

Abstract
The important anticancer drug paclitaxel binds to the β-subunit of the αβ-tubulin dimer in the microtubule in a stoichiometric ratio, promoting microtubule polymerization and stability. The conformation of microtubule-bound drug has been the subject of intense study, and various suggestions have been made for it. In previous work we presented experimental and theoretical evidence that paclitaxel adopts a T-shaped conformation when it is bound to tubulin. In this study we report additional experimental data and calculations that delineate the allowable parameters for effective paclitaxel-tubulin interactions.
Introduction
The natural product paclitaxel (PTX) (1a) and its closely related semisynthetic analog docetaxel (DTX, 1b) are clinically approved drugs for several tumor malignancies, including breast, lung, and ovarian carcinomas,1 while PTX has recently been shown to be effective in reducing the risk of restenosis following percutaneous coronary intervention.2 These molecules, in common with several other recently discovered natural products such as the epothilones,3 discodermolide,4 laulimalide,5 and eleutherobin,6 act by promoting the polymerization of tubulin to stabilized microtubules, leading to cell cycle arrest at the G2/M phase and hence to apoptotic cell death.7–10 Although PTX has been shown to influence the phosphorylation and translocation of cell signaling factors,11 its clinical activity is believed to be directly related to its microtubule-binding activity.10
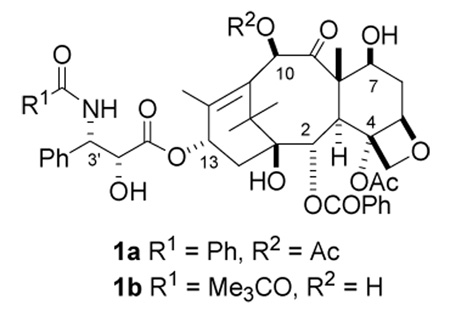
PTX is a complex molecule and is expensive to produce from natural sources. Ideally, future generations of this class of drugs should have much simpler structures, while retaining the activity of the parent drug. Any rational design of improved or simplified analogs would be greatly enhanced by knowledge of the “pharmacophore” of PTX, which can be approximately equated with the conformation of this compound in its binding site on β-tubulin. Thus the elucidation of this pharmacophore is a matter of great practical as well as theoretical significance.
The nature of the binding of PTX to tubulin polymer is not yet known in complete detail, although a significant advance was made by the determination of the electron crystallographic structure of α,β-tubulin, initially at 6.5 Å and later improved to 3.7 Å.12,13 Although the latter work was carried out with PTX-stabilized zinc-induced sheets, it lacks the resolution to define the detailed conformation of paclitaxel on the tubulin polymer. PTX is a large and conformationally mobile molecule, with rotatable groups at C-2, C-4, C-10, and C-13 appended to an essentially rigid tetracyclic ring system. The question of the preferred binding conformation of PTX on the binding site of the tubulin receptor is thus a complicated one.
Previous studies of the bioactive conformation have used evidence from NMR and modeling analyses to suggest three different models for the bioactive conformation of PTX. A “nonpolar” conformation with clustering of the C-2 benzoate and the C-3′ benzamido group of the side chain was proposed as the bioactive conformer on the basis of single conformation NMR studies in nonpolar solvents.14–16 Similar NMR studies in polar solvents revealed a hydrophobically collapsed conformation with clustering of the C-2 benzoate and the C-3′ phenyl group. This led to the proposal that the “polar” conformation is the bioactive form.17–21 Although most of the reports assumed a single conformation, deconvolution for PTX in CDCl322 and D2O/DMSO-d623 make it clear that the molecule adopts 8–14 conformations, no one of which achieves a population above 30%. A second approach has used spectroscopic studies of tubulin-bound PTX. One study using REDOR NMR provided F-13C distances of 9.8 and 10.3 Å between the fluorine of a 2-(p-fluorobenzoyl)PTX and the C-3′ amide carbonyl and C-3′ methine carbons, respectively.24 A related solid state study employing the NMR RFDR method reported a distance of 5.5 Å between the fluorines of 2-(p-fluorobenzoyl)-3′-(p-fluorophenyl)-10-acetyl-DTX.21 Both of the latter investigations singled out the polar conformation of PTX as the bound rotamer.
The “polar” and “nonpolar” conformations have inspired a number of elegant synthetic studies designed to generate constrained analogs which maintain these conformations. However, with one exception,25 none of the constrained analogs synthesized to date based on these conformations have yielded analogs with both tubulin-polymerization and cytotoxic activities equal to or greater than those of PTX itself. Thus, various compounds designed to mimic the polar conformation have been prepared, but these compounds were either inactive26 or less active than PTX.27 Similarly, constrained analogs based on the nonpolar conformation were also less active than PTX.28–31 Two recent studies have presented two bridged taxanes with microtubule stabilizing capacity equivalent to paclitaxel, but with diminished cytotoxicity.32 In a review of taxane bridging strategies, we have detailed the need for a new approach and outlined our initial efforts in this direction.33
Results and Discussion
Design and development of T-Taxol models
As outlined above, conception of various bridged analogs as candidates for binding at the taxane site on β-tubulin followed from hypotheses regarding the nature of the binding pose. In this respect, the T-Taxol conformation is no exception. The ground-breaking work of Nogales, Wolf and Downing that solved the structure of αβ-tubulin by electron crystallography (EC) was made possible, in part, by stabilizing sheets of polymerized tubulin with zinc cations and PTX.12 The 3.7 Å resolution structure, however, was unable to reveal paclitaxel’s conformation and binding pose. The molecule’s location was nonetheless represented by the single crystal X-ray structure of docetaxel introduced as a ligand place-holder.34 To overcome the paucity of structural detail for the ligand, a strategy combining the low resolution EC data and higher resolution small molecule structural data was employed.
Specifically, an analysis of the 2D NMR spectra of PTX in both CDCl322 and D2O/DMSO-d423 using NAMFIS methodology35 indicated that the compound adopts over a dozen conformations, among which a T-Taxol or butterfly conformation contributes mole fractions of 0.04 (4% population) and 0.02 (2%), respectively, in the two solvents. EC-density fitting of both the NMR conformations and a number of taxane X-ray structures led to the postulate that the T-Taxol conformation is the bioactive form.36 This model posits that the centroid of the C-2 benzoate phenyl of PTX is essentially equidistant from the centroids of the phenyl and benzamide-phenyl groups emanating from the C-3′ position; thus, the T-shaped conformation. A singular advantage of this structural motif is that when the T-Taxol conformer is nestled into the electron crystallographic density of the PTX-tubulin complex, His227 of the protein is interposed between the C-3′ benzamido and C-2 benzoyl phenyl rings of PTX. Hydrophobic collapse of the taxane phenyl groups in this sector of the binding pocket is thereby eliminated as a feature of ligand binding.36
Examination of the T-shape shows that the C-3′ phenyl and C4-OAc moieties are juxtaposed, the distance between the o-phenyl and methyl centroid hydrogens being only 2.5 Å (Figure 1).37 If the T-Taxol model accurately reflects the PTX-tubulin interaction, conformationally constrained analogs which maintain this juxtaposition should yield PTX analogs with improved tubulin-binding properties. The entropic penalty resulting from binding to tubulin is greatly reduced in the constrained analogs. As a result these analogs were expected to show a higher bioactivity than PTX itself, particularly when the bridge is short and emanates from the C-3′ phenyl ortho center. The predictions were vindicated by our preliminary results in this area, which demonstrated that taxoid design and synthesis based on the T-Taxol conformation yields compounds with improved activity in both tubulin polymerization and cytotoxicity assays.37,38 Very recently we have presented additional experimental evidence from REDOR NMR and molecular modeling for the T-Taxol model,39 and we have also shown that the model can guide the synthesis of cytotoxic analogs from non cytotoxic precursors.40 In this paper, we describe the synthesis, biological evaluation, and molecular modeling for an expanded set of highly active C-4 to C-3′ constrained analogs.
Figure 1.
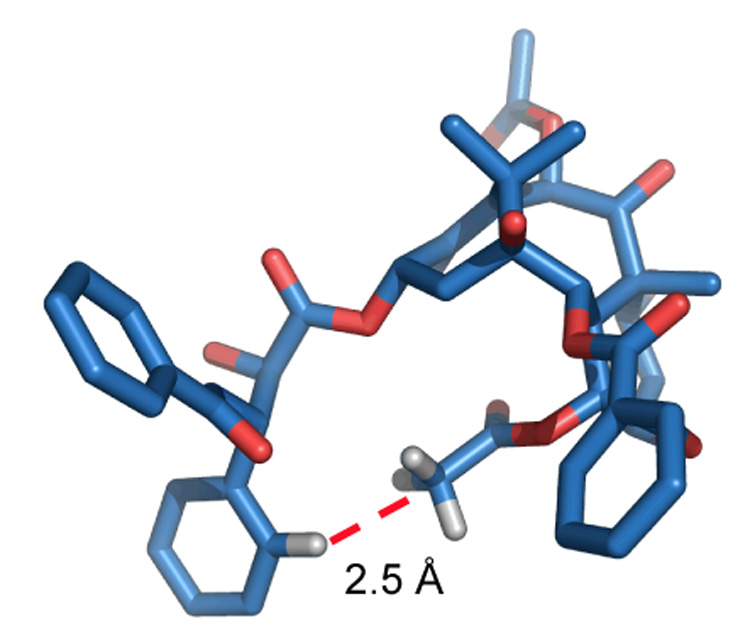
T-Taxol illustrating the short H---H distance between the centroid of the C-4 acetate methyl group and the ortho-position of the C-3′ phenyl ring.
Synthesis of Macrocyclic PTX analogs
We selected the well-established ring-closing metathesis strategy41 for the crucial macrocyclization step (Scheme 1), as it has proved highly efficacious for generation of macrocyclic taxoids.27 The proposed macrocyclic analogs (A) were envisioned to arise from the open chain ω,ω′-dienes B, which can be derived from modified β-lactam (C) and baccatin III (D) derivatives.
Scheme 1.
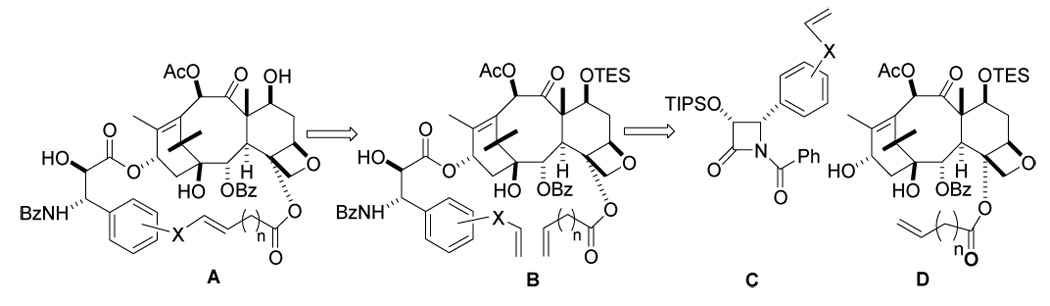
Retrosynthetic Logic for Preparation of Macrocyclic Taxanes A from ω,ω′-dienes B derived from β-Lactams C and Baccatins III D
The retrosynthesis shown in Scheme 1 required the preparation of several β-lactam derivatives with a substituent at either the ortho or meta position of the phenyl group and various C4 modified baccatin derivatives. The syntheses of the β-lactam derivatives were accomplished by application of literature procedures42,43 (Scheme 2). The syntheses of (±)-β-lactams (3a–e) were achieved starting from 3-allyloxy, 3-vinyl, 2-allyloxy, 2-butenyloxy and 2-bromo benzaldehydes (1a–e) through the N-(p-methoxy phenyl) (PMP) protected imines 2a–e. Resolution of the (±)-β-lactams (3a–e) was carried out with lipase PS (Amano) to yield the desired enantiomeric acetates (+)-(4a–e), along with undesired enantiomeric (−)-alcohols (not shown) in more than 95% yield.43,44 Functional group manipulations on 4a–e generated the triisopropylsilyl ether intermediates 5a–e. The (4)-2-bromophenyl derivative (5e) on Stille coupling45 with vinyltributyltin and allyltributyltin produced the (4)-2-vinylphenyl and (4)-2-allylphenyl lactam intermediates 5f–g respectively. The PMP-protected intermediates 5a–d and 5f–g were deprotected with ceric ammonium nitrate to produce secondary amides (6a–d, f–g), which were treated with benzoyl chloride in the presence of triethylamine and dimethylaminopyridine to result in the (+)-(3R,4S)-1-benzoyl-3-triisopropylsilyloxy-4-(aryl)-azetidin-2-ones 7a–d and 7f–g. (Scheme 2)
Scheme 2. Synthesis of β-Lactams 7a.
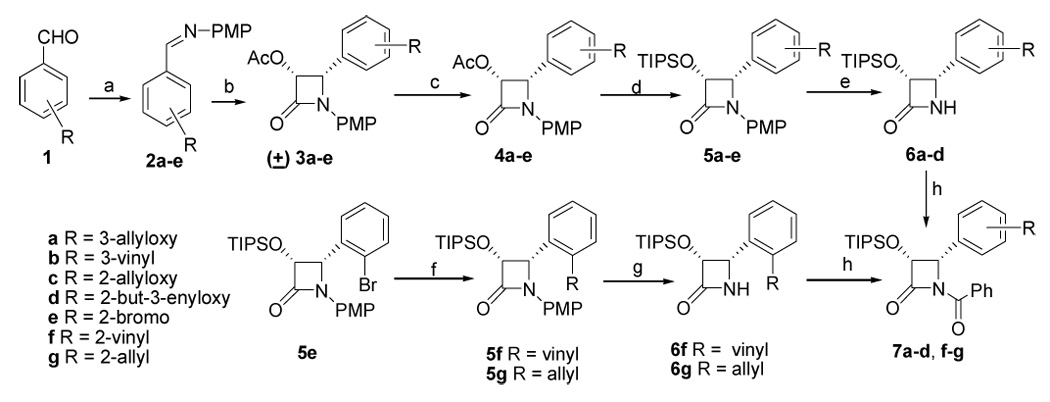
a a. p-MeOC6H4NH2, MgSO4, CH2Cl2 (100%); b. CH3COOCH2COCl, Et3N or Pri2EtN, −78 °C-room temp, 12 h (80–85%); c. lipase PS (Amano), phosphate buffer, pH = 7.2, CH3CN, 24 h - 12d (95–95%) d. i, 1 M, KOH, THF, 0 °C (quantitative); ii, TIPSCl, imidazole, DMF (90–94%); e. CAN, CH3CN, 0 °C (65–92%). f. Pd2(dba)3, Ph3P, dioxane, 80 °C, vinyl tributyl tin (5f, 80%), allyl tributyl tin (5g, 84%). g. (i) CAN, CH3CN, −5 °C (65–70%). (ii) PhCOCl, Et3N, DMAP, CH2Cl2 (7f, 86% and 7g, 90%); h. PhCOCl, Et3N, DMAP, CH2Cl2 (85–95%).
Synthesis of the baccatin derivatives 11a–d started with the known 4-deacetylbaccatin derivative 8.46 Initial attempts to acylate the hindered C4 hydroxy group using the DCC/DMAP method resulted in unacceptable yields of C4-acyl derivatives, but the use of acid chloride and LiHMDS in THF at 0 °C gave the C4-acyl baccatin derivatives 9a–d with yields ranging from 50–78%. A minor product acylated at the C4 position, but with a loss of the 1-dimethylsilyl group, was always obtained in this step, and it was used for the subsequent step. Global deprotection of 9a–d using HF.pyridine, followed by selective C-10 acetylation47 with 0.1 mol % of CeCl3 and acetic anhydride in tetrahydrofuran, gave greater than 90% yields of the desired 10-acetyl derivatives (10a–d). Selective protection of the C7 hydroxyl as its triethylsilyl ether afforded the C4 alkenoyl baccatins 11a–d in good yields (Scheme 3).
Scheme 3. Synthesis of Baccatins 11a–da.
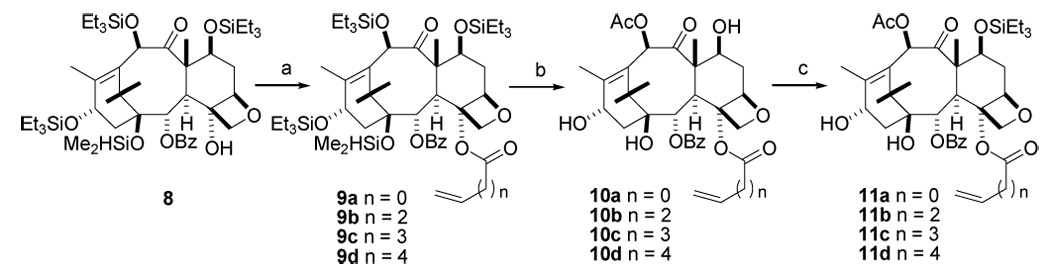
a a. LHMDS,THF, 0 °C, acryloyl chloride (9a, 52%), 4-pentenoyl chloride (9b, 78%), 5-hexenoyl chloride (9c, 71%), 6-heptenoyl chloride (9d, 50%). b. i, HF.Py, THF (n = 0, 60%, n = 2, 91%, n = 3, 80%, n = 4, 44%). ii, CeCl3, Ac2O, THF (10a, n = 0, 89%), (10b, n = 2, 92%), (10c, n = 3, 89%), (10d, n = 4, 95%). c. Triethylsilyl chloride, imidazole, DMF (11a, n = 0, 87%), (11b, n = 2, 85%), (11c, n = 3, 72%), (11d, n = 4, 83%).
Having prepared the desired baccatin III derivatives (11a–d) and β-lactam derivatives (7a–d, f–g), we synthesized the ω,ω′-diene taxoid precursors 12a–k, with yields ranging from 50–92% using the Holton-Ojima coupling protocols48 (Scheme 4). These dienes set the stage for the crucial ring closing metathesis reaction.
Scheme 4. Synthesis of bis-Alkenyl Baccatins 13a–ja.
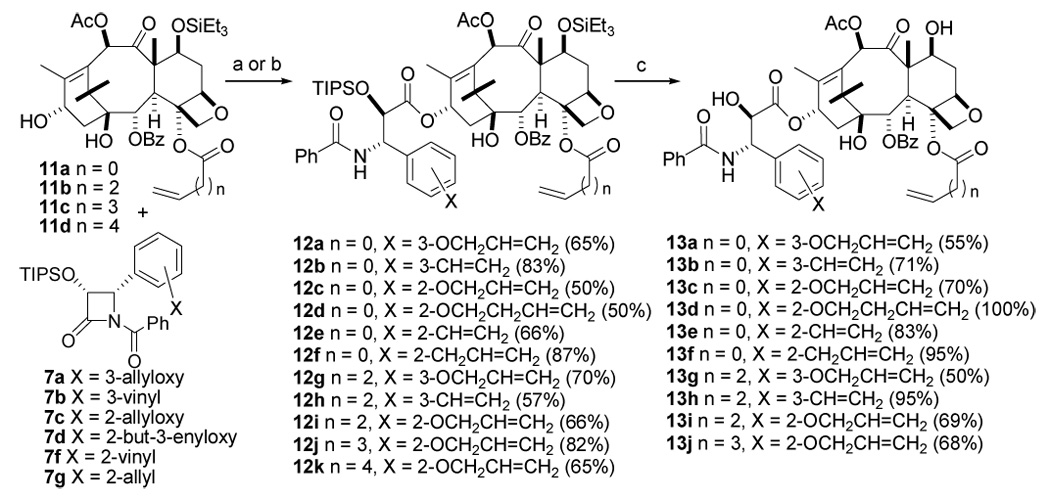
a a. LHMDS, THF, −40 °C b. NaH, THF, 0 °C-room temp c. HF.Py in THF, 0 °C-room temp, 12 h
Our initial studies of the cyclization reaction employed the ω,ω′-diene substrates 12g and 12h and Grubbs’s first generation catalyst in dichloromethane under high dilution condition,38 but in later studies the second generation Grubbs’s catalyst under the same high dilution conditions proved to give superior results. In this particular transformation the open chain diene substrates 12f–12h produce exclusively the Z alkenes 14f–14h, while the dienes 12a and 12c gave exclusively the E alkenes 14a and 14c. The remaining dienes 12d and 12i–12k yielded E/Z mixtures 14d, 14i–14k which were separable by silica gel chromatography. All the cyclic derivatives 14 were subjected to deprotection with HF.Py in tetrahydrofuran to produce the bridged paclitaxel analogs 15 in satisfactory yields. In most cases the more stable E forms of the bridging double bond were obtained, but for reasons we do not fully understand, only the Z form of compound 15f was obtained. The Z configuration was assigned on the basis of the coupling constant of 11.4 Hz for the olefinic protons of the bridging double bond. It is noteworthy that compound 23, with a m-methoxybenzoyl group at C2, gave a mixture of the Z-alkene 27 and the unconjugated E-alkene 24 on olefin metathesis (see below).
In a preliminary effort to explore the absence of E-15f from the reactions just described, we carried out density functional theory (DFT) single point calculations on MMFFs E and Z optimized structures (model; B3LYP/6-31G**). Indeed, in contrast to the experimental outcome, the E-form is predicted to be more stable than the Z-isomer. Preliminary optimization of truncated forms of Z-15f and E-15f provided a similar relative energy. We speculate that the outcome of ring closure metathesis for 15f is not governed by product thermodynamics, but by the geometry of the bulky Grubb’s catalyst in complex with the starting diene or the corresponding transition state.
Finally the saturated bridged paclitaxels 16 were prepared by hydrogenation of 15 (Scheme 5). It is worth noting here that neither the dienes 12b and 12e nor their 2′,7-silyl deprotected derivatives 13b and 13e yielded any of the expected olefin metathesis products 14b and 14e under any of the several ring closing metathesis conditions tested using either Grubb’s catalysts, presumably due to the ring stain inherent in forming the short bridges required for these compounds.
Scheme 5. Synthesis of Bridged Paclitaxels 15 and 16a.
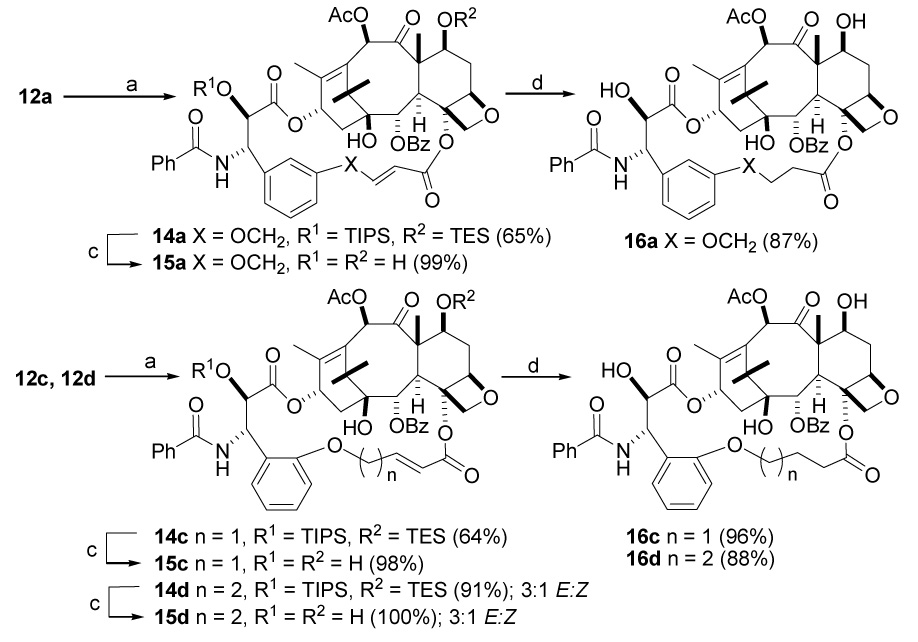
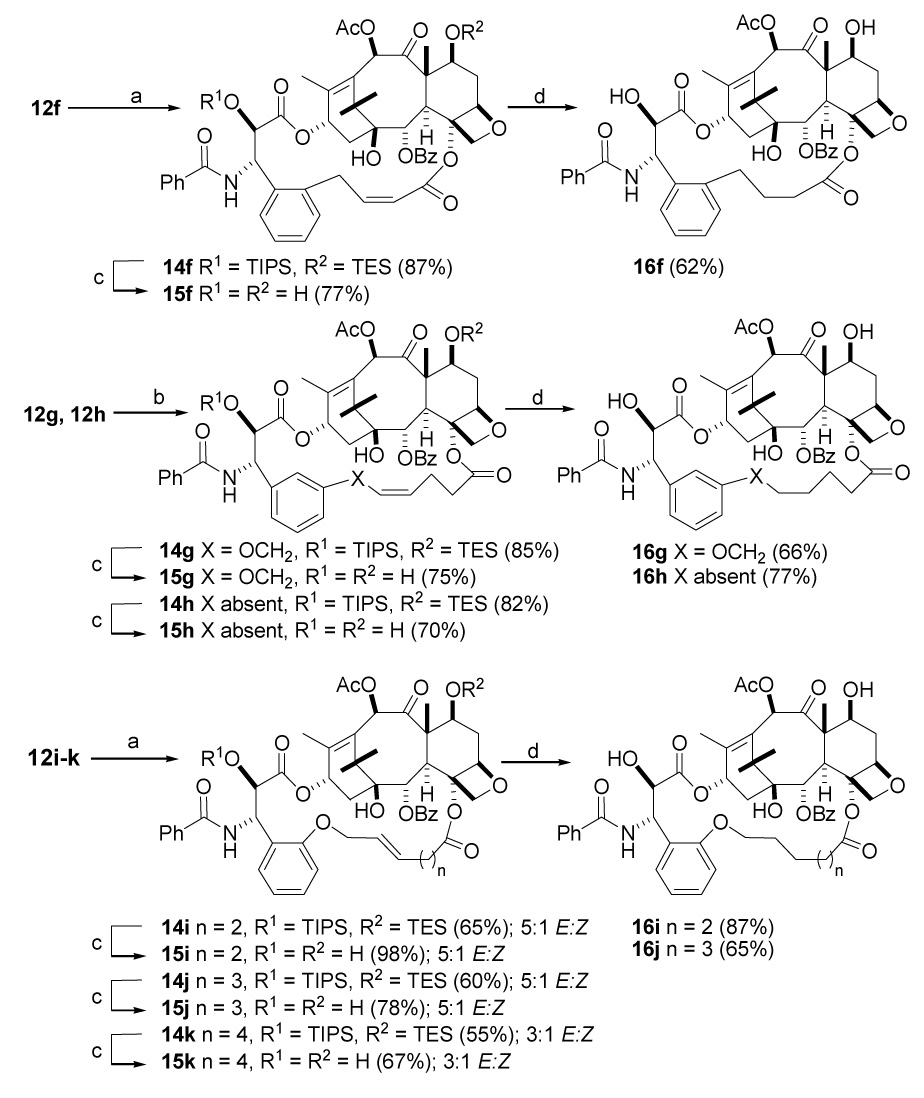
a a. (H2IMes)(PCy3)(Cl)2Ru=CHPh, DCM, room temp b. (PCy3)2(Cl2)Ru=CHPh, DCM, room temp c. HF, Py, THF, 0 °C - room temp d. H2, Pd/C, 35 psi.
Development of bridged PTX analogs with improved bioactivities
As pointed out in the introduction, a key test of the proposed bioactive conformations is the synthesis of a constrained analog that mimics this conformation and possesses an equal or greater bioactivity with respect to the parent compound. In our approach to the synthesis of such a conformationally constrained analog, we initially synthesized the 21- and 19-membered macrocyclic analogs 15g and 15h with 8 and 6 atoms, respectively, in a bridge between the C-4 acetyl group and the m-position of the C-3′-phenyl group of PTX.38 These analogs exhibited modest cytotoxicity against the A2780 ovarian cancer cell line and significant tubulin polymerization (TP) activity, but the activities were considerably less than those of PTX. The reduced activity of 15g was explained by a combination of NMR-NAMFIS analysis, which deconvolutes NMR virtual structures to individual conformers in solution, and a computational analysis of taxane-tubulin complexes. These studies indicated that compound 15g, although capable of adopting the bioactive T-form, is seated higher in the PTX- binding pocket as a result of close contact between the propene moiety of the tether and Phe270 of the protein.38,49
In the present study, we initially synthesized the dihydroderivatives 16g and 16h by hydrogenation of 15g and 15h, respectively, to evaluate whether increased flexibility of alkane-bridged macrocyclic taxoids might alleviate the unfavorable interaction with Phe270.49 However, the bioactivities of these compounds with saturated bridges were even less than those of the unsaturated compounds 15g and 15h. Surprisingly, the open chain analogs 13g and 13h showed better cytotoxicities than their macrocyclic counterparts 15g and 15h (Table 1).
Table 1.
Cytotoxicity and tubulin polymerization activity of macrocyclic and open chain paclitaxel analogs.
| Compound | IC50 values (nM)a,b |
Tubulin polym (ED50, µM)b,g | |
|---|---|---|---|
| A2780 | PC3 | ||
| PTX | 15c | 5 | 0.5 |
| 13a | 1900 | 550 | 1.0 |
| 13b | 1700 | 320 | 1.0 |
| 13c | 17800 | 2800c | 1.2c |
| 13d | 19800 | >13200 | 2.7c |
| 13e | 1600c | 275 | 1.4 |
| 13f | 1300e | 550 | 1.4 |
| 13g | 770c | 128 | 0.42 |
| 13h | 350 | 55 | 0.49 |
| 13i | 2520 | 2600 | 1.8 |
| 13j | 7400 | 4600c | 2.2 |
| 15a | 6300 | 580 | 4.6c |
| 15c | 14.5 | 15 | 0.28 |
| 15d | 20d | 16 | 0.67 |
| 15f | 0.3d | 2.5 | 0.3 |
| 15g | 650 | NDf | NDf |
| 15h | 680 | 34 | 1.6 |
| 15i | 830 | 55 | 0.9 |
| 15j | 440 | 90 | 0.53 |
| 15k | 1840c | 500 | 0.93 |
| 18f | 0.5d | 3.1 | 0.33 |
| 16a | 700d | 53 | 1.4 |
| 16c | 18.5d | 50c | 0.83 |
| 16d | 28 | 13 | 0.23 |
| 16f | 0.5d | 2.4 | 0.21 |
| 16g | 3600c | 1000 | 3.4c |
| 16h | 8800 | 570 | 1.4 |
| 16i | 980c | 51 | 0.76 |
| 16j | 2000 | 390c | 0.85 |
| 25 | 23.7d | 6 | 0.57h |
| 26 | 23.1e | NDf | NDf |
| 28 | 17.7d | 8.2 | 0.22h |
| 31a | 1.4d | 3.2 | 0.49h |
| 31b | >6i | 11 | 0.35h |
| 32a | 0.65d | 1.5 | 0.18h |
| 32b | 0.49d | 1.3 | 0.22h |
Average of three determinations unless otherwise stated
Standard error is less than 10% of the mean unless otherwise stated.
Standard error between 10% and 25% of the mean.
Standard error greater than 25% of the mean.
Single determination
ND: Samples were not tested.
ED50 values determined with GTP-tubulin unless otherwise stated.
ED50 values for these samples were determined with GDP-tubulin and were normalized to the GTP-tubulin scale.
The value cited is the lower of two widely different determinations, probably due to solubility problems.
Molecular modeling studies of the interaction of macrocyclic taxoids linked from the ortho position of the C-3″-phenyl group to the C-4 position indicated that these compounds are ideally suited to maintain the T-Taxol conformation while avoiding the unfavorable interaction with Phe270. Thus, these compounds were targeted as especially attractive candidates for synthesis. We adopted a similar ring closing metathesis strategy to that used for 15g and 15h, and subsequently synthesized the ortho-linked macrocyclic taxoids 15i–k and their dihydroderivatives 16i–j with 8–10 atoms in the bridge, respectively. These compounds were approximately 10–130 fold less active than PTX in both cell lines. Interestingly, and in contrast to 15g and 15h, the open chain analogs 13i–j were found to be inactive or only weakly active against the A2780 cancer cell line (Table 1).
At this point, we investigated the synthesis of compounds with shorter bridges. Thus, the two macrocyclic taxoids 15c and 15d and their saturated dihydroderivatives 16c and 16d, with 6 or 7 atoms in the bridge between C-4 and the C-3′ ortho-phenyl position of PTX were prepared. The compounds show approximately equal cytotoxicity to PTX against the A2780 ovarian cancer cell line, but reduced activity against the PC3 prostate cancer cell line. Comprehensive conformational NMR/NAMFIS and docking studies indicated that compound 15c was the best fit for the T-conformation of PTX. By modeling, this compound not only seats itself into the tubulin binding pocket, escaping the steric clash observed for meta bridged compounds 15g and 15h,37,38 but it also nicely accommodates His227 of β-tubulin, allowing the imidazole ring to insert itself between the stacked rings (cf. Figure 2).37
Figure 2.
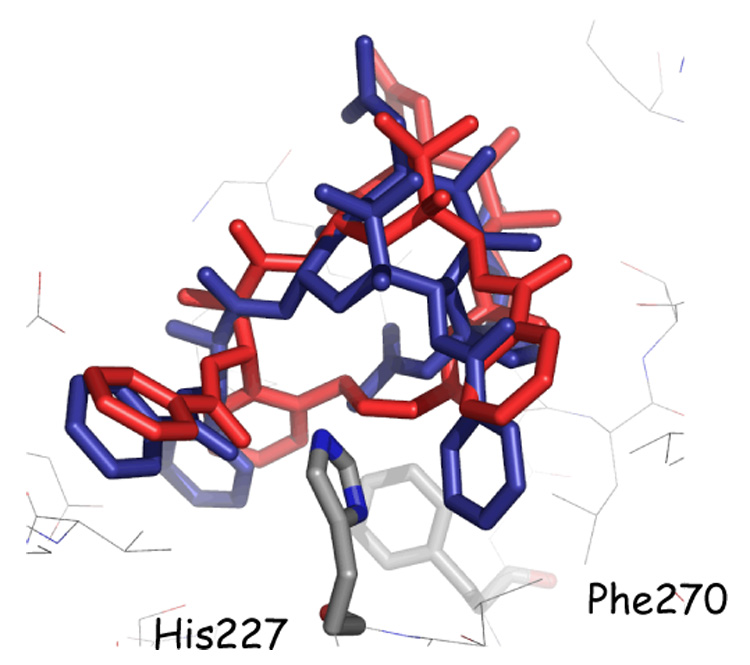
T-Conformations of PTX (blue) and 15f (red) in the β-tubulin binding site, the latter having been docked by the Glide software. Both conformations were derived by NAMFIS analyses. His227 is shown in front of the structures; the 15f bridge behind; Phe270 (Ref. 49) further behind.
We sought to refine the bridge in 15c further by shrinking the number of connecting atoms. Consequently the 17-membered macrocyclic taxoid 15f and its saturated dihydro derivative 16f, with 7 atoms in the bridge linking the ortho position of the C-3′ phenyl to the C-4 position, were synthesized. Gratifyingly, the bridged taxoid 15f exhibited excellent bioactivity. It is at least 50 times more potent than PTX against A2780, while its dihydro derivative 16f shows about 30 times more activity than PTX against the same cell line. Both bridged taxoids also show slightly increased cytotoxicity compared with PTX against the PC3 cell line. The unusual activities of 15c–d, 15f and 16c–d, 16f are not due to the substituents on the C-3′ phenyl or the C-4 positions, since the open chain analogs 13c–d and 13f are almost completely inactive or weakly active against both the A2780 and PC3 cell lines (Table 1). This indicates that the activity associated with the bridged taxoids must originate from their conformational restriction.
Molecular modeling studies suggested that the E bridged macrocyclic analog of 15f should be more stable than Z-15f, while minireceptor QSAR50 predicts this compound to be more active than PTX. Based on these projections, we investigated synthesis of the E macrocyclic bridged derivative of 15f. Subjection of diene 12f to ring closing metathesis with Grubbs second generation catalyst in dichloromethane at an elevated temperature of 55 °C produced exclusively the isomerized product 17f, which on deprotection with HF.Py furnished the isomerized E alkene 18f (Scheme 6). This compound showed excellent cytotoxicity, almost equal to that of the Z bridged derivative 15f, indicating that the precise stereochemistry of the bridging alkene linker causes only a small difference in the activity. The structure of 18f was confirmed by hydrogenation to give a dihydro derivative identical with 16f.
Scheme 6. Synthesis of Bridged Paclitaxel 18a.
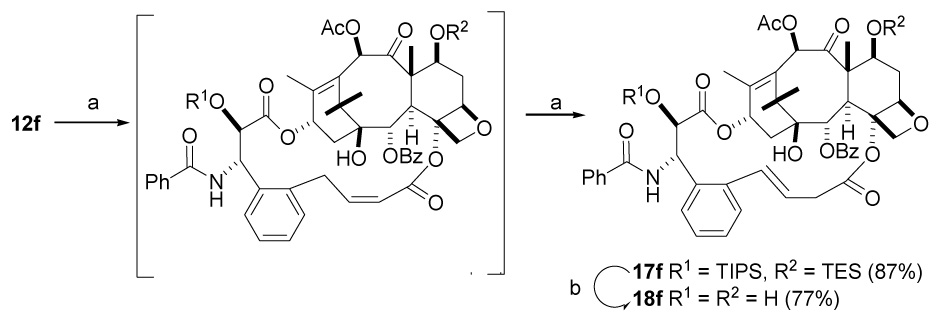
a a. Grubbs catalyst, CH2Cl2, 55 °C, 77%. b. HF/Py, THF, 77%
Synthesis of bridged paclitaxel derivatives with modified pendant groups
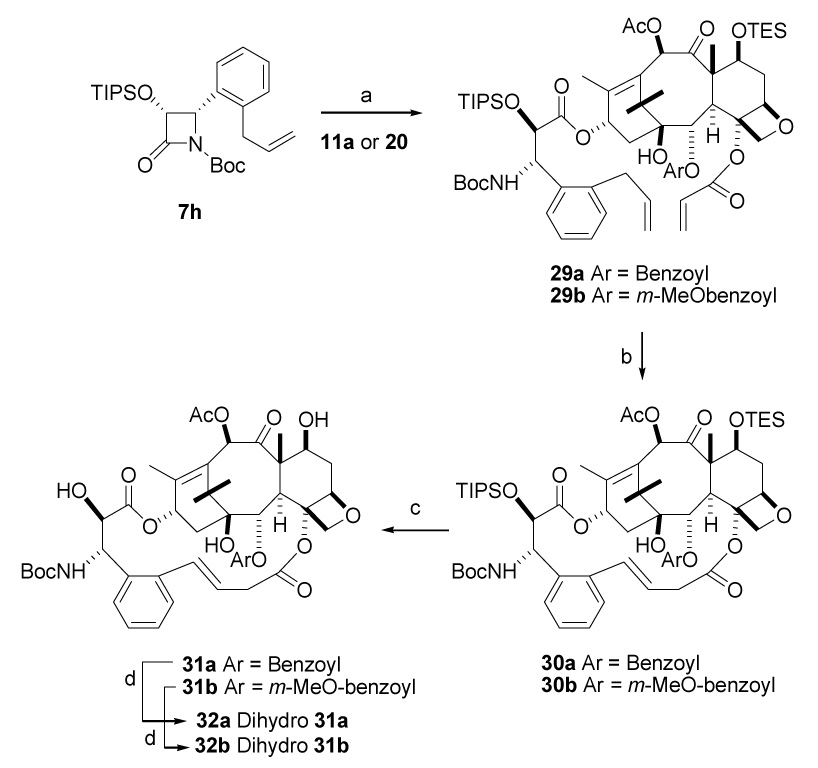
Having optimized the macrocyclic bridge between the C-4-acyl and the C-3′-ortho-phenyl positions, we next investigated the structure activity relationships at other sites of the bridged paclitaxel analogs in an attempt to identify additional paclitaxel analogs with further improved activity. We51 and others52 have previously reported the unusual cytotoxicity and in vitro tubulin polymerization activity of various C-2-aroyl substituted paclitaxel analogs, such as the C-2-m-methoxybenzoyl and m-azidobenzoyl derivatives. We thus embarked on the synthesis of the C-2 m-methoxybenzoyl macrocyclic paclitaxel analog 28. It was accomplished using modifications of known reactions. Thus 7,10,13-tris-(triethylsilyl)-10-deacetylbaccatin (III) 19 was debenzoylated with Red-Al followed by protection as its cyclic carbonate 20.27 Compound 20 was treated with m-methoxy phenyllithium at 0 °C, followed by dimethylsilyl chloride to give 21 in 70% overall yield. Finally 21 was converted to the key intermediate 22 by steps similar to those of Scheme 2. Coupling of the building blocks 22 and 7g under standard conditions48 furnished the diene 23 in 70% yield. Ring closing metathesis of diene 23 using Grubbs’ second generation catalyst yielded the bridged taxoids 24 and 27 in a 3:1 ratio, with no formation of E-alkene. Interestingly, the Grubbs first generation catalyst failed to bring about macrocyclisation under various solvent and temperature conditions. The final compounds 25 and 28 were obtained by deprotection with HF.Py in tetrahydrofuran, while hydrogenation of the two isomers delivered the same saturated compound 26. Disappointingly, the macrocyclic bridged m-methoxy derivative Z-28 and its isomer 25, both showed reduced activity against the A2780 and PC3 cell lines compared with the corresponding C2 benzoyl derivative Z-15f, and the dihydro derivative 26 was also less cytotoxic to the A2780 cell line (it was not tested against the PC3 cell line). The N-Boc derivatives 31a – 32b were however more promising, and in this series the C2 m-methoxybenzoyl group did not reduce activity significantly in compounds 31a and 32a and 32b, in contrast to its effect on the N-benzoyl series. The reasons for this difference between the effects of the C2 m-methoxybenzoyl group on the two different series is not clear. It is noteworthy that compounds 32a and 32b were the most potent compounds of the entire series against the PC3 cell line and also as promoters of tubulin polymerization.
We then elected to carry out structural modifications on the side chain of the bridged paclitaxel analogs, since docetaxel with an N-Boc substituent has improved activity as compared with PTX. The β-lactam derivative 7h was prepared by the procedure described Scheme 2. Coupling of 7h individually with 11a and 20 gave ω,ω′-dienes 29a and 29b, and these dienes upon ring closing metathesis produced exclusively the cyclic double bond isomerized products 30a and 30b. None of the normal Z or E unisomerized alkenes were obtained in this reaction. The usual HF.Py deprotection produced the products 31a and 31b, and hydrogenation furnished the dihydroderivatives 32a and 32b.
The bioactivity of compounds 31a is similar to that of PTX against the A2780 cell line, while the m-methoxy derivatives 31b and 32b showed 3–30 fold activity greater than PTX in the same cell line. In the PC3 cell line the compounds are 2-fold less and 4-fold greater than parent PTX, respectively. It thus appears that the additional activities generally conferred by the N-Boc group and the m-methoxybenzoyl group are unique to the unbridged compounds. Modeling suggests a modest reorientation of the terminal phenyl rings in bridged compounds within the taxane binding site.53 While the associated binding pose contributes to improved bridge-induced tubulin-binding, it apparently dampens these additive substitutent effects at the same time.
Historically, the search for microtubule modifiers has often been accompanied by a preliminary assessment of biological activity by examination of a drug’s ability to polymerize tubulin or to influence the stability of microtubules. More recently, this practice has been largely supplanted by a focus on cytotoxicity. The main reason, of course, is that an effective drug needs to penetrate the membrane of a cell to arrest microtubule function within its boundaries. Factors such as cell permeability, cytoplasmic metabolism and nonselective binding are, of course, unexamined by an in vitro appraisal of the state of microtubule polymerization. As a consequence, in many cases where both polymerization and cytotoxicity data are available, a correlation fails to emerge. This is the case in the present study. For example, numerous analogs are equal to or within a factor of two relative to PTX in the polymerization assay, but 20–1200 fold poorer in the cell-based assays (e.g. 13a,b,c,g,h, 15i,j and 16i,j). By the same token, other in vitro analogs whose activities differ by a factor of two from PTX (15c,d, 25, 28, 31a, 32b) are nearly equivalent to PTX in their cellular action. Measurements of critical concentration and Kp, the equilibrium constant for polymer growth, would appear to be a much more reliable approach to assessment of biological activity.37 Such measurements will be the subject of future work by our laboratories.
Resistance and the bridged analogs
An important discovery of the present work is that some of the bridged PTX analogs display promising activity against paclitaxel-resistant and epothilone A-resistant cell lines. The latter were derived by long-term exposure of 1A9 human ovarian carcinoma cells to PTX or epothilone-A (epo-A) in order to generate cells with drug-resistant clones. Subsequent molecular characterization of these clones revealed that the resistance phenotype was due to distinct acquired β-tubulin mutations at the taxane binding pocket.54,55 As a result, PTX’s ability to interact with the mutant tubulins in these clones is significantly impaired as evidenced by the lack of drug-induced microtubule-stabilizing activity and essentially lack of G2/M arrest.56 Since drug resistance is the factor that hampers most PTX’s clinical activity, we set out to test the activity of our analogs against the parental and drug-resistant cell lines (Table 2). While 13g and 15c exhibited similar or slightly improved activities over PTX, there are two compounds that stand out. Specifically the bridged taxoid 15f and the dihydro analog 16f exhibit a remarkable activity in that not only are they about 100-fold more active than PTX against the parental 1A9 cells, but they are also 1200- and 150- times more effective than PTX (respectively) towards the PTX-resistant cell line PTX10 (Table 2). As shown by their relative resistance values, compound 15f is able to completely overcome paclitaxel resistance (1.8 fold) as compared to 20-fold resistance exhibited by PTX. Compound 16f is able to overcome paclitaxel resistance by 50%, while both compounds are 50 to 90 times more active than PTX against the epothilone-resistant 1A9-A8 cells. The relative potencies of these two compounds are unique.
Table 2.
Bioactivity of bridged taxoids against paclitaxel and epothilone A resistant cell lines.a
| Bridged taxanes | IC50, nMb | RRc | IC50, nMb | RRc | |
|---|---|---|---|---|---|
| 1A9 | 1A9-PTX10 (Fβ270V) | PTX10/1A9 | 1A9-A8 (Tβ274I) | A8/1A9 | |
| PTX | 4.8 ± 4.5 | 157 | 20 | 21.5 ± 11.5 | 4.5 |
| 13g | 1.8 ± 2.5 | 65 | 18 | 148 ± 67.9 | 82 |
| 13h | 13.7 ± 8.6 | 42 | 5.5 | 137 ± 28.3 | 10 |
| 15c | 7.0 ± 0.85 | 126 | 17 | 26.5 ± 3.2 | 3.8 |
| 15d | 12.2 ± 7.0 | 157 | 9.2 | 46.4 ± 13.6 | 3.8 |
| 15f | 0.32 ± 0.35 | 0.13 | 1.8 | 0.27 ± 0.08 | 0.84 |
| 15g | 24.6 ± 9.0 | 196 | 6.3 | >300 ± 0.0 | >12 |
| 15h | 12.9 ± 7.5 | 157 | 21 | >300 ± 0.0 | >23 |
| 16e | 18.9 ± 1.27 | 35.9 | 1.8 | 66.5 ± 5.0 | 3.5 |
| 16f | 0.07 ± 0.02 | 1.03 | 12 | 0.44 ± 0.19 | 6.3 |
1A9 is the parental drug-sensitive cell line, 1A9- PTX10 is the paclitaxel resistant clone with an acquired Fβ270V (Ref. 49) mutation, and 1A9-A8 is the epothilone A resistant clone with an acquired Tβ274I mutation which also confers cross resistance to paclitaxel (5–10 times).
The IC50 values (nM) for each compound were determined in a 72-hr growth inhibition assay using the sulforhodamine-B method as previously described (Ref. 55). All values represent the average of three or four independent experiments.
RR, relative resistance = IC50 for resistant cell line/IC50 for parental cell line.
The relative resistance values (RR) for 1A9-PTX10 cells across the compounds listed in Table 2 range from 2–21; those for 1A9-A8, from 3–82. The same quantities for 15f and 16f are perfectly normal by comparison, similar to PTX, and found in the windows RR = 0.8–2 and 6.8–12, respectively. The implication is that the bridged compounds, from the point of view of acquired resistance, are normal taxanes. The remarkable improvements of 15f and 16f over paclitaxel in apparently overcoming resistance owe their origin to an unusually high potency rather than to subtle structural features of the bridged molecules that enable them to bypass the mutated binding site residues.
Bridged taxane conformations in solution
At the outset of our bridging studies, we hypothesized that creation of short bridges between the ortho-position of C-3′ and the methyl of C-4-OAc as depicted in Figure 1 would lead to bioactive conformations in the T-Taxol family and provide highly active taxane analogs. Table 1 provides ample support for the concept and reinforces the proposition that deviations from ortho substitution and short tethers evoke a reduction in both cytotoxicity and tubulin polymerization. It would appear that our initial conformational expectations have been fulfilled. However, we still lack an experimental structure of a bridged-taxane/tubulin complex and, thus, structural verification of the idea. We note that the two-carbon bridges between the C-3′ and C-4 positions now enforce a 17-membered ring. While the latter involves two lactones, two C=C units and seven bonds rigidified by the baccatin core, it is possible that the newly installed macrocyclic rings might sustain sufficient flexibility to require considerable conformational reorganization upon binding to tubulin. To examine this question, we performed empirical conformational analyses for two of our most active analogs, 15f and 18f. For both compounds, a quantitative NMR-ROESY determination was carried out. The corresponding cross-peaks were translated into intramolecular proton-proton separations based on internal distance standards. The corresponding distances were combined with taxane conformers derived by Monte Carlo conformational analyses within the NAMFIS framework.35 As described in numerous previous studies, this technique is able to deconvolute an averaged NMR spectrum into a description of the ensemble of contributing conformations along with an estimate of the individual conformer populations.22,57
As applied to 15f with a cis double bond within the bridge, NAMFIS analysis using 19 NMR-ROE-derived intramolecular atomic distances delivered five conformations with estimated populations of 40, 27, 17, 9 and 7% (see Experimental Section). The top populated conformer is the T-form (40%); while the second and fourth most populated structures (27 and 9%, respectively) are slightly collapsed forms of the T-conformation in which the C3’-benzamido phenyl to C2-phenyl centroid distance is 3 Å shorter. The third and fifth structures (17 and 7%, respectively) structures are somewhat extended structures that can be classified as neither T nor polar forms. Compound 18f delivers a similar result, namely only three conformations from 15 ROE cross peaks. One is the T-conformation (53%); the second, a slightly collapsed T-structure (33%); the third, the polar conformer (14%). A similar analysis for parent PTX (1a) delivered T-conformers with much reduced populations of 2–5%.37 Short-bridge ortho-tethering for this compound class increases the concentrations of the apparent bioactive conformers by 5–25 fold and thereby contributes to their exceptional activity. Figure 2 illustrates the superposition of PTX and 15f in the β-tubulin binding cleft. The conformations of both molecules place the C-3′ benzamido and C-2 benzoyl phenyls on either side of His227 (no π-stacking implied) and simultaneously avoid steric interaction with Phe270.49
Conclusions and Perspectives
Over the past decade numerous bridging strategies have been explored in the effort to restrain the conformation of the paclitaxel architecture to its bioactive tubulin-bound conformation. Most have led to the synthesis of compounds that are considerably less potent than PTX.33 A very few have demonstrated diminished cytotoxicity, but matched the latter in a tubulin/microtuble in vitro assay.32 Our strategy was directed from the start by analysis of the T-Taxol conformation proposed as the binding geometry of PTX on β-tubulin.36 Significantly, as depicted in Figure 1, it was predicted that a bridge between the methyl group of the C-4 acetate and the ortho-position of the C-3′ phenyl group would lead to compounds that are constrained to adopt the bioactive molecular shape.
In the context of killing A2780 ovarian cancer cells, the synthetic effort has led to seven compounds with cytotoxicities equal to that of PTX (15c, d, 16c, 25, 26, 28 and 31a), one that is 3-fold more active (32b), three that are 30-fold enhanced (16f, 18f, 31b), and one that shows 50-fold improvement (15f). In the PC3 prostate cell line, the activity enhancements are somewhat less but are still significant (Table 1). Equally important, a subset of these compounds has been tested against both PTX- and epothilone-resistant cell lines (1A9-PTX10 and 1A9-A8, respectively, Table 2). Two of the most active compounds, 15f and 16f, are 1200 and 160-fold more active than PTX with respect to PTX10, respectively, and 90 and 50-fold more cytotoxic, respectively, against cells raised against epothilone-resistant cells (A8). Clearly, significant increases in cancer cell-kill can have a dramatic effect on resistance stemming from acquired mutations.
We have examined the origin of the bioactivities by seeking to define the degree of rigidification introduced by the C-4 to C-3′ bridging principle. This has taken the form of extracting individual conformations associated with approximate populations from the averaged NMR spectrum of a compound. As applied to unconstrained PTX, the T-form was estimated to be present to the extent of 2–5% among eight diverse conformations.23,37 Constrained compounds 15f and 18f, on the other hand, appear in solution as five and three conformations, respectively, with 76% and 86% contributions from T-Taxol structures. While the significant activity increases measured for these substances cannot be attributed completely to conformational biasing, the results strongly suggest that this is a dominating factor.
If a given compound adopts the bioactive form, does this guarantee amplified activity? In a recent review of bridged taxanes we pointed out that enforcing the T-Taxol conformation is a necessary but not sufficient condition for eliciting high levels of drug potency.33 In addition to the appropriate molecular conformation, the tubulin-taxane ligand must also adopt a compatible molecular volume. Compound 15g with a 5-atom bridge between the meta position of the C-3′ phenyl and the C-4 methyl carbon (m- O-CH2-CH=CH-CH2-) illustrates the molecular dilemma. Like PTX it displays about 5% of the T-form in solution.38 Unlike PTX, the compound is 4–45 fold less cytotoxic depending on cell line and 10-fold weaker as a tubulin polymerization agent. Modeling demonstrates that a section of the long, suboptimally substituted bridge falls outside the molecular volume of PTX and competes with tubulin’s Phe27049 for the same space within the binding pocket as illustrated by Figure 3. Consequently, the ligand either rides higher in the pocket or is pushed out of it.38 By contrast, compound 15c (Table 1) with a three carbon ortho-bridge (o-O-CH2-C=) likewise presents the T-form in solution, but the truncated bridge avoids a steric clash with the protein and results in activities equivalent to that of PTX.37
Figure 3.
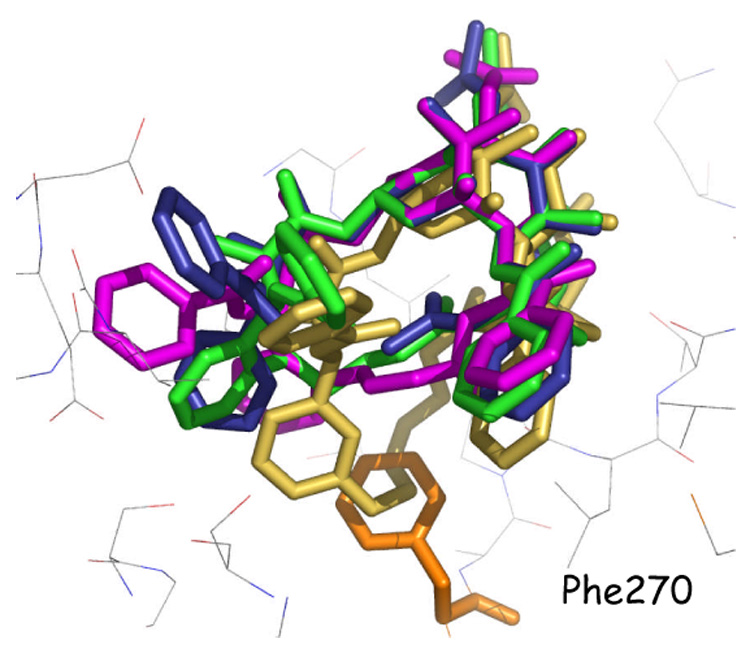
T-Conformation of PTX (blue) bound to β-tubulin. The T-conformers for the analogs E-15c (green) and Z-15g (medium brown) and E-18f (magenta) have been derived from NAMFIS analyses. The baccatin cores for PTX and the three analogs are superimposed. The meta-Z-bridged 15g (light brown) is pushed out of the taxane binding site because phenylalanine 270 of tubulin (orange) is in steric conflict with the extended bridge preventing effective binding to the protein. 15c and 18f avoid the Phe272 steric-clash and fit nicely into the PTX binding site.
Experimental Section
General Experimental Methods
All reagents and solvents received from commercial sources were used without further purification. 1H and 13C NMR spectra were obtained in CDCl3 on Varian Unity or Varian Inova spectrometers at 400 MHz or a JEOL Eclipse spectrometer at 500 MHz. High resolution FAB mass spectra were obtained on a JEOL HX-110 instrument. Specific rotations were measured on a Perkin-Elmer 241 polarimeter. Usual work-up was carried out by quenching the reaction, extracting the reaction mixture 3 times with EtOAc, combining the organic extracts and washing the combined extracts with H2O and brine, followed by drying over Na2SO4, filtration, and concentration of the organic solution to give crude product.
Representative Procedure for the Synthesis of β-Lactam Derivatives 7a–f
A typical procedure is described for the synthesis of these β-lactam derivatives.
(a) Synthesis of Racemic 1-(p-Methoxyphenyl)-3-acetyloxy-4-(m-allyloxyphenyl)azetidin-2-one (3a)
To a solution of 3-(allyloxy)benzaldehyde (1a, 5 g, 34 mmol) in CH2Cl2 (85 mL) was added p-anisidine (4.38 g, 35 mmol), followed by MgSO4 (50 g, in portions), and the resulting reaction mixture was stirred for 12 h. Removal of the MgSO4 by filtration and concentration of the CH2Cl2 solution yielded crude imine 2a (8.5 g). Diisopropylethylamine (100 mmol, 3 equiv.) followed by acetoxyacetyl chloride (40 mmol, 1.2 equiv.) were added to a solution of 2a (8.5 g, 33 mmol) in CH2Cl2 (75 mL) at −78 °C, and the resulting solution was brought to room temperature over 12 h. The reaction mixture was concentrated and the residue was purified by chromatography on silica gel using 10–20% EtOAc in hexane to furnish racemic 1-(p-methoxyphenyl)-3-acetyloxy-4-(m-allyloxyphenyl)azetidin-2-one (3a, 9.3g, 75%).
(b) Resolution of 3a
Lipase PS (Amano) (2 g) was added to a solution of 3a (4g, 10 mmol) in CH3CN and pH 7 phosphate buffer (1:2.5), and the resulting solution was stirred at rt for 74 h. After usual work-up the residue was separated by chromatography on silica gel to furnish (3R,4S)-1-methoxyphenyl-3-acetyloxy-4-(m-allyloxyphenyl)azetidin-2-one (4a) (1.92g, 48%) and (3S,4R)-1-methoxyphenyl-3-hydroxy-4-(m-allyloxyphenyl)azetidin-2-one (1.84 g, 46%).
(c) Synthesis of (3R,4S)-1-Methoxyphenyl-3-triisopropylsilyloxy-4-(m-allyloxyphenyl)-azetidin-2-one (5a)
A solution of 4a (920 mg, 2.5 mmol) in tetrahydrofuran (20 mL) was added dropwise to a stirred solution of 1M KOH (1M, 80 mL) and tetrahydrofuran (20 mL), and the resulting solution was stirred for 15 min. Addition of H2O followed by usual work-up produced (3R,4S)-1-methoxyphenyl-3-hydroxy-4-(m-allyloxyphenyl)azetidin-2-one (800 mg, 99%). To a solution of this alcohol (800 mg, 2.48 mmol) in N,N-dimethyl formamide (3 mL) was added imidazole (500 mg, 7.4 mmol, 3 equiv.) followed by Pri3SiCl (508 mg, 2.65 mmol, 1.1 equiv.) and the resulting solution was stirred at room temperature for 8 h. EtOAc was added to quench the reaction, and usual work-up followed by chromatography over silica gel with 2% EtOAc in hexane furnished (3R,4S)-1-methoxyphenyl-3-triisopropylsilyloxy-4-(m-allyloxyphenyl)azetidin-2-one (5a) (1.15 g, 99%).
(d) Synthesis of (3R,4S)-1-Benzoyl-3-triisopropylsilyloxy-4-(m-allyloxyphenyl)azetidin-2-one (7a)
To a solution of 5a (980 mg, 2.3 mmol) in CH3CN (35 mL) was added ceric ammonium nitrate (3.35 g, 6.1 mmol, 3 equiv.) in H2O (17 mL). The mixture was allowed to stand for 20 min at 0 °C and was then stirred for 45 min. EtOAc was added and the organic phase was washed with saturated sodium metabisulfite. Usual work-up followed by chromatography over silica gel with 10% EtOAc in hexane furnished the imide (3R,4S)-3-triisopropylsilyloxy-4-(m-allyloxyphenyl)azetidin-2-one (6a, 600 mg, 78%). To a solution of 6a (568 mg, 1.5 mmol) in CH2Cl2 (20 mL) was added triethylamine (0.63 mL, 4.5 mmol, 3 equiv.), dimethylaminopyridine (20 mg) and benzoyl chloride (0.52 mL, 4.5 mmol, 3 equiv.) at 0 °C and the resulting solution was stirred for 1 h. Usual work-up followed by chromatography over silica gel with 2–5% EtOAc furnished (3R,4S)-1-benzoyl-3-O-triisopropylsilyloxy-4-(m-allyloxyphenyl)azetidin-2-one 7a (600 mg, 82%). 7a: [α]D + 97 (c = 0.4, CHCl3). 1H NMR (400 MHz) δ= 8.02 (2H, d, J = 7.3 Hz), 7.58 (1H, t, J = 7.3 Hz), 7.47 (2H, t, J = 7.4 Hz), 7.26 (1H, m), 6.98 (1H, d, J = 7.8 Hz), 6.94 (1H, dd, J = 1.9, 1.8 Hz), 6.86 (1H, dd, J = 7.3, 2.5 Hz), 6.02 (1H, m), 5.39 (1H, d, J = 6.1 Hz), 5.38 (1H, 2 × dd, J = 16, 6 Hz), 5.26 (1H, 2 × dd, J = 10.3, 1.6 Hz), 5.23 (1H, d, J = 6.1 Hz), 4.51 (2H, m), 1.00 (3H, m), 0.91 (4 s, 18H). 13C NMR (125 MHz) δ= 166.3, 165.4, 158.6, 135.4, 133.4, 133.3, 132.1, 129.9, 129.2, 128.2, 120.9, 117.6, 114.8, 114.7, 76.7, 68.9, 61.1, 17.5, 17.4, 11.7. HRFABMS: m/z calcd for C28H37NO4Si+ 479.2492, found 479.2494 (Δ = 0.5 ppm). Characterization data for compounds 7b–d and 7f–g are proved in the Supporting Information.
Representative Procedure for the Synthesis of Baccatin III Derivatives 11a–d. Synthesis of 4-Deacetyl-4-acryloyl-7-O-triethylsilylbaccatin III (11a)
(a) Synthesis of 1-Dimethylsilyl-4-deacetyl-4-acryloyl-4-deacetyl-7-O,10-O,13-O-tris-triethylsilylbaccatin III (9a)
Lithium hexamethyldisilazide (LiHMDS) (1M, 1.3 ml, 1.3 mmol, 1.2 equiv.) was added dropwise at 0 °C to a solution of 1-dimethylsilyl-4-deacetyl-7-O,10-O,13-O-tris-triethylsilylbaccatin III46 (8) (1 g, 1.1 mmol) in THF (6 mL), and the resulting solution was stirred for 45 min. Acryloyl chloride (0.123 ml, 1.54 mmol, 1.3 equiv.) was added to the above solution at 0 °C and the reaction mixture stirred for 3 h. Saturated NH4Cl solution (10 mL) was added, and the aqueous phase was subjected to the usual work-up. The crude product as purified by chromatography over silica gel with 4% EtOAc in hexane to furnish 1-dimethylsilyl-4-deacetyl-4-acryloyl-7-O,10-O,13-O-tris-triethylsilylbaccatin III (9a) as a white solid (550 mg, 52%).
(b) Synthesis of 4-Deacetyl-4-acryloyl-10-deacetylbaccatin III
To a solution of 1-dimethylsilyl-4-deacetyl-4-acryloyl-7-O,10-O,13-O-tris-triethylsilylbaccatin III (9a) (470 mg, 0.49 mmol) in THF (30 mL) was added dropwise HF.Py (70% HF, 2.37 mL) at 0 °C, and the resulting solution was brought to room temperature over 24 h, after which saturated NaHCO3 solution (50 mL) was added carefully to quench the reaction. The aqueous phase was subjected to the usual work-up, and the crude product was purified by preparative thin layer chromatography on silica gel developed with 70% EtOAc in hexane to give 4-deacetyl-4-acryloyl-10-deacetyl-baccatin III as a white solid (193 mg, 70%).
(c) Synthesis of 4-Deacetyl-4-acryloylbaccatin III (10a)
Anhydrous CeCl3 (20 mg) was added to a solution of 4-deacetyl-4-acryloyl-10-deacetyl-baccatin III (210 mg, 0.377 mmol) in THF (10 mL) and the resulting solution was stirred for 5 min. Acetic anhydride (0.65 mL, 14-fold excess) was added to the above solution and the reaction mixture was stirred for 4 h. EtOAc (100 mL) was added and the solution was washed with saturated NaHCO3, water, and brine, dried over Na2SO4, and concentrated. The crude product was subjected to preparative thin layer chromatography on silica gel with 60% EtOAc in hexane to give 4-deacetyl-4-acryloyl-baccatin III (10a) as a white solid (200 mg, 96%).
(d) Synthesis of 4-Deacetyl-4-acryloyl-7-O-triethylsilylbaccatin III (11a)
To a solution of 4-deacetyl-4-acryloylbaccatin III (10a) (50 mg, 0.083 mmol) in dichloromethane (5 mL) was added imidazole (56 mg, 0.836 mmol, 10 equiv.) followed by triethylsilylchloride (0.50 mmol, 6 equiv.) at 0 °C, and the resulting solution was stirred for 3 h. Dilute HCl (0.05M, 5 mL) solution was added to quench the reaction followed by EtOAc (40 mL). The organic phase was worked up in the usual way to give a crude product which was subjected to preparative thin layer chromatography over silica gel with 45% EtOAc in hexane to give 11a as a white solid (40 mg, 72%). (11a): 1H NMR (500 MHz, CDCl3) δ = 8.11 (2H, d, J = 7.1 Hz), 7.59 (1H, t, J = 7.2 Hz), 7.46 (2H, t, J = 7.2 Hz), 6.52 (1H, dd, J = 17.4, 1.2 Hz, 1H), 6.47 (1H, s), 6.28 (1H, dd, J = 17.4, 10.5 Hz), 6.01 (1H, dd, J = 17.4, 1.2 Hz), 5.64 (1H, d, J = 6.8 Hz), 4.94 (1H, d, J = 7.7 Hz), 4.76 (1H, m), 4.53 (1H, dd, J = 10, 6.7 Hz), 4.33 (1H, d, J = 8.4 Hz), 4.20 (1H, d, J = 8.4 Hz), 3.94 (1H, d, J = 6.8 Hz), 2.55 (1H, m), 2.20 (3H, s), 2.22-2.10 (2H, m), 2.18 (3H, s), 1.90 (1H, m), 1.70 (3H, s), 1.18 (3H, s), 1.02 (3H, s), 0.91 (9H, t, J = 7.2 Hz), 0.60 (6H, m). 13C NMR (125 MHz) δ = 202.2, 169.4, 167.1, 165.3, 144.0, 133.7, 131.2, 130.1, 129.8, 129.6, 128.6, 84.2, 81.3, 78.8, 76.6, 75.8, 74.8, 72.4, 68.1, 58.8, 47.2, 42.8, 38.9, 37.3, 26.9, 21.0, 20.1, 14.9, 10.0, 6.84, 5.36. HRFABMS m/z calcd for C38H53O11Si+ 713.3357, found 713.3326 (Δ = 4.4 ppm). Characterization data for compounds 11b–d are proved in the Supporting Information.
Representative Procedures for the Coupling of Baccatin III Derivatives 11a–d with β-Lactam Derivatives 7a–d and 7f–g
A typical procedure is described for the synthesis of 3′-dephenyl-3′-(m-allyloxyphenyl)-4-deacetyl-4-acryloyl-7-O-triethylsilyl-2′-O-triisopropylsilylpaclitaxel (12a).
Synthesis of 3′-Dephenyl-3′-(m-allyloxyphenyl)-4-deacetyl-4-acryloyl-7-O-triethylsilyl-2′-O-triisopropylsilylpaclitaxel (12a)
To a solution of baccatin III derivative 11a (25 mg, 0.035 mmol), β-lactam derivative 7a (34 mg, 0.07 mmol, 2 equiv.) in THF (6.5 mL) was added LiHMDS at −40 °C and the resulting solution was stirred for 3 h. Saturated aqueous NH4Cl (2 mL) was added to quench the reaction, and usual work-up gave crude product which was purified by preparative TLC using 20% EtOAc in hexane as solvent to give 12a (17 mg, 65%). 1H NMR (400 MHz) δ = 8.10 (2H, dd, J = 7.2, 1.6 Hz), 8.00 (1H, m), 7.73 (2H, dd, J = 7.2, 1.6 Hz), 7.70-7.30 (8H, m), 7.04 (1H, d, J = 8.8 Hz), 6.98 (1H, d, J = 8 Hz), 6.90 (1H, bs), 6.85 (1H, d, J = 8 Hz), 6.63(1H, d, J = 17.6 Hz), 6.52 (1H, d, J = 18.4 Hz), 6.47 (1H, s), 6.44 (1H, d, J = 10.4 Hz), 6.16 (1H, t, J = 7 Hz), 6.03 (2H, m), 5.93 (1H, d, J = 10.8 Hz), 5.72 (1H, d, J = 8 Hz), 5.59 (1H, d, J = 8.8 Hz), 5.42(1H, dt, J = 17.2, 1 Hz), 5.29 (1H, dt, J = 10.4, 1 Hz), 4.90 (1H, d, J = 9.6 Hz), 4.87 (1H, s), 4.65 (2H, m), 4.33 (1H, d, J = 8.8 Hz), 4.26 (1H, d, J = 8.8 Hz), 3.90 (1H, d, J = 7.2 Hz), 2.58 (1H, m), 2.40 (1H, m), 2.17 (3H, s), 2.10(1H, m), 2.08 (3H, s), 1.92 (1H, m), 1.74 (3H, s), 1.62 (3H, s), 1.22 (3H, s), 1.16 (3H, s), 0.95 (30H, m), 0.59 (6H, m). 13C NMR (100 MHz) δ = 202.0, 172.0, 169.5, 167.2, 167.0, 165.3, 159.2, 140.5, 140.4, 134.2, 133.7, 133.6, 133.3, 133.0, 131.9, 130.4, 130.1, 129.8, 129.6, 129.2, 128.9, 128.7, 127.7, 127.1, 118.9, 117.9, 114.5, 113.2, 84.4, 81.5, 79.0, 76.8, 75.3, 75.28, 75.22, 72.4, 71.7, 69.1, 58.6, 55.9, 47.0, 43.5, 37.4, 26.7, 21.8, 21.1, 18.0, 14.4, 12.7, 10.4, 6.9, 5.5. HRFABMS m/z calcd for C66H89NO15Si2Na+ 1214.5668, found 1214.5668 (Δ = 0 ppm). Characterization data for compounds 12b–f. are proved in the Supporting Information.
Representative Procedure for the Deprotection of 12a–i. Synthesis of 3′-Dephenyl-3′-(m-allyloxyphenyl)-4-deacetyl-4-acryloylpaclitaxel (13a)
To a solution of 3′-dephenyl-3′-(m-allyloxyphenyl)-4-deacetyl-4-acryloyl-7-O-triethylsilyl-2′-O-triisopropylsilylpaclitaxel (12a, 15 mg, 0.012 mmol) in THF (5 mL) was added hydrogen fluoride-pyridine complex (70%, 0.2 mL) at 0 °C, and the resulting solution was brought to room temperature overnight. Saturated NaHCO3 solution (5 mL) was added carefully to quench the reaction. Usual work-up followed by preparative TLC (silica gel, 50% EtOAc in hexane) furnished 13a (5.5 mg, 55%). (13a): 1H NMR (400 MHz, CDCl3) δ = 8.15 (2H, d, J = 7.2 Hz), 7.72 (2H, d, J = 8 Hz), 7.61 (1H, t, J = 7 Hz), 7.50 (3H, m), 7.40 (3H, m), 7.10 (2H, m), 6.91(1H, d, J = 7.6 Hz,) 6.83 (1H, d, J = 9.2 Hz), 6.50 (1H, dd, J = 17.6, 1.2 Hz), 6.35 (1H, d, J = 10.4 Hz), 6.29 (1H, s), 6.1 (1H, m), 6.05 (1H, m), 5.68 (3H, m), 5.47 (1H, dd, J = 9.2, 1.6 Hz), 5.30 (1H, dq, J = 17.2, 1.6 Hz), 5.30 (1H, dq, J = 10.5, 1.2 Hz), 4.92 (1H, dd, J = 9.4, 1.6 Hz), 4.74 (1H, d, J = 2 Hz), 4.56 (2H, m), 4.48 (1H, dd, J = 10.4, 6.8 Hz), 4.34 (1H, d, J = 8.4 Hz), 4.25 (1H, d, J = 8.4 Hz), 3.86 (1H, d, J = 6.8 Hz), 2.58 (1H, m), 2.42 (1H, m), 2.30 (1H, m), 2.24 (3H, s), 1.90 (1H, m), 1.83 (3H, s), 1.70 (3H, s), 1.62 (1H, m), 1.22 (3H, s), 1.14 (3H, s). 13C NMR (100 MHz) δ = 203.8, 173.3, 171.5, 167.2, 167.1, 165.4, 159.3, 142.3, 140.1, 133.9, 133.4, 133.2, 132.4, 132.1, 130.4, 130.2, 129.5, 129.3, 128.94, 128.91, 127.2, 119.6, 118.2, 114.5, 114.0, 84.6, 81.6, 79.1, 76.7, 75.8, 75.2, 72.7, 72.3, 72.2, 69.1, 58.8, 54.5, 45.9, 43.3, 35.9, 35.7, 32.1, 29.9, 27.0, 24.9, 22.9, 22.0, 21.0, 15.1, 9.8. HRFABMS m/z calcd for C51H56NO15+ 922.3650, found 922.36676 (Δ = 2.9 ppm). Similar procedures were employed to prepare compounds 13b–j; characterization data for these compounds are proved in the Supporting Information.
Representative Procedure for Ring-closing Metathesis of 12a–i. Synthesis of 7-O-triethylsilyl-2′-O-triisopropylsilyl Bridged Paclitaxel 14a
Grubbs second generation catalyst (2 mg) in CH2Cl2 (2.5 mL) was added over 3 h to a solution of 12a (16 mg, 0.013 mmol) in CH2Cl2 (4 mL), and the resulting solution was stirred for 1 h. The reaction mixture was concentrated and the crude mass was applied to preparative thin layer chromatography using 25% EtOAc in hexane as solvent to furnish 14a (10 mg, 65%). 14a: 1H NMR (500 MHz) δ = 8.10 (2H, d, J = 7.1 Hz), 7.82 (2H, d, J = 7.1 Hz), 7.65 (1H, t, J = 7.3 Hz), 7.53 (2H, m), 7.45 (3H, m), 7.35 (1H, m), 7.21 (2H, m), 6.94 (2H, m), 6.46 (1H, s), 6.14 (1H, m), 6.12 (1H, d, J = 16 Hz), 5.73 (1H, d, J = 7.3 Hz), 5.5 (1H, d, J = 6.6 Hz), 4.93 (2H, m), 4.85 (1H, d, J = 8 Hz), 4.56 (1H, d, J = 2 Hz), 4.48 (1H, dd, J = 10.5, 6.6 Hz), 4.28 (2H, s), 3.81 (1H, d, J = 7.1 Hz), 2.50 (1H, m), 2.29 (1H, m), 2.18 (3H, s), 2.16 (1H, m), 2.08 (3H, s), 1.99 (3H, m), 1.91 (1H, m), 1.74 (3H, s), 1.24 (3H, s), 1.17 (3H, s), 1.10 (21H, m), 0.9 (9H, t, J = 7.8Hz), 0.57 (6H, m). 13C NMR (125 MHz) δ = 201.8, 171.5, 169.5, 167.3, 167.1, 164.1, 159.7, 147.0, 142.6, 140.5, 134.1, 134.0, 133.6, 132.0, 130.1, 130.0, 129.7, 128.9, 127.2, 123.8, 118.9, 118.2, 113.7, 84.4, 81.9, 79.1, 77.0, 75.2, 75.1, 72.7, 70.8, 70.0, 58.7, 57.6, 47.6, 43.4, 37.4, 35.9, 26.8, 21.6, 21.1, 18.1, 17.9, 14.5, 12.9, 10.3, 6.9, 5.5. HRFABMS m/z calcd for C64H85NO15Si2Na+ 1186.5355, found 1185.5330 (Δ = 2.2 ppm). Similar procedures were employed to prepare compounds 14b–i; characterization data for these compounds are proved in the Supporting Information.
Representative Procedure for the Deprotection of Macrocyclic Paclitaxels 14a–j. Synthesis of Bridged Paclitaxel 15a
To a solution of 14a (9 mg, 0.0077 mmol) in THF (3.5 ml) was added hydrogen fluoride-pyridine complex (70%, 0.15 mL) at 0 °C, and the resulting solution was brought to room temperature overnight. Saturated NaHCO3 solution (5 mL) was added carefully to quench the reaction and after the usual work-up resulted the crude product was purified by preparative TLC (silica gel, 60% EtOAc in hexane) to furnish 15a (6.3 mg, 91%). 15a: 1H NMR (400 MHz) δ = 7.98 (2H, d, J = 7.6 Hz), 7.80 (2H, d, J = 7.6 Hz), 7.69 (1H, t, J = 7.6 Hz), 7.61 (1H, t, J = 6.8 Hz), 7.53 (1H, m), 7.44 (2H, m), 7.22 (1H, dd, J = 7.6 Hz), 7.10 (1H, m), 7.05 (1H, bs), 6.98 (1H, m), 6.80 (1H, bs), 6.26 (s, 1H), 6.13 (1H, d, J = 16 Hz) 6.11 (1H, d, J = 9.2 Hz), 5.60 (1H, d, J = 6.8 Hz), 5.31 (1H, dd, J = 9.3, 6.4 Hz), 5.04 (1H, dd, J = 14.4, 9.6 Hz), 4.88 (2H, m), 4.48 (1H, d, J = 9.6 Hz), 4.37 (1H, m), 4.21 (2H, s), 3.68 (1H, d, J = 6.8 Hz), 2.58 (2H, m), 2.22 (3H, s), 1.90 (1H, m), 1.84 (3H, s), 1.78 (1H, m), 1.68 (3H, s), 1.53 (1H, m), 1.25-1.22 (1H, m), 1.21 (3H, s), 1.0 (3H, s). 13C NMR (100 MHz) δ = 203.7, 173.0, 171.5, 169.3, 167.0, 163.6, 155.7, 143.3, 142.9, 138.4, 134.1, 133.6, 132.6, 132.4, 131.2, 130.1, 129.6, 128.9, 127.3, 126.8, 122.5, 119.3, 111.2, 84.3, 81.5, 79.4, 77.4, 76.7, 76.4, 75.7, 75.1, 72.5, 70.5, 66.1, 58.8, 58.6, 45.9, 43.2, 35.7, 35.0, 27.0, 22.3, 21.0, 14.8, 9.7. HRFABMS m/z calcd for C49H52NO15 894.3337, found 894.3294 (Δ = 4.8 ppm). Similar procedures were employed to prepare compounds 15b–j; characterization data for these compounds are proved in the Supporting Information.
Representative Procedure for the Hydrogenation of Macrocyclic Paclitaxels 15a–j. Synthesis of Dihydro Bridged Paclitaxel 16a
To a solution of 15a (6 mg, 0.0067 mmol) in methanol (5 ml) was added 10%Pd-C (10 mg) and hydrogenated at 35psi for 6h. The reaction mixture was filtered a through short plug of silica gel, eluting with 20% methanol in EtOAc. The filtrate was concentrated to crude mass, which was applied to silica gel preparative plate using 65% EtOAc in hexane as solvent provided the dihydro bridged paclitaxel 16a (5 mg, 83% yield). 16a: 1H NMR (400 MHz) δ = 8.06 (2H, d, J = 7.2 Hz), 7.82 (2H, d, J = 7.2 Hz), 7.10-7.03 (4H, m), 7.55-7.45 (4H, m), 7.19 (1H, t, J = 7.6 Hz), 7.08 (1H, bs), 6.96-6.88 (2H, m), 6.19 (1H, s), 6.14 (1H, t, J = 8.8 Hz) 5.57 (1H, d, J = 7.2 Hz), 5.24 (1H, dd, J = 10.6, 6.8 Hz), 4.89 (1H, d, J = 8.4 Hz), 4.62 (1H, m), 4.42 (2H, m), 4.32 (1H, m), 4.22 (1H, d, J = 7.2 Hz), 3.52 (1H, d, J = 6.8Hz), 2.70 (1H, m), 2.50 (5H, m), 2.23 (1H, m), 2.22 (3H, s), 2.04 (3H, s), 1.88 (1H, m), 1.65 (3H, s), 1.60 (4H, m), 1.42 (3H, s), 1.10 (3H, s). 13C NMR (100 MHz) δ = 203.6, 172.9, 171.5, 170.7, 167.0, 159.7, 142.7, 137.6, 134.2, 133.6, 133.5, 132.8, 132.4, 131.3, 130.4, 129.4, 129.07, 129.0, 127.4, 127.3, 122.1, 119.0, 109.6, 84.4, 80.9, 79.6, 76.3, 75.6, 74.9, 72.4, 70.5, 64.9, 64.5, 60.6, 59.7, 58.4, 45.7, 43.2, 38.3, 35.5, 35.1, 32.1, 31.4, 30.8, 29.92, 29.89, 29.5, 27.8, 27.1, 22.5, 21.0, 19.3, 14.7, 14.3, 13.9, 9.7. HRFABMS m/z calcd for C49H54NO15Na+ 918.3313, found 918.3337 (Δ = 2.6 ppm).
NAMFIS Analysis: 2D NMR-ROESY Spectra for 15f and 18f
The 1D 1H assignments of 15f and 18f were accomplished by interpretation of the corresponding COSY, HMBC and HSQC spectra. The 2D NMR ROESY analysis for both compounds was performed on an INOVA 400 MHz NMR spectrometer with 70, 100, 125, 150, 180 ms mixing times to check linearity of the cross-relaxation buildup rates. Interproton distances were calculated from the integrated cross peak volumes using the initial rate approximation and an internal calibration distance between H-6a and H-6b of 1.77 Å. This provided 19 and 15 ROE-based interproton distances for 15f and 18f, respectively.
Solution conformation deconvolution for 15f and 18f
A 10,000 step conformational search was performed on both molecules with the MMFF94s force field and the GBSA/H20 solvation model in MacroModel v7.2. An energy cutoff of 12 kcal/mol resulted in a pool of 93 and 374 unique conformations for 15f and 18f, respectively, with the global minimum found 62 and 29 times, respectively. The NAMFIS methodology integrated 19 ROE-determined distances measured in CDCl3 and the 93 conformers of 15f to deconvolute the NMR data into four conformations. One of these, however, places the C-4 acetate C=O beneath the oxetane ring. Inspection of literature X-ray structures of PTX and analogs shows that the carbonyl group consistently resides beneath the 6-membered C-ring. Quantum chemical calculations (B3LYP/6-31G*, not shown) also demonstrate that oxetane orientation is ca. 4 kcal/mol higher in energy. Thus the 93 conformers were depleted of the latter conformation, and NAMFIS was rerun to give five conformations with populations of 40, 27, 17, 9 and 7%. (SSD=59). The dominant conformer is the T-form (40%); while the second and fourth most populated structures (27 and 9%, respectively) are slightly collapsed variants of the T-conformation in which the C3’- benzamido phenyl to C2-phenyl centroid distance is 3 Å shorter. The third and fifth conformers (17 and 7%, respectively) are somewhat extended structures that can be classified as neither T nor polar forms. For 18f, the 374 conformations likewise included a subset of carbonyl/oxetane structures, one of which appeared in the initial NAMFIS run employing 15 ROE-determined distances. These high-energy structures were removed from the dataset. A second NAMFIS run yielded three conformations with populations of 53, 33 and 14%(SSD=96). One is the T-conformation (53%); the second, a slightly collapsed T-structure (33%); the third, the polar conformer (14%).
Glide docking
The top populated NAMFIS derived conformation of 15f was docked into the 1JFF tubulin pocket using Glide in Maestro. An initial rigid dock of the ligand did not allow it to be situated in the pocket, so a follow-up flexible dock using the extra position (XP) option was performed. It resulted in three very similar poses, all in the T-shape form, with the top pose differing from the NAMFIS conformation with an RMS deviation of only 0.6 Å when all heavy atoms (C,N,O) were superposed.
Biological Data
Cytotoxicities were determined using the published method58 for A2780 cells, the MTT assay59 for PC3 cells, and the sulforhodamine-B method for the paclitaxel-resistant cell lines.55 ED50 values for induction of tubulin assembly were determined using either light scattering at 350 nm in a Hewlett-Packard 8453 absorption spectrometer or fluorescence in a 96-well plate format.60
Supplementary Material
Scheme 7. Synthesis of Bridged Paclitaxels 25 – 28a.
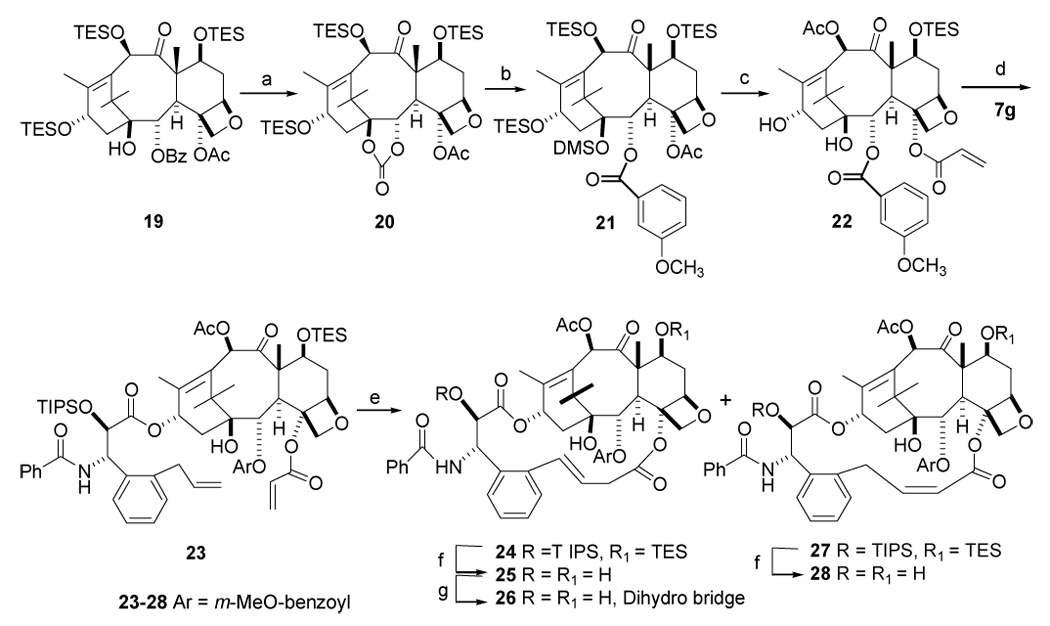
a a. i, Red-Al, THF, 0 °C, ii, carbonyldiimidazole, imidazole, 33%(overal 2 steps) b. i, m-methoxyl phenyl-MgBr, Et2O, O °C. ii, DMSCl, imidazole, 70%( oveal 2 steps). c. i, Red-Al, THF, 0 °C, 55% ii, LHMDS, CH2=CHCOCl, THF, 43%, iii, HF/Py, THF, 0 °C-RT, 70%. iv, Ac2O, CeCl3, THF, RT, 90%, v, TESCl, imidazole, DCM, 84%. d. LHMDS, THF, 90%. e. Grubbs catalyst (2nd), CH2Cl2, RT, 70%. f. HF/Py, THF, 80%. g. H2, Pd/C, MeOH, 60%.
Scheme 8. Synthesis of Bridged Paclitaxels 31 and 32a.
a a. LHMDS, THF, 90%. b. Second generation Grubbs' catalyst, CH2Cl2, RT, 70%. c. HF/Py, THF, 80% d. H2, Pd/C, MeOH, 60%.
Acknowledgements
This work was supported by the National Cancer Institute, National Institutes of Health (Grant CA-69571) and we are grateful for this support. We are likewise grateful to William Bebout and Rebecca Guza in the Department of Chemistry, Virginia Polytechnic Institute and State University, for mass spectra and preliminary cytotoxicity determinations, respectively. We are pleased to acknowledge the support and encouragement of Prof. Dennis Liotta (Department of Chemistry, Emory University).
Abbreviations
- PTX
paclitaxel
- NAMFIS
NMR analysis of molecular flexibility in solution
Footnotes
Supporting Information Available: Characterization data for new compounds 7b–d, 7f–g, 11b–d, 12b–k, 13b–j, 14c–d, 14f–k, 15c–d, 15f–k, 16c–d, 16f–j, 25, 26, 28, 31a, 31b, 32a, and 32b. 1H NMR spectra of compounds 15c, 15d, 15f, 15f (expanded), 15f(COSY), 16c, 16f, 25, 31a, 31b, 32a, and 32b. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Rowinsky EK. The Development and Clinical Utility of the Taxane Class of Antimicrotuble Chemotherapy Agents. Ann. Rev. Med. 1997;48:353–374. doi: 10.1146/annurev.med.48.1.353. [DOI] [PubMed] [Google Scholar]; (b) Crown J, O'Leary M. The Taxanes: an Update. The Lancet. 2000;355:1176–1178. doi: 10.1016/S0140-6736(00)02074-2. [DOI] [PubMed] [Google Scholar]
- 2.Halkin A, Stone GW. Polymer-based Paclitaxel-eluting Stents in Percutaneous Coronary Intervention: A Review of the TAXUS Trials. J. Intervent. Cardiol. 2004;17:271–282. doi: 10.1111/j.1540-8183.2004.04040.x. [DOI] [PubMed] [Google Scholar]
- 3.(a) Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Epothilones, a New Class of Microtubule-stabilizing Agents with a Taxol-like Mechanism of Action. Cancer Res. 1995;55:2325–2333. [PubMed] [Google Scholar]; (b) Kowalski RJ, Giannakakou P, Hamel E. Activities of the Microtubule-stabilizing Agents Epothilones A and B with Purified Tubulin and in Cells Resistant to Paclitaxel (Taxol) J. Biol. Chem. 1997;272:2534–2541. doi: 10.1074/jbc.272.4.2534. [DOI] [PubMed] [Google Scholar]
- 4.ter Haar E, Kowalski RJ, Hamel E, Lin CM, Longley RE, Gunasekera SP, Rosenkranz HS, Day BW. Discodermolide, a Cytotoxic Marine Agent that Stabilizes Microtubules More Potently than Taxol. Biochemistry. 1996;35:243–250. doi: 10.1021/bi9515127. [DOI] [PubMed] [Google Scholar]
- 5.Gapud EJ, Bai R, Ghosh AK, Hamel E. Laulimalide and Paclitaxel: A Comparison of their Effects on Tubulin Assembly and their Synergistic Action when Present Simultaneously. Mol. Pharmacol. 2004;66:113–121. doi: 10.1124/mol.66.1.113. [DOI] [PubMed] [Google Scholar]
- 6.Lindel T, Jensen PR, Fenical W, Long BH, Casazza AM, Carboni J, Fairchild CR. Eleutherobin, a New Cytotoxin that Mimics Paclitaxel (Taxol) by Stabilizing Microtubules. J. Am. Chem. Soc. 1997;119:8744–8745. [Google Scholar]
- 7.Schiff PB, Fant J, Horwitz SB. Promotion of Microtubule Assembly in vitro by Taxol. Nature. 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- 8.Horwitz SB. Mechanism of Action of Taxol. Trends Pharmacol. Sci. 1992;13:134–136. doi: 10.1016/0165-6147(92)90048-b. [DOI] [PubMed] [Google Scholar]
- 9.Jordan MA, Toso RJ, Thrower D, Wilson L. Mechanism of Mitotic Block and Inhibition of Cell Proliferation by Taxol at Low Concentrations. Proc. Natl. Acad. Sci. 1993;90:9552–9556. doi: 10.1073/pnas.90.20.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blagosklonny MV, Fojo T. Molecular Effects of Paclitaxel: Myths and Reality (a Critical Review) Int. J. Cancer. 1999;83:151–156. doi: 10.1002/(sici)1097-0215(19991008)83:2<151::aid-ijc1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 11.a) Sunters A, Madureira PA, Pomeranz KM, Aubert M, Brosens JJ, Cook SJ, Burgering BM, Coombes RC, Lam EW. Paclitaxel-induced Nuclear Translocation of FOXO3a in Breast Cancer Cells is Mediated by c-Jun NH2-terminal Kinase and Akt. Cancer Res. 2006;66:212–220. doi: 10.1158/0008-5472.CAN-05-1997. [DOI] [PubMed] [Google Scholar]; b) Faried LS, Faried A, Kanuma T, Nakazato T, Tamura T, Kuwano H, Minegishi T. Inhibition of the Mammalian Target of Rapamycin (mTOR) by Rapamycin Increases Chemosensitivity of CaSki Cells to Paclitaxel. Eur. J. Cancer. 2006;42:934–947. doi: 10.1016/j.ejca.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 12.Nogales E, Wolf SG, Downing KH. Structure of the αβ. Tubulin Dimer by Electron Crystallography. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- 13.Lowe J, Li H, Downing KH, Nogales E. Refined Structure of αβ-Tubulin at 3.5 Å Resolution. J. Mol. Biol. 2001;313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- 14.Dubois J, Guenard D, Gueritte-Voeglein F, Guedira N, Potier P, Gillet B, Betoeil J-C. Conformation of Taxotere and Analogues Determined by NMR Spectroscopy and Molecular Modeling Studies. Tetrahedron. 1993;49:6533–6544. [Google Scholar]
- 15.Williams HJ, Scott AI, Dieden RA, Swindell CS, Chirlian LE, Francl MM, Heerding JM, Krauss NE. NMR and Molecular Modeling Study of Active and Inactive Taxol Analogues in Aqueous and Non Aqueous Solution. Can. J. Chem. 1994;72:252–260. [Google Scholar]
- 16.Cachau RE, Gussio R, Beutler JA, Chmurny GN, Hilton BD, Muschik GM, Erickson JW. Solution Structure of Taxol Determined Using a Novel Feedback-scaling Procedure for NOE-restrained Molecular Dynamics. Supercomput Appl. High Perform. Comput. 1994;8:24–34. [Google Scholar]
- 17.Vander Velde DG, Georg GI, Grunewald GL, Gunn CW, Mitscher LA. "Hydrophobic Collapse" of Taxol and Taxotere Solution Conformations in Mixtures of Water and Organic Solvent. J. Am. Chem. Soc. 1993;115:11650–11651. [Google Scholar]
- 18.Paloma LG, Guy RK, Wrasidlo W, Nicolaou KC. Conformation of a Water-soluble Derivative of Taxol in Water by 2D-NMR Spectroscopy. Chem. Biol. 1994;1:107–112. doi: 10.1016/1074-5521(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 19.Ojima I, Chakravarty S, Inoue T, Lin S, He L, Horwitz SB, Kuduk SC, Danishefsky SJ. A Common Pharmacophore for Cytotoxic Natural Products that Stabilize Microtubules. Proc. Natl. Acad. Sci. USA. 1999;96:4256–4261. doi: 10.1073/pnas.96.8.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ojima I, Kuduk SD, Chakravarty S, Ourevitch M, Begue J-P. A Novel Approach to the Study of Solution Structures and Dynamic Behavior of Paclitaxel and Docetaxel Using Fluorine-containing Analogs as Probes. J. Am. Chem. Soc. 1997;119:5519–5527. [Google Scholar]
- 21.Ojima I, Inoue T, Chakravarty S. Enantiopure Fluorine-containing Taxoids: Potent Anticancer Agents and Versatile Probes for Biomedical Problems. J. Fluorine Chem. 1999;97:3–10. [Google Scholar]
- 22.Snyder JP, Nevins N, Cicero DO, Jansen J. The Conformations of Taxol in Chloroform. J. Am. Chem. Soc. 2000;122:724–725. [Google Scholar]
- 23.Snyder JP, Nevins N, Jimenez-Barbero Cicero D, Jansen JM. Unpublished. [Google Scholar]
- 24.Li Y, Poliks B, Cegelski L, Poliks M, Gryczynski Z, Piszczek G, Jagtap PG, Studelska DR, Kingston DGI, Schaefer J, Bane S. Conformation of Microtubule-Bound Paclitaxel Determined by Fluorescence Spectroscopy and REDOR NMR. Biochemistry. 2000;39:281–291. doi: 10.1021/bi991936r. [DOI] [PubMed] [Google Scholar]
- 25.Barboni L, Lambertucci C, Appendino G, Vander Velde DG, Himes RH, Bombardelli E, Wang M, Snyder JP. Synthesis and NMR-Driven Conformational Analysis of Taxol Analogoues Conformationally Constrained on the C13 Side Chain. J. Med. Chem. 2001;44:1576–1587. doi: 10.1021/jm001103v. [DOI] [PubMed] [Google Scholar]
- 26.Boge TC, Wu Z-J, Himes RH, Vander Velde DG, Georg GI. Conformationally Restricted Paclitaxel Analogues: Macrocyclic Mimics of the "Hydrophobic Collapse" Conformation. Bioorg. Med. Chem. Lett. 1999;9:3047–3052. doi: 10.1016/s0960-894x(99)00522-3. [DOI] [PubMed] [Google Scholar]
- 27.Ojima I, Lin S, Inoue T, Miller ML, Borella CP, Geng X, Walsh JJ. Macrocycle Formation by Ring-closing Metathesis. Application to the Syntheses of Novel Macrocyclic Taxoids. J. Am. Chem. Soc. 2000;122:5343–5353. [Google Scholar]
- 28.Ojima I, Geng X, Lin S, Pera P, Bernacki RJ. Design, Synthesis and Biological Activity of Novel C2-C3′ N-Linked Macrocyclic Taxoids. Bioorg. Med. Chem. Lett. 2002;12:349–352. doi: 10.1016/s0960-894x(01)00747-8. [DOI] [PubMed] [Google Scholar]
- 29.Geng X, Miller ML, Lin S, Ojima I. Synthesis of Novel C2-C3′N-Linked Macrocyclic Taxoids by Means of Highly Regioselective Heck Macrocyclization. Org. Lett. 2003;5:3733–3736. doi: 10.1021/ol0354627. [DOI] [PubMed] [Google Scholar]
- 30.Querolle O, Dubois J, Thoret S, Dupont C, Guéritte F, Guénard D. Synthesis of Novel 2-O,3′-N-Linked Macrocyclic Taxoids with Variable Ring Size. Eur. J. Org. Chem. 2003:542–550. [Google Scholar]
- 31.Querolle O, Dubois J, Thoret S, Roussi F, Montiel-Smith S, Guéritte F, Guénard D. Synthesis of Novel Macrocyclic Docetaxel Analogues. Influence of Their Macrocyclic Ring Size on Tubulin Activity. J. Med. Chem. 2003;46:3623–3630. doi: 10.1021/jm030770w. [DOI] [PubMed] [Google Scholar]
- 32.a) Querolle O, Dubois J, Thoret S, Roussi F, Guéritte F, Guénard D. Synthesis of C2-C3’ N-Linked Macrocyclic Taxoids. Novel Docetaxel Analogs with High Tubulin Activity. J. Med. Chem. 2004;47:5937–5944. doi: 10.1021/jm0497996. [DOI] [PubMed] [Google Scholar]; b) Geney R, Sun L, Pera P, Bernacki RJ, Xia S, Horwitz SB, Simmerling CL, Ojima I. Use of the Tubulin Bound Paclitaxel Conformation for Structure-Based Rational Drug Design. Chem. Biol. 2005;12:339–348. doi: 10.1016/j.chembiol.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 33.Kingston DGI, Bane S, Snyder JP. The Taxol Pharmacophore and the T-Taxol Bridging Principle. Cell Cycle. 2005;4:279–289. [PubMed] [Google Scholar]
- 34.Guéritte-Voegelein F, Guénard D, Mangatal L, Potier P, Guilhem J, Césario M, Pascard C. Structure of a Synthetic Taxol Precursor: N-tert-Butoxycarbonyl-10-deacetyl-N-debenzoyltaxol. Acta Crystallogr. C. 1990;46:781–784. [Google Scholar]
- 35.Cicero DO, Barbato G, Bazzo R. NMR Analysis of Molecular Flexibility in Solution: A New Method for the Study of Complex Distributions of Rapidly Exchanging Conformations. Application to a 13-Residue Peptide with an 8-Residue Loop. J. Am. Chem. Soc. 1995;117:1027–1033. [Google Scholar]
- 36.Snyder JP, Nettles JH, Cornett B, Downing KH, Nogales E. The Binding Conformation of Taxol in β-Tubulin: A Model Based on the Electron Crystallographic Density. Proc. Natl. Acad. Sci. USA. 2001;98:5312–5316. doi: 10.1073/pnas.051309398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ganesh T, Guza RC, Bane S, Ravindra R, Shanker N, Lakdawala AS, Snyder JP, Kingston DGI. The Bioactive Taxol Conformation on β-tubulin: Experimental Evidence from Highly Active Constrained Analogs. Proc. Natl. Acad. Sci. USA. 2004;101:10006–10011. doi: 10.1073/pnas.0403459101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Metaferia BB, Hoch J, Glass TE, Bane SL, Chatterjee SK, Snyder JP, Lakdawala A, Cornett B, Kingston DGI. Synthesis and Biological Evaluation of Novel Macrocyclic Paclitaxel Analogues. Org. Lett. 2001;3:2461–2464. doi: 10.1021/ol016124d. [DOI] [PubMed] [Google Scholar]
- 39.Paik Y, Yang C, Metaferia B, Tang S, Bane S, Ravindra R, Shanker N, Alcaraz AA, Snyder JP, Schaefer J, O’Connor RD, Cegelski L, Kingston DGI. REDOR NMR Distance Measurements for the Tubulin-Bound Paclitaxel Conformation. J. Am. Chem. Soc. 2007 doi: 10.1021/ja0656604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang S, Yang C, Brodie P, Bane S, Ravindra R, Sharma S, Jiang Y, Snyder JP, Kingston DGI. Bridging Converts a Noncytotoxic nor-Paclitaxel Derivative to a Cytotoxic Analog by Constraining it to the T-Taxol Conformation. Org. Lett. 2006;8:3983–3986. doi: 10.1021/ol061438s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.For the leading references, see:Trnka TM, Grubbs RH. The Development of L2X2Ru=CHR Olefin Metathesis Catalysts: An Organometallic Success Story. Acc. Chem. Res. 2001;34:18–29. doi: 10.1021/ar000114f., and references cited thereinFürstner A. Olefin Metathesis and Beyond. Angew. Chem. Int. Ed. Engl. 2000;39:3012–3043.Grubbs RH, Chang S. Recent Advances in Olefin Metathesis and its Applications in Organic Synthesis. Tetrahedron. 1998;54:4413–4450.and references cited therein.
- 42.For the synthesis of various substituted racemic β-lactams, see:Carr JA, Al-Azemi TF, Long TE, Shim J-Y, Coates CM, Turos E, Bisht KS. Lipase Catalyzed Resolution of 4-Aryl Substituted β-Lactams: Effect of Substitution on the 4-Aryl Ring. Tetrahedron. 2003;59:9147–9160., and references cited therein.
- 43.Brieva R, Crich JZ, Sih CJ. Chemoenzymatic Synthesis of the C-13 Side Chain of Taxol: Optically Active 3-Hydroxy-4-phenyl β-lactam Derivatives. J. Org. Chem. 1993;58:1068–1075. [Google Scholar]
- 44.Substrates 4a–d took 3–7 days for resolution using lipase PS, and substrate 4e took 35 days for complete resolution.
- 45.For leading references, see:Littke AF, Schwarz L, Fu GC. Pd/P(t-Bu3): A Mild and General Catalyst for Stille Reactions of Aryl Chlorides and Aryl Bromides. J. Am. Chem. Soc. 2002;124:6343–6348. doi: 10.1021/ja020012f., and references cited therein.
- 46.Chen S-H, Kadow JF, Farina V, Fairchild CR, Johnston KA. First Synthesis of Novel Paclitaxel (Taxol) Analogs Modified at the C4-Position. J. Org. Chem. 1994;59:6156–6158. [Google Scholar]
- 47.Holton RA, Zhang Z, Clarke PA, Nadizadeh H, Procter DJ. Selective Protection of the C(7) and C(10) Hydroxyl Groups in 10-Deacetyl Baccatin III. Tetrahedron Lett. 1998;39:2883–2886. [Google Scholar]
- 48.(a) Holton RA, Biediger RJ, Boatman PD. Semisynthesis of Taxol and Taxotere. In: Suffness M, editor. Taxol: Science and Application. New York: CRC Press; 1995. pp. 97–121. [Google Scholar]; (b) Georg GI, Boge TC, Cheruvallath ZS, Clowers JS, Harriman GCB, Hepperle M, Park H. The Medicinal Chemistry of Taxol. In: Suffness M, editor. Taxol: Science and Application. New York: CRC Press; 1995. pp. 317–375. [Google Scholar]; (c) Ojima I, Habus I, Zhao M, Zucco M, Park YH, Sun CM, Brigaud T. New and Efficient Approaches to the Semisynthesis of Taxol and its C-13 Side Chain Analogs by Means of β-Lactam Synthon Method. Tetrahedron. 1992;48:6985–7012. [Google Scholar]; (d) Ojima I, Lin S, Chakravarty S, Fenoglio I, Park YH, Sun C-M, Appendino G, Pera P, Veith JM, Bernacki RJ. Syntheses and Structure-Activity Relationships of Novel Nor-seco Taxoids. J. Org. Chem. 1998;63:1637–1645. [Google Scholar]
- 49.The reader should note that Phe272 is the pdb structure code for the Phe 270 sequence code (Fβ270V) in Table 2.
- 50.Wang M, Xia X, Kim Y, Hwang D, Jansen JM, Botta M, Liotta DC, Snyder JP. A Unified and Quantitative Receptor Model for the Microtubule Binding of Paclitaxel and Epothilone. Org. Lett. 1999;1:43–46. doi: 10.1021/ol990521v. [DOI] [PubMed] [Google Scholar]
- 51.Chaudhary AG, Gharpure MM, Rimoldi JM, Chordia MD, Gunatilak AAL, Kingston DGI, Grover S, Lin CM, Hamel E. Unexpectedly Facile Hydrolysis of the 2-Benzoate Group of Taxol and Synthesis of Analogs With Improved Activities. 1994;116:4097–4098. [Google Scholar]
- 52.(a) Ojima I, Kuduk SD, Pera P, Veith JM, Bernacki RJ. Syntheses and Structure-Activity Relationships of Nonaromatic Taxoids: Effects of Alkyl and Alkenyl Ester Groups on Cytotoxicity. J. Med. Chem. 1997;40:279–285. doi: 10.1021/jm9606711. [DOI] [PubMed] [Google Scholar]; (b) Georg GI, Harriman GCB, Ali SM, Datta A, Hepperle M, Himes RH. Synthesis of 2-O-Heteroaroyl Taxanes: Evaluation of Microtubule Assembly Promotion and Cytotoxicity. Bioorg. Med. Chem. Lett. 1995;5(2):115–118. [Google Scholar]; (c) Nicolaou KC, Renaud J, Nantermet PG, Couladouros EA, Guy RK, Wrasidlo W. Chemical Synthesis and Biological Evaluation of C-2 Taxoids. J. Am. Chem. Soc. 1995;117:2409–2420. [Google Scholar]
- 53.Alcaraz AA, Mehta AK, Snyder JP. The T-Taxol Conformation. J. Med. Chem. 2006;49:2478–2488. doi: 10.1021/jm051119r. [DOI] [PubMed] [Google Scholar]
- 54.Giannakakou P, Gussio R, Nogales E, Downing KH, Zaharevitz D, Bollbuck B, Poy G, Sackett D, Nicolaou KC, Fojo TA. A common pharmacophore for epothilone and taxanes: Molecular basis for drug resistance conferred by tubulin mutations in human cancer cells. Proc. Natl. Acad. Sci. USA. 2000;97:2904–2909. doi: 10.1073/pnas.040546297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giannakakou P, Sackett D, Kang Y-K, Zhan Z, Buters JTM, Fojo T, Poruchynsky MS. Paclitaxel-resistant Human Ovarian Cancer Cells have Mutant β-Tubulins That Exhibit Impaired Palitaxel-driven Polymerization. J. Biol. Chem. 1997;272:17118–17125. doi: 10.1074/jbc.272.27.17118. [DOI] [PubMed] [Google Scholar]
- 56.Zhou J, O’Brate A, Zelnak A, Giannakakou P. Survivin Deregulation in β-Tubulin Mutant Ovarian Cancer Cells Underlies Their Compromised Mitotic Response to Taxol. Cancer Res. 2004;64:8708–8714. doi: 10.1158/0008-5472.CAN-04-2538. [DOI] [PubMed] [Google Scholar]
- 57.a) Nevins N, Cicero D, Snyder JP. A Test of the Single-Conformation Hypothesis in the Analysis of NMR Data for Small Polar Molecules: A Force Field Comparison. J. Org. Chem. 1999;64:3979–3986. [Google Scholar]; b) Monteagudo E, Cicero DO, Cornett B, Myles DC, Snyder JP. The Conformations of Discodermolide in DMSO. J. Am. Chem. Soc. 2001;123:6929–6930. doi: 10.1021/ja015569u. [DOI] [PubMed] [Google Scholar]; c) Thepchatri P, Cicero DO, Monteagudo E, Ghosh AK, Cornett B, Snyder JP. Conformations of Laulimalide in DMSO-d6. J. Amer. Chem. Soc. 2005;127:12838–12846. doi: 10.1021/ja042890e. [DOI] [PubMed] [Google Scholar]
- 58.Louie KG, Behrens BC, Kinsella TJ, Hamilton TC, Grotzinger KR, McKoy WM, Winker MA, Ozols RF. Radiation Survival Parameters of Antineoplastic Drug-sensitive and Resistant Human Ovarian Cancer Cell Lines and Their Modification by Buthionine Sulfoximine. Cancer Res. 1985;45:2110–2115. [PubMed] [Google Scholar]
- 59.Chang MC, Uang BJ, Wu HL, Lee JJ, Hahn LJ, Jeng JH. Inducing The Cell Cycle Arrest and Apoptosis of Oral KB Carcinoma Cells by Hydroxychavicol: Roles of Glutathione and Reactive Oxygen Species. Br. J. Pharmacol. 2002;135:619–630. doi: 10.1038/sj.bjp.0704492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.(a) Chatterjee SK, Barron DM, Vos S, Bane S. Baccatin III Induces Tubulin to Assemble into Long Microtubules. Biochemistry. 2001;40:6964–6970. doi: 10.1021/bi002816i. [DOI] [PubMed] [Google Scholar]; (b) Barron DM, Chatterjee SK, Ravindra R, Roof R, Baloglu E, Kingston DGI, Bane S. A Fluorescence-Based High-Throughput Assay for Antimicrotubule Drugs. Anal. Biochem. 2003;315:49–56. doi: 10.1016/s0003-2697(02)00691-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.