mutations in the different populations of bone marrow stromal cells (BMSCs) as described previously (Fig. 1).
Direct DNA sequencing can detect only one mutant cell out of two‐three cells. Consequently, a peptide nucleic acid (PNA) sequence complementary to the wildtype sequence was added to the reaction mixture to prevent the binding of the sense primer to the normal allele. This allowed for the selective amplification and sequencing of the mutant allele and is estimated to detect one mutant cell out of 200 cells. The target DNA (200–500 ng) was amplified in a standard 100‐μl PCR reaction mixture using 2.5 U of Ampli Taq Gold polymerase (Perkin Elmer, Boston, MA, USA) and 1 μg of each primer, forward primer, 5′‐GTTTCAGGACCTGCTTCGC‐3′; reverse primer, 5′‐GCAAAGCCAAGAGCGTGAG‐3′, and 2 μg of the PNA sequence amino‐terminal 5′‐CGCTGCCGTGTC carboxy‐terminal‐3′ (bold indicates overlap of the forward primer with the PNA sequence).
The samples were heated to 94°C for 15 min, cycled 40 times (94°C for 30 s, 68°C for 60 s [to allow the PNA to bind specifically to any nonmutant allele and block the annealing of the forward primer], 55°C for 30 s, and 72°C for 60 s), and terminated for 7 min at 72°C. The 325‐bp PCR products were purified using the Promega Wizard PCR Preps DNA Purification System (Madison, WI, USA) and sequenced using a Perkin Elmer Applied Biosystem 377 Automated Sequencer, and the sequence was read from the 3′‐5′ (reverse) strand.
For direct DNA sequencing, the extracted DNA was amplified in a 100‐μl reaction by standard PCR with primers that generate a 270‐bp product spanning the mutation. For each reaction, 10 μg of each primer was used: forward primer, 5′‐TGACTATGTGCCGAGCGA‐3′; reverse primer, 5′‐AACCATGATCTCTGTTATATAA‐3′.
The samples were heated to 94°C for 15 min, cycled 35 times (94°C for 30 s, 55°C for 30 s, 72°C for 30 s), and terminated for 7 min at 72°C. The PCR products were purified and sequenced as described, and the sequence was read from the 5′‐3′ strand.
Assessment of the number of CFU‐Fs by colony‐forming efficiency assays
Colony‐forming efficiency (CFE), which enumerates the number of CFU‐Fs present in the original marrow cell suspension, was determined by plating single cell suspensions at low density as described above. On days 10–14, cultures were washed with Hank's balanced salt solution, fixed with methanol, and stained with an aqueous solution of saturated methyl violet. Colonies containing 50 or more cells were counted using a dissecting microscope, and CFE (number of colonies per 1 × 105 marrow cells plated) was determined (Fig. 1).
Histological analysis
All specimens were fixed in freshly prepared phosphate‐buffered 4% formalin for 24 h. After washing in PBS, samples were decalcified in buffered 10% EDTA and paraffin embedded. Additional fixed, nondecalcified specimens were embedded in methyl methacrylate (MMA). Five‐micrometer‐thick sections were cut from paraffin blocks and stained with H&E; 3‐μm‐thick sections were cut from MMA blocks and stained either with Goldner's trichrome or von Kossa and Giemsa stains.
Analysis of apoptosis
For detection of apoptosis in situ in paraffin sections of FD tissue, a modification of the TUNEL procedure using reagents from a commercially available kit (TA4628; R&D Systems, Minneapolis, MN, USA) was performed. After digestion with 1 mg/ml proteinase K for 15 min at room temperature, endogenous peroxidase was quenched with 3% hydrogen peroxide. After placement in equilibration buffer, terminal deoxynucleotidyl transferase and digoxigenin‐labeled dUTP were added, and sections were incubated for 1 h at 37°C. After rinsing in stop‐wash buffer, TUNEL signal was detected with peroxidase‐labeled anti‐digoxigenin antibody and developed with diaminobenzidine.
In vivo transplantation assay
Non‐clonal strains of bone marrow stromal cells were transplanted into immunocompromised mice as described previously. Briefly, cells were expanded ex vivo and passaged, and 2 × 106 cells of the second or third passage were resuspended in 1 ml of nutrient medium. Cell suspensions were incubated with 40 mg of hydroxyapatite/tricalcium phosphate ceramic particles (Zimmer, Warsaw, IN, USA) for 30 min at 37°C with slow mixing to allow adhesion of cells to the particles and transplanted into the subcutaneous tissue of NIH‐bg‐nu‐nu‐xidBR (beige) mice (Harlan Sprague Dawley, Indianapolis, IN, USA). All procedures were performed under an institutionally approved protocol for the use of animals in research (02‐222). All transplants were harvested at 8 wk, fixed with 4% buffered formalin, and processed for paraffin or methyl methacrylate embedding.
Statistical analyses
All statistical analyses were performed using the Mann‐Whitney test (nonpaired, nonparametric, two‐tailed p values).
RESULTS
Variable frequency of mutant BMSCs in FD lesions
To evaluate the frequency of mutant cells among total BMSCs, non‐clonal cultures were established from each of 18 samples from affected sites from 15 different patients (patients 1–15) with PFD/MAS (6 males, 9 females; age range, 7–52 yr; average age, 24.5 yr; Table 1). Genomic DNA extracted from monolayers of BMSC cultures was analyzed using both the PNA clamping method (estimated sensitivity 1:200 cells), and the direct PCR/sequencing method (estimated sensitivity 1:2 or 3 cells caused by the inability to detect small changes from the baseline of the DNA sequencer chart; Fig. 2). GNAS mutations were shown by the PNA‐based method in all samples (R201H in 3 patients and R201C in 12; Figs. 2A and 2B). When the standard PCR/sequencing method was used on the same samples, however, evidence of mutations could only be obtained in 11 samples (patients 1–10a; Fig. 2D, for example), and no mutation could be detected in the remaining 7 samples (patients 10b–15; Fig. 2E, for example). Given the difference in sensitivity of the PNA‐based method compared with the standard PCR/sequencing procedure, we concluded that the frequency of total mutant stromal cells was comparatively high in 11/18 and comparatively low in 7/18 of the FD lesions analyzed.
Figure FIG. 2.
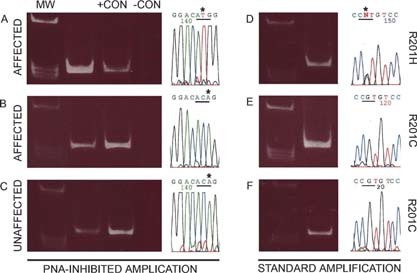
Identification of mutations by PNA‐inhibited amplification (A‐C, 3′‐5′ strand sequenced) and direct DNA sequencing (D‐F, 5′‐3′ strand sequenced) (MW, molecular weight markers; +CON, positive control [DNA isolated from a mutant clone]; −CON, negative control [DNA isolated from a normal clone]). In some cultures of BMSCs derived from affected tissue, mutation (in this case, R201H) could be detected by both methods (A and D). In other cultures of BMSCs derived from affected tissue, mutation was detectable by PNA‐inhibited amplification (in this case, R201C) (B), but no mutation was found by direct DNA sequencing (E). BMSCs were also derived from clinically defined normal bone. In some cases, mutation was detected by PNA‐inhibited amplification (C), but no mutation was detected by direct DNA sequencing (F).
Number of CFU‐Fs, and mutant CFU‐Fs, are depleted with aging in FD lesions
Analysis of clonogenic efficiency of cells (CFE; number of CFU‐F/105 nucleated cells) in the fibrotic marrow from radiographically abnormal bone from 12 of the 15 patients showed a significantly higher CFE in nine of the samples (patients 1–4 and 6–10a) with high mutational load compared with four of the samples (patients 10b, 12a, 13, and 15) with low mutational load (766 ± 274.3 versus 74 ± 18.3 SE, respectively; p = 0.045). The CFE in the nine samples with high mutational load was similar to what we previously observed in other patients with FD and ∼18‐fold higher than our series of normal donors (N = 40, 1–76 yr of age, 42 ± 4.6 SE, p = 0.0001). Interestingly, CFE observed in samples with low mutational load was not statistically different from CFE of samples from normal donors (74 ± 18.3 versus 42 ± 4.6 SE, p = 0.1405; Fig. 3A).
Figure FIG. 3.
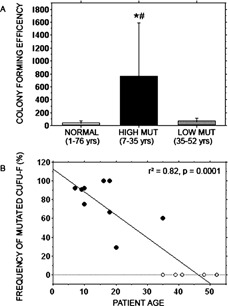
Colony‐forming efficiency (CFE) and frequency of mutated clones. (A) Colony‐forming efficiency of single cell suspensions of bone marrow nucleated cells was determined for a series of normal donors (age range, 1–76 yr) from patients in which high levels of mutation were detected (age range, 7–35 yr) and from patients in which low levels of mutation were detected (age range, 35–52 yr). The average CFE was ∼18‐fold higher in samples from patients with a high level of mutation compared with that of normal donors (*p = 0.0001) and significantly higher compared with that of patients with a low level of mutation (# p = 0.045). There was no statistical difference between samples from normal donors compared with those from patients with a low level of mutation. (B) Clones (∼20 from each patient) isolated from low‐density cultures of single cell suspensions of bone marrow mononuclear cells were genotyped by direct DNA sequencing. The percent of mutated clones (representative of the percent of mutated CFU‐Fs) was compared with the age of the patient by linear regression and was found to have a tight negative correlation.
From 13 of the 15 patients (patients 1–4, 6–10b, and 12a–15), we also analyzed the frequency of mutant stromal cells belonging to the clonogenic subset, which includes skeletal (mesenchymal) progenitors and stem cells. A fraction of each cell suspension made from the parent tissue sample was separately plated at clonal density (1–3 × 105 nucleated cells per 150‐mm dish). In this type of culture, discrete colonies are formed, each representing the progeny of a single colony‐initiating cell (CFU‐F). From each sample, ∼20 individual clones were separately expanded in culture, and mutant clones (representative of a mutant CFU‐F) were identified by analyzing genomic DNA using the standard PCR/direct DNA sequencing method.
This analysis showed a variable frequency (ranging from 0% to 100%) of mutant clones (i.e., of mutant CFU‐Fs) in the different samples (Table 1). All nine samples from patients found to contain mutant BMSCs at relatively high frequency (positive by standard PCR/direct DNA sequencing) in non‐clonal cultures also contained detectable mutant CFU‐Fs, although in variable proportions. Conversely, five samples characterized by a relatively low frequency of mutant BMSCs (positive by PNA clamping only) in non‐clonal cultures were found to contain no detectable mutant CFU‐Fs. This suggested that the frequency of total mutant cells was related to the frequency of mutant clonogenic cells in each sample and that these were absent or exceedingly rare in some cultures.
More striking was the finding that all samples with a high frequency of mutant cells, high CFE, and detectable mutant clonogenic cells came from patients of age range 7–35 yr. All of those with a lower frequency of mutant cells, low CFE, and no detectable mutant, clonogenic cells, came from patients older than the age of skeletal maturity (range, 35–52 yr). Linear regression analysis showed a highly significant negative correlation between the frequencies of mutant CFU‐Fs (expressed as percentage of positive clones) versus patient age (Fig. 3B; p = 0.0001). These data strongly suggested a change in the composition of the stromal cell populations between the second and fourth decade of life.
Histological normalization of FD lesions
To study potential differences in the clinical characteristics of the lesions from which our samples were derived, scintigraphic and radiographic materials were reviewed in each case. All 18 samples were derived from obviously affected skeletal sites showing enhanced signal on bone scans (Fig. 4A), marked deformity, abnormalities in density and architecture, and abnormal trabeculation patterns on radiographs (Fig. 4B). On closer inspection, however, the occurrence of multiple dense rinds, and the absence of typical ground‐glass appearances (Fig. 4B), were noted to be recurrent among the seven sites with low mutational load and were also noted to be from older patients (35–52 yr of age).
Figure FIG. 4.
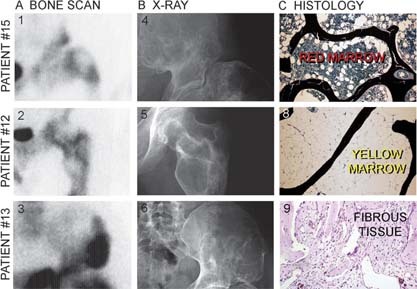
Bone scintigraphy (A), X‐ray images (B). and histology of FD lesions (C) in three different patients >35 yr of age. Whereas both scintigraphy (panels 1–3) and X‐ray images (panels 4–6) show the presence of lesions, the histology of putatively lesional bone shows normal histology in two of three patients. In one patient, histology shows normal bone and hematopoietically active marrow, consistent with the histology of the iliac crest in a normal adult subject (panel 7). In the second patient, histology shows normal bone trabeculae and adipose marrow, consistent with the histology of the femur in a normal adult (panel 8). In the third patient, nondescript fibrous tissue is seen along with normal bone trabeculae (panel 9). The trabeculae showed normal lamellar structure, instead of the woven texture typical of FD, on examination by polarized light microscopy (data not shown).
Surprisingly, specimens from four of seven of these specimens (patients 10c, 11, 12a, and 15) showed a completely normal bone and bone marrow histology. In particular, hematopoiesis and adipocytes populated the bone marrow space, which was free of any fibrotic change. The structure of bone trabeculae was lamellar. Mineralization as assessed by von Kossa staining of undecalcified sections was normal, and osteoblasts exhibited a normal cuboidal morphology (Fig. 4C, panels 7 and 8). In the remaining three of seven cases (patients 10b, 13, and 14), a non‐descript fibrotic change was observed focally in the bone marrow space (Figs. 4C, panel 9, B, and 5D). However, bone trabeculae were again consistently lamellar in structure as shown by polarized light microscopy (data not shown) and lined by osteoblasts of normal morphology (Figs. 5B and 5D). This was in sharp contrast with the histological features seen in typical FD lesions, characterized by woven bone texture, frequent mineralization defects, and distinctive shape changes in osteoblasts (Figs. 5A and 5C, 9, 10).
Figure FIG. 5.
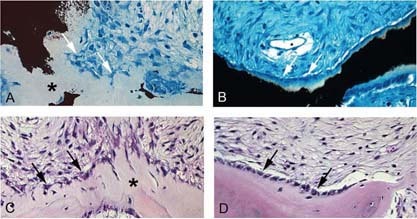
Histological changes in the “normalization” of fibrous dysplastic lesions. von Kossa and Giemsa (A and B) and H&E (C and D) staining of lesional tissue from patient 7 (18 yr old) with high mutation levels (A and C) and from patient 10 (sample b, 35 yr old) with low mutation levels (B and D). In patient 7, bone is poorly mineralized, and there is excessive osteoid (* in A). Osteoblasts display a distinctive retracted, stellate morphology (white arrows in A, black arrows in C), characteristics that are typically found in FD lesions. In patient 10, mineralization is normal (B), and normal‐appearing cuboidal osteoblasts cover the bone‐forming surfaces (white arrows in B, black arrows in D).
High levels of apoptosis in FD lesions
The observation that a partial “sterilization” of the bone environment with respect to the disease genotype could occur as a function of age indicated that progressive demise of mutant osteogenic cells and their clonogenic progenitors could take place within FD lesions. Because apoptosis is the standard mechanism by which defective/redundant cells are eliminated in development, we asked whether a similar mechanism could operate in FD and lead to progressive elimination of mutant cells. TUNEL analysis of tissue sections from four patients with typical FD histology and high mutational load showed an unexpectedly high frequency of TUNEL+ cells (Fig. 6A), which were mostly concentrated along the surfaces of abnormal bone trabeculae, but also included osteocytes contained therein (Fig. 6B). This observation was matched by a high number of readily apparent, typical apoptotic bodies in FD osteoblasts and osteocytes in parallel sections stained with von Kossa and Giemsa (Figs. 6C–6E) and H&E (Figs. 6F–6I). Both the number of TUNEL+ cells and the number of apoptotic bodies dramatically exceeded the reported frequencies of apoptotic events in bone tissue sections in different species.
Figure FIG. 6.
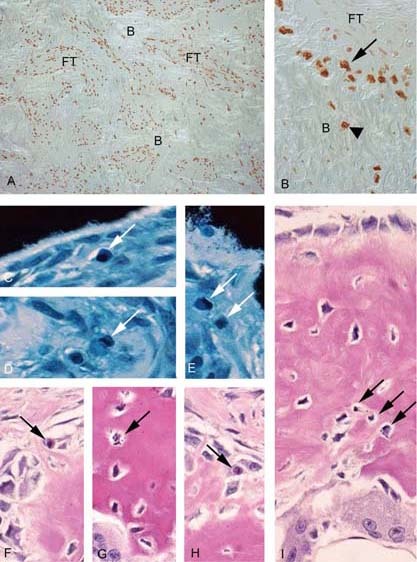
Apoptosis in fibrous dysplastic tissue. (A) TUNEL analysis of tissue from patients with typical FD histology and high mutational load shows extremely high frequencies of apoptotic cells throughout the fibrotic marrow (FT) and bone (B). (B) High levels of TUNEL+ cells are found on the bone surfaces (arrow) and within the abnormal bone (arrowhead). (C–E) Apoptotic bodies in nondecalcified sections stained with von Kossa and Giemsa (white arrows). (F–I) Apoptotic bodies in decalcified sections stained with H&E (black arrows).
Stromal cell strains from unaffected bone in FD patients may contain mutant BMSCs but do not contain mutant CFU‐Fs
We next asked whether a mutational profile characterized by low frequency of mutant cells and absence/rarity of mutant CFU‐Fs (which we observed in the “normalized” sites) could also be found at radiographically and histologically normal skeletal sites in FD patients (i.e., outside of sites of FD involvement). Using the PNA assay, capable of detecting ∼1:200 mutant cells, this was found to be the case for four of seven samples from uninvolved sites (patients 5c, 16a, 17, and 18; Table 1; Fig. 2C, for example), whereas direct DNA sequencing was negative (Fig. 2F). In three of these samples, sufficient numbers of cells were available for determining CFE and for isolating clones (patients 16a, 17, and 18). The CFE was not statistically different from normal (89 ± 52.5 versus 42 ± 4.6 SE, p = 0.45), and no mutant CFU‐Fs were detected (Table 1). We concluded that a low frequency of mutant BMSCs, in the absence of detectable mutant CFU‐Fs, could be consistent with the lack of development of FD lesions. In a similar fashion, the same profile of mutational load could be consistent with a secondary “normalization” of previously active FD lesions over time. However, this would result in continued remodeling activity (reflected in positive findings by bone scintigraphy; Fig. 4A), but on an abnormal architectural scaffold, testifying to the history of the lesion and reflected in the persistence of radiographic changes (Fig. 4B).
Stromal cell strains depleted of mutant CFU‐Fs, but not of mutant BMSCs, fail to transfer FD on heterotopic transplantation into mice
Because we had previously shown that FD‐like tissue can be recreated in immunocompromised mice by transplantation of stromal cell strains containing mutant CFU‐Fs, we asked whether cell strains depleted in mutant CFU‐Fs, but containing some mutant BMSCs, were effective in regenerating FD‐like tissue in vivo. Because the same profile of mutation frequency can be observed both in normalized skeletal sites and uninvolved sites, cell strains derived from these two types of anatomical locations were expanded in culture and transplanted into the subcutaneous tissue of immunocompromised mice. These experiments showed that cell strains with high mutation levels and mutant CFU‐Fs reproduced FD‐like tissues at heterotopic sites (Fig. 7A). Cell strains from “normalized” sites (Fig. 7B) or normal sites (Fig. 7C), both of which contain detectable mutant BMSCs but undetectable mutant CFU‐Fs, generated normal bone that supported the formation of hematopoietic marrow.
Figure FIG. 7.
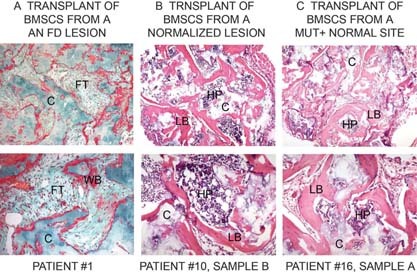
Transplantation of BMSCs in vivo. BMSCs derived from FD lesions and normal sites were combined with HA/TCP as a carrier (C) and transplanted subcutaneously into immunocompromised mice. (A) BMSCs from lesions with high levels of mutation (patient 1) reproduced a fibrous dysplastic ossicle characterized by formation of poorly mineralized woven bone (WB) and fibrous tissue (FT), reminiscent of a native lesion. (B) BMSCs from lesions in which the level of mutation was only detectable by PNA‐inhibited amplification (patient 10, sample b) formed normal lamellar bone (LB) and established a normal hematopoietic marrow (HP). (C) BMSCs derived from a normal site, but mutation positive by the PNA assay (patient 16, sample a) also established a normal ossicle.
DISCUSSION
Our study of the frequency of mutant BMSCs and of their progenitors (CFU‐Fs) as a determinant of the natural history of FD lesions was prompted by the observation that each individual FD lesion encrypts a somatic mosaic of normal and mutant osteogenic cells and their clonogenic precursors. We hypothesized that there might be a critical threshold of mutational load, above which a lesion would develop and below which (perhaps) a normal bone structure would be preserved. In particular, we focused on the rapidly adherent clonogenic subset of BMSCs (CFU‐Fs), of which 10–20% are multipotent skeletal (mesenchymal) stem cells, able to recreate a bone/marrow organ in vivo and to maintain skeletal homeostasis. Analysis of the clonogenic population is a reliable approximation to a numerical assessment of the skeletal (mesenchymal) stem cell population.
Analysis of BMSC cultures from a series of radiographically identifiable FD lesions confirmed that the proportion of mutant cells varies greatly. Our analysis also indicated that the clonogenic subset of stromal cells was depleted of mutant CFU‐Fs in the same lesions with an overall low frequency of mutant BMSCs. These types of lesions were observed in a group of older patients, and surprisingly, were characterized by a partially or completely normalized histology. This shows that a low frequency of non‐clonogenic mutant BMSCs does not preclude the formation of normal bone by surviving normal CFU‐Fs. This concept is specifically reaffirmed by the detection of rare, nonclonogenic mutant BMSCs in some normal skeletal sites of FD patients and that this low level of nonclonogenic mutant BMSCs remains compatible with formation of a normal bone structure. The notion that FD lesions “improve” or “stabilize” at skeletal maturity (or with age) is not novel in the literature18, 19 but is based on clinical observations rather than on adequate documentation of pathological changes or establishment of a physiological reason. Our data consolidate this notion and link the occurrence of lesion “burnout” to a clear change in the distribution of the disease genotype specifically among BMSCs and their clonogenic progenitors. With respect to the non‐skeletal manifestations of GNAS mutations in MAS, it does not seem that café au lait hyperpigmentation or endocrine hyperfunction regress with age, with the one exception of hyperphosphaturia, caused by elevated production of fibroblast growth factor (FGF)23 by the abundant and active (albeit misfunctioning) osteoblastic cells in FD lesions. Hyperphosphaturia does seem to abate with aging (unpublished data), most likely because of the normalization of the FD lesions that we describe here.
Fibrous tissue in the abnormal marrow spaces of FD lesions is composed of cells with phenotypic resemblances to BMSCs. Stromal cells cultures from the abnormal FD bone marrow carry the causative GNAS mutation, and they recapitulate FD in vivo when transplanted into immunocompromised mice, establishing the concept that GNAS mutations cause development of FD lesions through their impact on skeletal progenitor/stem cells. Our current observations extend the implications of the concept. First, paucity or absence of mutant CFU‐Fs in bone is compatible with preservation of a normal bone structure and with the lack of development of FD lesions. Second, loss of mutant CFU‐Fs is coupled to partial or total restoration of normal histological structure of bone and marrow within a site where an FD lesion previously existed. The persistence of overt radiographic changes in lesions that seem to be “sterilized” of the disease genotype testifies to the fact that those lesions were once truly active morbid sites. Importantly, restoration of a normal internal structure therein implies a role for bone remodeling by a non‐mutant pool of local skeletal progenitors that ultimately predominate and replace FD bone with normal bone. Hence, loss of mutant skeletal stem cells is a selective event, sparing their normal counterparts. Finally, stromal cell populations that do contain mutant BMSCs, but not mutant CFU‐Fs, do not transfer FD to immunocompromised mice on transplantation, and generate normal bone instead, indicating that mutant clonogenic skeletal stem cells must be present to transfer FD in the transplantation assay.
Demise of mutant CFU‐Fs and BMSCs within FD lesions is consistent with Happle's classical prediction that mutant cells arising early in development would only survive through mosaicism, whereas mutations transmitted through the germ line would result in lethality. Although this view has never been directly proven, several pieces of evidence contribute to validate it: (1) somatic mosaicism is observed in all cases of FD and MAS; (2) individual FD lesions are themselves somatic mosaics; (3) no instance of inherited disease is recorded in the literature, either for FD or for MAS; (4) only mixtures of normal and mutant FD‐derived stromal cells are able to generate FD‐like ossicles in immunocompromised mice, whereas pure mutant strains do not survive and fail to generate FD‐like ossicles. These data imply that cell lethality remains associated with the disease genotype, as shown by the unusually high level of apoptosis that may ultimately lead to lesion “sterilization.” Others have suggested that an enhanced osteogenic cell proliferation is associated with FD tissue.22, 23 Taken together with our data, this implies that FD lesions result from the accelerated birth and accelerated death of mutant osteogenic cells. It is important to note here that overt FD lesions are not congenital, but develop in infancy and early childhood, during active skeletal growth. Thus, increased proliferation of mutant BMSCs and subsequent negative selection of mutant osteoblastic and osteocytic cells may occur at distinct times in an patient's lifespan, specifically during skeletal growth and in adult life, respectively. Consistent with this view, FD lesions in young individuals contain a greatly expanded pool of clonogenic cells compared with age‐matched normal subjects (∼18‐fold higher), and a high proportion of the clonogenic stromal cells carry GNAS mutations. Hence, the expansion of the mutant CFU‐F pool relative to the pool of normal CFUF‐Fs is related to the expansion of stromal tissue in situ at the expense of hematopoietic marrow. In the older lesions, the pool of mutant CFU‐Fs shrinks relative to the pool of normal CFU‐Fs, the overall population of clonogenic cells reverts to a normal size (reflected in a normal CFE), and in situ, hematopoiesis regains space at the expense of the stromal tissue.
In this study, dysfunctional osteoblasts and osteocytes were found to be highly apoptotic, which may also lead to accelerated turnover (bone formation and resorption) of FD bone, as evidenced by the presence of large numbers of osteoclasts in some instances24, 25 and an extensive system of cement/arrest lines (Schmorl's mosaic) that are often noted in FD bone. Furthermore, levels of bone resorption may also be dependent on mutational load within a lesion because of the fact that interleukin 6 (IL‐6), known to modulate osteoclastogenesis, is constitutively expressed at high levels by mutant BMSCs, whereas normal BMSCs express very low IL‐6 levels without induction by factors such as PTH.
Our data point to the consumption of mutant skeletal stem cells within FD lesions over time. Because stem cells are known to self‐renew, consumption may dispel the notion of a skeletal stem cell. However, the actual rate of self‐renewal of stem cells is relative to the turnover rate of the tissue in which they reside. In bone, which turnovers much more slowly than blood, for example, self‐renewal for the life span of the organism may be associated with a limited number of stem cell divisions that could be exceeded because of accelerated remodeling of FD bone. Nonetheless, nonmutant skeletal stem cells do remain capable of fueling bone turnover to regenerate bone and bone marrow stromal tissue in situ and also to generate bone and bone marrow stromal tissues in vivo in the mouse transplantation model, after extensive proliferation in vitro. The rarefication of mutant skeletal stem cells relative to normal ones over time suggests differential self‐renewal for GNAS‐mutant and nonmutant skeletal stem cells by an‐alteration of growth kinetics. This may result in either (1) a highly accelerated rate of proliferation of mutated skeletal stem cells leading to their consumption at an earlier point in time compared with their normal counter parts and/or (2) a switch from a conservative, asymmetric population kinetics (self‐renewal and maintenance of a skeletal stem cell) to a more symmetric population kinetics, leading to an early depletion of mutated skeletal stem cells. The latter would generate a pool of transiently amplifying progenitors, consistent with the observed expansion of the clonogenic and the stromal cell pools in FD. This would also be consistent with a true depletion of mutant skeletal stem cells over time. This hypothesis implies that a regulatory pathway disrupted by activating GNAS mutations may play a significant role in regulating stem cell self‐renewal. This notion is supported by recent findings of skeletal stem cell depletion in a transgenic mouse model with a constitutively active PTH/PTHrP receptor under control of the 2.3‐kb Col1A1 promoter, which is upstream of Gsα in the same pathway, and leads to development of a FD‐like osteogenic phenotype.
Our data have several implications for the therapy of FD lesions. First, and most simply, they show that the natural history of FD is anchored to specific, and biologically quite distinct, temporal phases, which must be taken into account when considering surgical, medical, or other (e.g., cell therapy) modes of intervention. Second, they show that the normal process of bone remodeling can turn FD bone into normal bone over time. Because abating bone remodeling is a basic tenet of current medical approaches to FD treatment (use of bisphosphonates), further work is warranted to clarify whether in fact this is beneficial in the long term. Third, the very notion of skeletal stem cells opens the prospect of stem cell therapy for genetic diseases of the skeleton, as for other genetic diseases of connective tissues. Our data show an example of stem cell therapy for a genetic disease of the skeleton, brought about by nature itself. Eradication of mutant stem cells and their replacement by normal ones (that is, what one would wish to do in a cell therapy approach) occurs spontaneously in (some, at least) FD lesions. However, this spontaneous process spans several decades, possibly as a reflection of the naturally slow rate of tissue turnover in bone compared with other tissues. Correction of FD lesions through stem cells should then not only aim for long‐term reconstitution of the skeletal tissues but also that this would take place faster than what nature can do itself. How to accomplish these two goals remains the challenge in treatment of this disease.
Acknowledgements
This work was supported by Telethon Grant GGP0463 (to PB), MIUR (PB, MR), AIRL (PB), and the DIR, NIDCR of the IRP, NII, DHHS (to SAK, NC, MTC, and PGR).
The authors state that they have no conflicts of interest.
REFERENCES
- 1. Bianco P, Riminucci M, Majolagbe A, Kuznetsov SA, Collins MT, Mankani MH, Corsi A, Bone HG, Wientroub S, Spiegel AM, Fisher LW, Robey PG 2000. Mutations of the GNAS1 gene, stromal cell dysfunction, and osteomalacic changes in non‐McCune‐Albright fibrous dysplasia of bone. J Bone Miner Res 15: 120–128. [DOI] [PubMed] [Google Scholar]
- 2. Albright F, Butler AM, Hampton AO, Smith P 1937. Syndrome characterized by osteitis fibrosa disseminata, areas of pigmentation and endocrine dysfunction with precocious puberty in females. N Engl J Med 216: 727–746. [Google Scholar]
- 3. Schwindinger WF, Francomano CA, Levine MA 1992. Identification of a mutation in the gene encoding the alpha subunit of the stimulatory G protein of adenylyl cyclase in McCune‐Albright syndrome. Proc Natl Acad Sci USA 89: 5152–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM 1991. Activating mutations of the stimulatory G protein in the McCune‐Albright syndrome. N Engl J Med 325: 1688–1695. [DOI] [PubMed] [Google Scholar]
- 5. Shenker A, Weinstein LS, Sweet DE, Spiegel AM 1994. An activating Gs alpha mutation is present in fibrous dysplasia of bone in the McCune‐Albright syndrome. J Clin Endocrinol Metab 79: 750–755. [DOI] [PubMed] [Google Scholar]
- 6. Happle R 1986. The McCune‐Albright syndrome: A lethal gene surviving by mosaicism. Clin Genet 29: 321–324. [DOI] [PubMed] [Google Scholar]
- 7. Bianco P, Robey PG, Wientroub S 2003. Fibrous dysplasia of bone Glorieux FH, Pettifor JM, Juppner H. (eds.) Pediatric Bone: Biology and Disease. Academic Press, New York, NY, USA, 509–540. [Google Scholar]
- 8. Bianco P, Kuznetsov SA, Riminucci M, Fisher LW, Spiegel AM, Robey PG 1998. Reproduction of human fibrous dysplasia of bone in immunocompromised mice by transplanted mosaics of normal and Gsalpha‐mutated skeletal progenitor cells. J Clin Invest 101: 1737–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Riminucci M, Fisher LW, Shenker A, Spiegel AM, Bianco P, Gehron Robey P 1997. Fibrous dysplasia of bone in the McCune‐Albright syndrome: Abnormalities in bone formation. Am J Pathol 151: 1587–1600. [PMC free article] [PubMed] [Google Scholar]
- 10. Riminucci M, Liu B, Corsi A, Shenker A, Spiegel AM, Robey PG, Bianco P 1999. The histopathology of fibrous dysplasia of bone in patients with activating mutations of the Gs alpha gene: Site‐specific patterns and recurrent histological hallmarks. J Pathol 187: 249–258. [DOI] [PubMed] [Google Scholar]
- 11. Friedenstein AJ 1980. Stromal mechanisms of bone marrow: Cloning in vitro and retransplantation in vivo. Hamatol Bluttransfus 25: 19–29. [DOI] [PubMed] [Google Scholar]
- 12. Owen M, Friedenstein AJ 1988. Stromal stem cells: Marrow‐derived osteogenic precursors. Ciba Found Symp 136: 42–60. [DOI] [PubMed] [Google Scholar]
- 13. Bianco P, Gehron Robey P 2000. Marrow stromal stem cells. J Clin Invest 105: 1663–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krebsbach PH, Kuznetsov SA, Satomura K, Emmons RV, Rowe DW, Gehron Robey P 1997. Bone formation in vivo: Comparison of osteogenesis by transplanted mouse and human marrow stromal fibroblasts. Transplantation 63: 1059–1069. [DOI] [PubMed] [Google Scholar]
- 15. Jilka RL, Weinstein RS, Bellido T, Parfitt AM, Manolagas SC 1998. Osteoblast programmed cell death (apoptosis): Modulation by growth factors and cytokines. J Bone Miner Res 13: 793–802. [DOI] [PubMed] [Google Scholar]
- 16. Kuznetsov SA, Krebsbach PH, Satomura K, Kerr J, Riminucci M, Benayahu D, Robey PG 1997. Single‐colony derived strains of human marrow stromal fibroblasts form bone after transplantation in vivo. J Bone Miner Res 12: 1335–1347. [DOI] [PubMed] [Google Scholar]
- 17. Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I, Tagliafico E, Ferrari S, Robey PG, Riminucci M, Bianco P 2007. Self‐renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 131: 324–336. [DOI] [PubMed] [Google Scholar]
- 18. Lichtenstein L, Jaffe HL 1942. Fibrous dysplasia of bone: A condition affecting one, several or many bones, the graver cases of which may present abnormal pigmentation of skin, premature sexual development, hyperthyroidism and still other extraskeletal abnormalities. Arch Pathol 33: 777–797. [Google Scholar]
- 19. Grabias SL, Campbell CJ 1977. Fibrous dysplasia. Orthop Clin North Am 8: 771–783. [PubMed] [Google Scholar]
- 20. Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron Robey P 2003. FGF‐23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest 112: 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bianco P, Robey PG 1999. An animal model of fibrous dysplasia. Mol Med Today 5: 322–323. [DOI] [PubMed] [Google Scholar]
- 22. Alman BA, Greel DA, Wolfe HJ 1996. Activating mutations of Gs protein in monostotic fibrous lesions of bone. J Orthop Res 14: 311–315. [DOI] [PubMed] [Google Scholar]
- 23. Marie PJ, de Pollak C, Chanson P, Lomri A 1997. Increased proliferation of osteoblastic cells expressing the activating Gs alpha mutation in monostotic and polyostotic fibrous dysplasia. Am J Pathol 150: 1059–1069. [PMC free article] [PubMed] [Google Scholar]
- 24. Riminucci M, Kuznetsov SA, Cherman N, Corsi A, Bianco P, Gehron Robey P 2003. Osteoclastogenesis in fibrous dysplasia of bone: In situ and in vitro analysis of IL‐6 expression. Bone 33: 434–442. [DOI] [PubMed] [Google Scholar]
- 25. Corsi A, Collins MT, Riminucci M, Howell PG, Boyde A, Robey PG, Bianco P 2003. Osteomalacic and hyperparathyroid changes in fibrous dysplasia of bone: Core biopsy studies and clinical correlations. J Bone Miner Res 18: 1235–1246. [DOI] [PubMed] [Google Scholar]
- 26. Kuznetsov SA, Riminucci M, Ziran N, Tsutsui TW, Corsi A, Calvi L, Kronenberg HM, Schipani E, Robey PG, Bianco P 2004. The interplay of osteogenesis and hematopoiesis: Expression of a constitutively active PTH/PTHrP receptor in osteogenic cells perturbs the establishment of hematopoiesis in bone and of skeletal stem cells in the bone marrow. J Cell Biol 167: 1113–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]