

Abstract
PDGFR inhibitors are successfully used in a number of cancer treatments. The standard treatment for acute promyelocytic leukemia (APL) involves differentiation therapy with retinoic acid (RA). However, the relapse rates are significant. In the present work we evaluated the effects of RA therapy in the presence of PDGFR inhibitor, AG1296. Adding AG1296 with RA increased secretion of TNF-α, IL-8, and MMP-9 expression. This treatment induced higher levels of ICAM-1 endothelial cell expression, and increased cellular mobility. Inhibiting PDGFR enhanced RA-induced expression of integrin. Integrin ligand increased differentiation markers CD11b, inducible oxidative metabolism and PDGFR-â phosphorylation. While the neutrophil- endothelial cell interactions are strengthened by the combined treatment, the endotheliumsubstratum interactions are weakened, a situation common in RAS.
Introduction
The standard clinical treatment of acute promyelocytic leukemia (APL) is differentiation induction therapy using all trans-retinoic acid (RA). Differentiation therapy results in phenotypic conversion of promyelocytic cells into mature neutrophils. This conversion is characterized by CD38 and CD11b expression, cell cycle arrest in G0 and ability of mature neutrophils to perform a respiratory burst. RA treatment results in complete remission in 92 – 95% of patients [1].However, this therapeutic approach has several limitations such as the development of resistance to RA, retinoic acid syndrome (RAS) or hyperleukocytosis [2]. RAS, also known as APL differentiation syndrome, currently occurs in 10% – 15% of patients and is fatal in approximately 10% of them [3]. It is caused by the RA-mediated conversion of APL blast cells to mature neutrophils. These neutrophils are abundant and express cytokines, such as interleukin (IL)-1β, IL-8, interferon-γ (INF-γ) and tumor necrosis factor-α (TNF-α) [4]. Endothelial cells are exposed to these cytokines in the blood stream and respond by expressing adhesion molecules which subsequently facilitate the binding of neutrophils to endothelial cells (reviewed in [5]) and by increasing the endothelial cell’s mobility leading to increased leukocyte infiltration and lung capillary leak. CD11b expressed on neutrophils are key components for neutrophil binding to endothelial cells expressing ICAM. Neutrophils migrate through the endothelium by expressing extracellular matrix degrading enzymes, such as matrix metallo-proteinases (MMPs), and increasing their motility on and through endothelial cells via cytoskeletal rearrangements that are triggered by signals from the focal adhesion kinase (FAK) upon adhesion [6] This process leads to the impairment of the endothelial barrier with leukocyte infiltration characterizing the cardio-pulmonary distress during RAS.
HL-60 cells are human myeloblastic leukemia cells that serve as a model for studying differentiation induction therapy [7]. These cells undergo growth arrest and myeloid differentiation in response to retinoic acid (RA) [8]. We have recently reported that AG1296, a potent and selective inhibitor for PDGFR and its family members, Kit and Flt3 [9], enhances various facets of both RA induced myeloid differentiation, including inducible oxidative metabolism [10]. However, other features typical of these induced differentiation programs were diminished, including induced G0 cell cycle arrest. Because we observed that PDGFR inhibition during RA induced differentiation resulted in increased adherence of cells to tissue culture surfaces and an increase in CD11b expression, which is a cell surface molecule that heterodimerizes with the integrin receptor, we hypothesized that the effect of AG1296 on differentiation was partly mediated by the cross talk of the PDGFR with the integrin receptor and might have implications during RAS.
To achieve a higher rate of complete remission in clinical treatment of APL, RA is administered in combination with chemotherapy [3]. However, the impact of the drug – drug interaction on RAS is unclear. Here, we report that the presence of the platelet derived growth factor receptor (PDGFR) inhibitor AG1296 during RA-induced myeloid differentiation results in neutrophils that excrete higher levels of IL-8 and TNF-α, express higher levels of MMP-9 and subsequently induce higher levels of ICAM-1 expression on endothelial cells. The results of this study suggest a higher risk for RAS in patients that receive PDGFR inhibitors during differentiation induction therapy and that the PDGFR is involved in suppressing features that contribute to RAS. The findings also suggest a potential in vitro model for RAS.
Materials and Methods
Cell culture
HL-60 human myeloblastic leukemia cells and EAhy 926 human macrovascular endothelial cells were grown in RPMI 1640 and DMEM, respectively. Media were supplemented with 5% heat-inactivated fetal bovine serum (both: Invitrogen, Carlsbad, CA) and 1x antibiotic/antimicotic (Sigma, St. Louis, MO) in a 5% CO2 humidified atmosphere at 37°C. RA (Sigma, St. Louis, MO) was added from a 0.5 mM stock solution in ethanol with a final concentration of 2 µM in culture. AG1296 and integrin αMβ2 ligand stabilizing integrin αMβ2 in its active state [11] (Calbiochem, La Jolla, CA) were added as carrier blanks to cell cultures at the same time as RA treatment at a concentration of 10 µM from stock solutions (20 mM) in DMSO. Equal amounts of DMSO were added to all treatment groups not receiving AG1296 or integrin ligand. Experimental cultures were initiated at a density of 0.2 × 106 cells/ml. Viability was monitored by 0.2% trypan blue (Invitrogen, Calsbad, CA) exclusion and routinely exceeded 95% throughout the time frame of the experiments.
CD11b, CD18, CD38, MMP-9, and pPDGFR-β expression studies
0.5 × 106 cells were collected from cultures and centrifuged at 1000 rpm in a microfuge for 5 min. Cell pellets were resuspended in 100 µl 37°C PBS containing 5 µl of the respective antibody (APC conjugated CD11b, FITC conjugated CD18, PE conjugated CD38 (all from BD Biosciences, San Jose, CA). Following 1h incubation at 37°C cell surface expression levels were analyzed with a BD LSRII flow cytometer (BD Biosciences, San Jose, CA). To stain for MMP-9, PDGFR pY1021 we used primary antibodies from Biovision, Mountain View, CA, Cell Signaling, Danvers, MA, and Santa Cruz Biotechnology, CA, respectively. 0.5 × 106 cells were collected from cultures and centrifuged at 1000 rpm in a microfuge for 5 min, fixed, and permeabilized by resuspension of the pellet in 100 µl 2% paraformaldehyde in PBS for 10 min at room temperature followed by the addition of 900 µl 100% ice cold methanol to obtain a 90% methanol solution. Following an incubation for 20 min at −20°C samples were washed 2 times in PBS and resuspended in 100 µl PBS containing 1µl of respective primary antibody. Following a 1h incubation period cells were washed once with 1 ml PBS and staining with secondary FITC-conjugated anti-rabbit antibody (BD Biosciences, CA) for 1h and analyzed by flow cytometry. For CD11b, CD18, and CD38 undifferentiated control cells were used to determine the fluorescence intensity of cells negative for the respective surface antigen. The gate to determine percent increase of expression was set to exclude 95% of the control population. Results for MMP-9, and pPDGFR-β are given as mean fluorescence intensity as determined by quantifying the fluorescence intensity of the entire cell population sample (30,000 cells) by flow cytometry. Cells unstained with the primary antibody but incubated with the secondary antibody were used to generate the background signal.
Measurement of inducible oxidative metabolism
0.5 × 106 cells were collected and centrifuged at 1000 rpm for 5 min in a microfuge. Cell pellets were resuspended in 200 µl 37°C PBS containing 5 µM 5-(and-6)-chloromethyl-2’,7’-dichlorodihydro–fluorescein diacetate acetyl ester (H2-DCF, Molecular Probes, Eugene, OR) and 0.2 µg/ml 12-o-tetradecanoylphorbol-13-acetate (TPA, Sigma, St. Louis, MO). Both, H2-DCF and TPA stock solutions were made in DMSO at concentrations of 0.2 mg/ml and 5 mM, respectively. A control group incubated in H2-DCF and DMSO only was included. Cells were incubated for 20 min at 37°C prior to analysis by flow cytometry. Oxidized DCF was excited by a 488 nm laser and emission collected through a 505 long pass dichroic mirror and a 530/30 nm band pass filter. The shift in fluorescence intensity in response to TPA was used to determine the percent cells with the capability to generate inducible oxidative metabolites. Gates to determine percent positive cells were set to exclude 95% of control cells not stimulated with TPA.
TNF-α and IL-8 secretion
Cells were treated or not with 2 µM RA plus or minus AG1296 for 72 h. 2 million cells were pelleted and resuspended in fresh cell culture media to account for cell treatment groups growing at slightly different rates. After 5 h cells were pelleted again and the cell culture media was stored at −80°C until further analysis. ELISAs were performed according to the manufacturer’s instructions of ELISA MAX™ Set Standard reagent sets for human TNF-α and human IL-8 (BioLegend, San Diego, CA). We used 100 µl of collected media for TNF-α and 10 µl for IL-8 analyses. HRP-mediated conversion of TMB substrate (BioLegend, San Diego, CA) was determined reading absorbance at 450 nm using a plate reader (BioRad 680). Values obtained from optical absorbance were converted to values in pg/ml using the optical absorbances obtained from standards with known concentrations.
ICAM-1 expression on endothelial cells and Endothelial cell migration
To determine cytotoxic effects of neutrophils on endothelial cells we grew endothelial cells for 3 days in 6 well plates to form a 90% confluent endothelial cell layer. Endothelial cells were subsequently exposed to 2 million HL-60 cells in 2 ml of fresh cell culture media. HL-60 cells had been treated or not with 2 µM RA for 72 h plus or minus 10 µM AG1296. After 24 h of co-culturing endothelial and HL-60 cells, endothelial cells were collected using an enzyme-free cell dissociation buffer (Invitrogen, Carlsbad, CA). For the ICAM expression, equal amounts of cells were stained with APC-conjugated ICAM-1 (Biolegend, San Diego, CA) and PE-conjugated endothelial cell marker CD144 to distinguish between endothelial and HL-60 cells (eBioscience, San Diego, CA). Cells were analyzed using a BD Biosciences LSRII flow cytometer. Only cells staining positive for CD144 were included in the quantification of ICAM-1.
A characteristic of RAS is the weakening of the endothelial cell-cell and cell-substratum interactions. In order to assess these phenomena, an endothelial cell migration assay (wounding assay) was performed. Confluent endothelial cell monolayers (in 6 well plates) were wounded with a pipette tip, each well was washed with PBS and then the HL-60 cells were added as described above. After 24h of coculture, the cell suspension was removed, the monolayer was washed with PBS and the pictures were taken. Cells that had migrated to the wounded areas were counted for quantification of cell migration.
Statistical Analysis
Statistical analyses were performed using SPSS 11.0 for Windows, Student Edition. Means of treatment groups of interest were compared using the Paired-Samples T Test. All treatment groups were compared to control cells. The effect of AG1296 was determined by comparing groups receiving RA with groups receiving RA plus AG1296. A p-value of < 0.05 was considered significant.
Results
AG1296 treated HL-60 undergoing myeloid differentiation have enhanced secretion of TNF-α and IL-8 and increased expression of MMP-9
During RAS neutrophils elicit their cytotoxic effects by releasing cytokines such as TNF-α and IL-8. In addition, neutrophils express extracellular matrix degrading enzymes, such as metalloproteinase-9 (MMP-9) to facilitate their invasion into subendothelial tissues.
We quantified TNF-α and IL-8 secretion of HL-60 cells treated for 72 h with RA plus and minus AG1296. RA treated cells showed significantly increased TNF-α (p=0.034; Figure 1A) and IL-8 (p=0.009; Figure 1B) secretion. When cells received AG1296 in addition to RA we found a significant increase in TNF-α (p=0.012) and IL-8 (p=0.001) secretion when compared to cells receiving RA alone and when compared to control cells (p< 0.010).
Figure 1. AG1296 increases the secretion of TNF-α and IL-8, and MMP-9 protein expression in myeloid differentiated HL-60 cells.



TNF-a (A) and IL-8 (B) secretion was determined by ELISA from cells that were treated or not with RA plus or minus 10 µM AG1296 for 72 h. MMP-9 (C) expression was determined by flow cytometry following 72 h RA-meditated myeloid differentiation in the absence or presence of 10 µM AG1296. Shown are means with error bars representing standard errors of means from 3 independent experiments. The insert shows MMP-9 histograms for a typical experiment. (*) indicates treatment groups that were significantly different from control and (#) indicates RA plus AG1296 treated groups that were significantly different from cells receiving RA only. C, control; A, 10 µM AG1296; R, 2 µM retinoic acid; R + A, 10 µM AG1296 plus 2 µM retinoic acid; R sus, retinoic acid treated suspension cells; R adh, retinoic acid treated adherent cells; R + A sus, retinoic acid plus AG1296 treated suspension cells; RA adh, retinoic acid plus AG1296 treated adherent cells;
MMP-9 expression was measured in cells treated for 72 h with RA plus or minus AG1296. We separated cells in suspension from adherent cells as MMP-9 is predominantly expressed in adherent cells. We have determined previously [10] that AG1296 causes a higher percentage of differentiated HL-60 cells to adhere to substrates and hence we wanted to account for that difference by separating suspension from adhering cells rather than taking a pooled sample. Cells treated with RA plus AG1296 that adhered to substrate 72 h post treatment had significantly (p=0.029) higher MMP-9 protein expression compared to adherent cells treated with RA alone (Figure 1C). Suspension cells that were differentiated with RA or RA plus AG1296 did not have MMP-9 expression above control levels.
These data suggest that inhibiting the PDGFR during RA-mediated differentiation results in neutrophils that are potentially more cytotoxic to endothelial cells and could hence increase the risk of resulting in RAS.
HL-60 cells treated with AG1296 during myeloid differentiation induce higher ICAM-1 levels in endothelial cells
Because the AG1296 mediated increase in TNF-α and IL8 secretion and MMP-9 expression suggested increased cytotoxic effects, we aimed to determine endothelial cell activation after exposure to HL-60 cells. When endothelial cells grown to form a monolayer for 72 h were exposed for 24 h to undifferentiated HL-60 cells (untreated control HL-60 cells and AG1296 treated HL-60 cells), endothelial cells did not express ICAM-1 significantly above endothelial cells that had not been exposed to HL-60 cells (Figure 2A). However, HL-60 cells, induced to differentiate for 72 h with RA, induced ICAM-1 expression in 30.02% endothelial cells. HL-60 cells induced to differentiate by treating for 72 h with RA plus AG1296 induced ICAM-1 expression in 48.88% endothelial cells which was significantly higher than in endothelial cells exposed to HL-60 cells treated with RA alone (p=0.0003).
Figure 2. HL-60 cells treated with AG1296 during myeloid differentiation induce ICAM-1 expression in endothelial cells.

HL-60 cells were treated with RA in absence or presence of 10 µM AG1296 for 72 h. A total of 2 million HL-60 cells in 2 ml of media were pipetted on a monolayer of EAhy926 endothelial cells that were grown in a 6 well plate for 72h. Following 24h of co-culture both cell types were removed from the culture dish and stained with ICAM-1 and endothelial cell marker CD144 to distinguish the two cell types. Error bars representing standard errors of means from 6 independent experiments. The insert shows ICAM-1 histograms for a typical experiment. (*) indicates treatment groups that were significantly different from control and (#) indicates RA plus AG1296 treated groups that were significantly different from cells receiving RA only. C, control; A, 10 µM AG1296; R, 2 µM retinoic acid; R + A, 10 µM AG1296 plus 2 µM retinoic acid;
ICAM-1 is expressed in activated endothelial cells and is involved in mediating the migration of neutrophils into subendothelial tissues [5]. AG1296 treatment during myeloid differentiation might therefore lead to enhanced migration of neutrophils through the endothelial barrier.
Stabilizing integrin αMβ2 had similar effects on differentiation as AG1296
Because the PDGFR inhibitor AG1296 caused adhesion, we suspected integrins, molecules that mediate cell adhesion, may mediate some of the effects we observed using AG1296 during myeloid differentitation. To evaluate the role of integrins during myeloid differentiation of HL-60 cells, we used an integrin ligand that stabilizes the active form of integrin αMβ2 by binding to its intracellular domain. Cells that were treated with RA plus integrin ligand had slightly lower CD38 expression after 24 h when compared to cells that received RA only (Figure 3A). CD38 is the earliest marker of differentiation along the neutrophil lineage. 72 h co-treatment of HL-60 cells with RA and integrin ligand significantly (p=0.0238) reduced the induced G0 arrest when compared to cells treated with RA alone (Figure 3B). In contrast, CD11b expression after 48 h was significantly (p=0.032) higher in cells that were treated with RA plus integrin ligand when compared to cells that were treated with RA alone (Figure 3C). The integrin ligand also significantly (p=0.023) enhanced RA-induced capability for inducible oxidative metabolism compared to cells that received only RA for 48h (Figure 3D). The mature neutrophils are characterized by ability to perform inducible oxidative metabolism.
Figure 3. HL-60 cells treated with integrin αMβ2 ligand during myeloid differentiation have enhanced features of myeloid differentiation.




HL-60 cells were treated or not with RA plus or minus integrin ligand for 24 h, 48 h, and 72 h, respectively. CD38 expression at 24 h (A) and G0 arrest at 72 h (B) were decreased in cells treated with RA plus integrin ligand when compared to cells treated with RA alone. CD11b (C) expression at 48 h and the capability of oxidative metabolism at 48 h (D) were increased in cells treated with RA plus integrin ligand when compared to cells treated with RA alone. Shown are means with error bars representing standard errors of means from 3 independent experiments. The insert shows CD38 histograms (panel A), DNA histograms (panel B), CD11b histograms (panel C) for a typical experiment. (*) indicates treatment groups that were significantly different from control and (#) indicates RA plus AG1296 treated groups that were significantly different from cells receiving RA only. C, control; L, 5 µM integrin αMβ2 ligand; R, 2 µM retinoic acid; R + L, 5 µM integrin αMβ2 ligand plus 2 µM retinoic acid.
These data revealed that the integrin ligand had the same effect on differentiation markers as AG1296 has, namely an increase in CD11b and oxidative metabolism. This further indicates that some of the effects we observed with the PDGFR inhibitor AG1296 were mediated through adhesion molecules such as integrin αMβ2 and points to a negative cross talk between PDGFR and integrin regulated pathways.
AG1296 increases CD18 expression and integrin αMβ2 ligand enhances PDGFR-β phosphorylation
Since using a PDGFR inhibitor or an integrin ligand resulted in the same outcome in regards to myeloid differentiation we decided to further investigate a possible negative crosstalk between PDGFR and integrin. When we measured integrin β2 expression in RA treated HL-60 cells receiving AG1296 during differentiation we found integrin expression to be significantly increased (p=0.0147) compared to cells that received RA alone (Figure 4A). However, when cells were treated with the integrin ligand in addition to RA, PDGFR-β phosphorylation was enhanced at Y1021 activation site when compared to cells that were treated with RA alone (Figure 4B).
Figure 4. Inhibiting PDGFR enhances integrin 2β expression while activating integrin enhances PDGFR-β phosphorylation.


Panel (A) depicts integrin 2β expression following treatment with RA plus or minus AG1296 for 48 h. Panel (B) shows PDGFR-β Y1021 phosphorylation in HL-60 cells following treatment with RA plus or minus integrin αMβ2 ligand for 72 h. Shown are means with error bars representing standard errors of means from 3 independent experiments. The insert shows PDGFR-β Y1021 phosphorylation histograms for a typical experiment. (*) indicates treatment groups that were significantly different from control and (#) indicates RA plus AG1296 treated groups that were significantly different from cells receiving RA only. C, control; A, 10 µM AG1296; R, 2 µM retinoic acid; R + A, 10 µM AG1296 plus 2 µM retinoic acid; L, 5 µM integrin αMβ2 ligand; R + L, 5 µM integrin αMβ2 ligand plus 2 µM retinoic acid.
These data show that a potential negative crosstalk between the PDGF and integrin receptors is not reciprocal. Enhancing the activated state of the integrin receptor enhanced PDGFR-β phosphorylation but inhibiting the PDGFR using AG1296 enhanced the expression of integrin β2.
AG1296 in conjunction with retinoic acid increases the mobility of endothelial cells
Treating the leukemic HL-60 with PDGFR inhibitor AG-1296 along with retinoic acid caused increased endothelial cell motility, a feature of the lung capillary leak. RA-or AG1296- treated HL-60 cells tended to increase endothelial motility compared to the motility induced by the control HL-60, however statistical significance was not reached (p= 0.08 when comparing control to RA treated cells). The motility induced by both agents is significantly higher than the mobility induced by the control HL-60 (p= 0.002) or by the retinoic acid alone (p= 0.03) - Figure 5.
Figure 5. Inhibiting PDGFR simultaneously with RA induced differentiation enhances endothelium mobility.
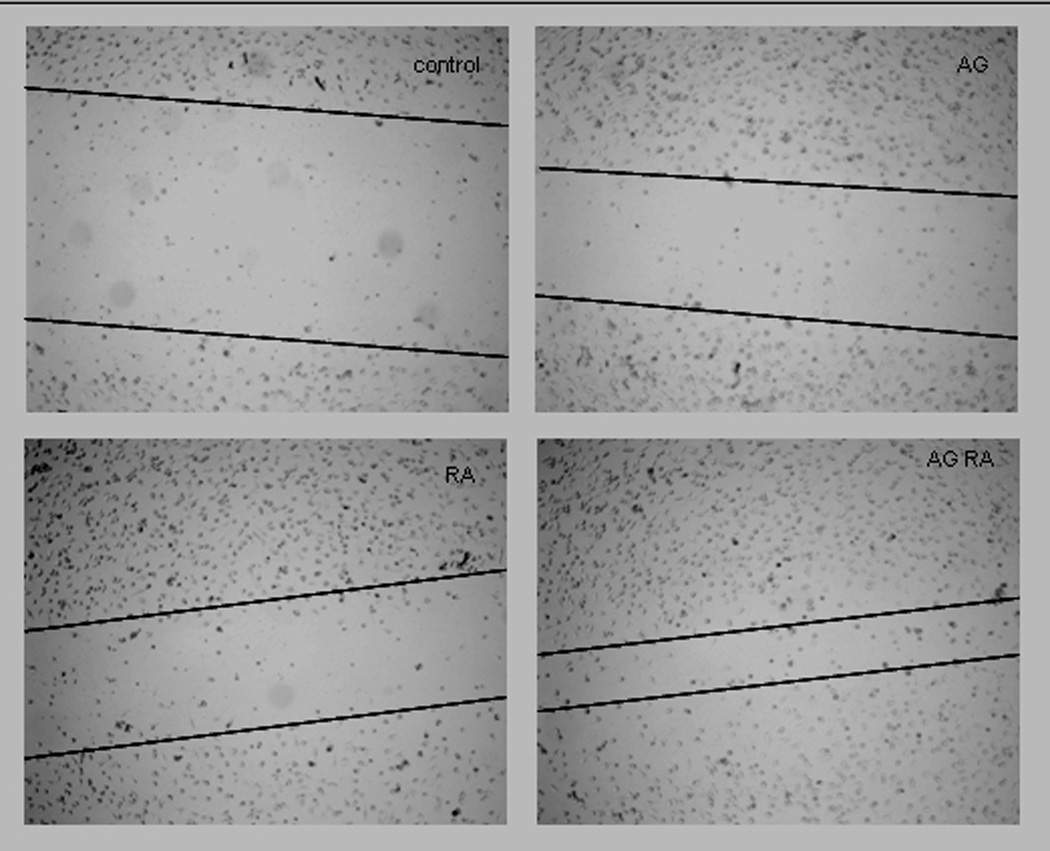

HL-60 cells were treated with RA in absence or presence of 10 µM AG1296 for 72 h. A total of 2 million HL-60 cells in 2 ml of fresh media were pipetted on a monolayer of EAhy926 endothelial cells that were grown in a 6 well plate for 72h, wounded with a pipette tip and washed extensively with PBS prior to addition of HL-60 cells. Following 24h of co-culture, the cell suspension was removed, the endothelial layer was washed with PBS at 37C. One milliliter of the warm PBS was added to the wells in order to take the pictures. Figure 5A shows endothelial cell migration, in a representative experiment, after exposure to control, AG1296, RA, or RA plus AG1296 treated HL-60 and the figure 5B presents the quantization resulting from 3 independent experiments.
Discussion
Here we report that the presence of the PDGFR inhibitor AG1296 increases signs of RAS and that the effects seen with the PDGFR inhibitor could be, at least in part, due to the interaction of the PDGFR receptor with the integrin receptor signaling pathway.
A negative cross talk between the PDGFR and adhesion has been suggested before. For example, it was reported that cell adhesion induces ubiquitin-mediated degradation of the PDGFR-β [12]. Zaslavsky et al. found that spingosin-1-phosphate induced cell detachment in HEK293 cells which could be reversed by AG1296 or engaging β1 integrin into an active state [13]. The precise mechanism of the PDGFR – integrin cross talk is not known. However, growth factors, such as the macrophage colony-stimulating factor and the hepatocyte growth factor have been reported to induce inside-out signaling of integrins via their intracellular tails [14].
Using a ligand that stabilizes the active form of αMβ2 integrin we found a distinct effect of integrin signaling during myeloid differentiation. Integrins have been implicated in the differentiation of cell types that are adhesion dependent. For example, the differentiation of myofibroblasts [15] and chondrocytes [16] has been shown to be dependent on integrin signaling. The role of the integrin in the differentiation of non-adherent cells is less obvious. However in the hematopoetic linage the integrin receptor has been found to be important in regulating the differentiation of monocytes to macrophages [17] and CD16 positive monocytes to osteoclasts [18]. Because some differentiation markers, such as inducible oxidative metabolism and CD11b expression were enhanced while some, such as CD38 expression and G0/G1 arrest were marginally affected in the presence of the integrin ligand, we can not assume that integrins by themselves are instrumental during RA-induced myeloid differentiation. Rather, integrin signaling might interfere with or enhance certain aspects of the differentiation program.
As for the effect of the integrin ligand, we believe that the PDGFR signaling is interfering with RA-induced myeloid differentiation that results in the upregulation of differentiation markers such as CD11b or oxidative metabolism. The net effect however, appears to increase signs of RAS. Here we report that inhibiting the PDGFR with AG1296 during RA-induced differentiation increases integrin CD18 and MMP-9 expression. CD18 on neutrophils can firmly bind to ICAM-1 on endothelial cells [19]. MMP-9 is a metalloproteinase capable of basement membrane degradation and hence facilitates neutrophil migration through the endothelium (reviewed in [20]. The overall effect of PDGFR inhibitor and RA coadministration is an increased attachement of the activated neutrophils to the endothelium and an increased cell mobility for the endothelium. AG1296 enhances the expression and secretion of cytokines TNF-α and IL-8, both of which have been implicated in RAS [4]. These cytokines act on endothelial cells, causing them to express adhesion molecules that neutrophils subsequently bind to. Here we show that endothelial cells exposed to HL-60 cells receiving AG1296 during RA-mediated myeloid differentiation had elevated ICAM-1 expression levels. ICAM-1 is one of the main integrin ligands involved in leukocyte transendothelial migration. It also acts as a signal transducer leading to actin cytoskeleton remodeling in endothelial cells which in turn facilitates neutrophil invasion [5]. Collectively these could contribute to the pathology of RAS.
The platelet derived growth factor receptor (PDGFR) has now been implicated in a variety of cancers and leukemias [21–24] and is targeted by pharmaceuticals such as imatinib mesylate (Gleevec), an inhibitor of the PDGFR family, Abl and Arg tyrosine kinases [25]. The data of this study indicate that the presence of PDGFR inhibitors during differentiation induction therapy in APL might increase the risk of the patient to suffer from RAS that are mediated through enhanced adherence of neutrophils to endothelial cells, cytokine expression and invasion of the endothelium.
Acknowledgments
Supported by grants from USPHS NIH (CA33505) and NYSTEM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang ZY, Chen Z. Differentiation and apoptosis induction therapy in acute promyelocytic leukaemia. Lancet Oncol. 2000;1:101–106. doi: 10.1016/s1470-2045(00)00017-6. [DOI] [PubMed] [Google Scholar]
- 2.Tallman MS. Differentiating therapy with all-trans retinoic acid in acute myeloid leukemia. Leukemia. 1996;10 Suppl 1:S12–S15. [PubMed] [Google Scholar]
- 3.Fenaux P, Wang ZZ, Degos L. Treatment of acute promyelocytic leukemia by retinoids. Curr Top Microbiol Immunol. 2007;313:101–128. doi: 10.1007/978-3-540-34594-7_7. [DOI] [PubMed] [Google Scholar]
- 4.Hsu HC, Tsai WH, Chen PG, Hsu ML, Ho CK, Wang SY. In vitro effect of granulocyte-colony stimulating factor and all-trans retinoic acid on the expression of inflammatory cytokines and adhesion molecules in acute promyelocytic leukemic cells. Eur J Haematol. 1999;63:11–18. doi: 10.1111/j.1600-0609.1999.tb01844.x. [DOI] [PubMed] [Google Scholar]
- 5.Hordijk PL. Endothelial signalling events during leukocyte transmigration. Febs J. 2006;273:4408–4415. doi: 10.1111/j.1742-4658.2006.05440.x. [DOI] [PubMed] [Google Scholar]
- 6.Cox BD, Natarajan M, Stettner MR, Gladson CL. New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. J Cell Biochem. 2006;99:35–52. doi: 10.1002/jcb.20956. [DOI] [PubMed] [Google Scholar]
- 7.Yen A, Forbes M, DeGala G, Fishbaugh J. Control of HL-60 cell differentiation lineage specificity, a late event occurring after precommitment. Cancer Res. 1987;47:129–134. [PubMed] [Google Scholar]
- 8.Yen A, Brown D, Fishbaugh J. Control of HL-60 monocytic differentiation. Different pathways and uncoupled expression of differentiation markers. Exp Cell Res. 1987;168:247–254. doi: 10.1016/0014-4827(87)90432-0. [DOI] [PubMed] [Google Scholar]
- 9.Levitzki A. PDGF receptor kinase inhibitors for the treatment of PDGF driven diseases. Cytokine Growth Factor Rev. 2004;15:229–235. doi: 10.1016/j.cytogfr.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 10.Reiterer G, Yen A. Platelet-derived growth factor receptor regulates myeloid and monocytic differentiation of HL-60 cells. Cancer Res. 2007;67:7765–7772. doi: 10.1158/0008-5472.CAN-07-0014. [DOI] [PubMed] [Google Scholar]
- 11.Bjorklund M, Aitio O, Stefanidakis M, Suojanen J, Salo T, Sorsa T, Koivunen E. Stabilization of the activated alphaMbeta2 integrin by a small molecule inhibits leukocyte migration and recruitment. Biochemistry. 2006;45:2862–2871. doi: 10.1021/bi052238b. [DOI] [PubMed] [Google Scholar]
- 12.Baron V, Schwartz M. Cell adhesion regulates ubiquitin-mediated degradation of the platelet-derived growth factor receptor beta. J Biol Chem. 2000;275:39318–39323. doi: 10.1074/jbc.M003618200. [DOI] [PubMed] [Google Scholar]
- 13.Zaslavsky A, Li S, Xu Y. Sphingosine-1-phosphate induces a PDGFR-dependent cell detachment via inhibiting beta1 integrin in HEK293 cells. FEBS Lett. 2005;579:3899–3906. doi: 10.1016/j.febslet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 14.Faccio R, Novack DV, Zallone A, Ross FP, Teitelbaum SL. Dynamic changes in the osteoclast cytoskeleton in response to growth factors and cell attachment are controlled by beta3 integrin. J Cell Biol. 2003;162:499–509. doi: 10.1083/jcb.200212082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lygoe KA, Wall I, Stephens P, Lewis MP. Role of vitronectin and fibronectin receptors in oral mucosal and dermal myofibroblast differentiation. Biol Cell. 2007;99:601–614. doi: 10.1042/BC20070008. [DOI] [PubMed] [Google Scholar]
- 16.Woods A, Wang G, Beier F. Regulation of chondrocyte differentiation by the actin cytoskeleton and adhesive interactions. J Cell Physiol. 2007;213:1–8. doi: 10.1002/jcp.21110. [DOI] [PubMed] [Google Scholar]
- 17.Sudhakaran PR, Radhika A, Jacob SS. Monocyte macrophage differentiation in vitro: Fibronectin-dependent upregulation of certain macrophage-specific activities. Glycoconj J. 2007;24:49–55. doi: 10.1007/s10719-006-9011-2. [DOI] [PubMed] [Google Scholar]
- 18.Komano Y, Nanki T, Hayashida K, Taniguchi K, Miyasaka N. Identification of a human peripheral blood monocyte subset that differentiates into osteoclasts. Arthritis Res Ther. 2006;8:R152. doi: 10.1186/ar2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma YQ, Plow EF, Geng JG. P-selectin binding to P-selectin glycoprotein ligand-1 induces an intermediate state of alphaMbeta2 activation and acts cooperatively with extracellular stimuli to support maximal adhesion of human neutrophils. Blood. 2004;104:2549–2556. doi: 10.1182/blood-2004-03-1108. [DOI] [PubMed] [Google Scholar]
- 20.Verma RP, Hansch C. Matrix metalloproteinases (MMPs): chemical-biological functions and (Q)SARs. Bioorg Med Chem. 2007;15:2223–2268. doi: 10.1016/j.bmc.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 21.Cools J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201–1214. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- 22.Golub TR, Barker GF, Lovett M, Gilliland DG. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell. 1994;77:307–316. doi: 10.1016/0092-8674(94)90322-0. [DOI] [PubMed] [Google Scholar]
- 23.Magnusson MK, Meade KE, Brown KE, Arthur DC, Krueger LA, Barrett AJ, Dunbar CE. Rabaptin-5 is a novel fusion partner to platelet-derived growth factor beta receptor in chronic myelomonocytic leukemia. Blood. 2001;98:2518–2525. doi: 10.1182/blood.v98.8.2518. [DOI] [PubMed] [Google Scholar]
- 24.Ostman A, Heldin CH. Involvement of platelet-derived growth factor in disease: development of specific antagonists. Adv Cancer Res. 2001;80:1–38. doi: 10.1016/s0065-230x(01)80010-5. [DOI] [PubMed] [Google Scholar]
- 25.Pietras K, Sjoblom T, Rubin K, Heldin CH, Ostman A. PDGF receptors as cancer drug targets. Cancer Cell. 2003;3:439–443. doi: 10.1016/s1535-6108(03)00089-8. [DOI] [PubMed] [Google Scholar]