

Abstract
We demonstrated that peripheral T cell tolerance toward murine melanoma self-antigens gp100 and TRP-2 can be broken by an autologous oral DNA vaccine containing the murine ubiquitin gene fused to minigenes encoding peptide epitopes gp10025–33 and TRP-2181–188. These epitopes contain dominant anchor residues for MHC class I antigen alleles H-2Db and H-2Kb, respectively. The DNA vaccine was delivered by oral gavage by using an attenuated strain of Salmonella typhimurium as carrier. Tumor-protective immunity was mediated by MHC class I antigen-restricted CD8+ T cells that secreted TH1 cytokine IFN-γ and induced tumor rejection and growth suppression after a lethal challenge with B16G3.26 murine melanoma cells. Importantly, the protective immunity induced by this autologous DNA vaccine against murine melanoma cells was at least equal to that achieved through xenoimmunization with the human gp10025–33 peptide, which differs in its three NH2-terminal amino acid residues from its murine counterpart and was previously reported to be clearly superior to an autologous vaccine in inducing protective immunity. The presence of ubiquitin upstream of the minigene proved to be essential for achieving this tumor-protective immunity, suggesting that effective antigen processing and presentation may make it possible to break peripheral T cell tolerance to a self-antigen. This vaccine design might prove useful for future rational designs of other recombinant DNA vaccines targeting tissue differentiation antigens expressed by tumors.
The development of antigen-specific cancer vaccines was stimulated by the recent identification and cloning of several unaltered melanoma self-antigens that can be recognized by T cells isolated from mice or human cancer patients (1–3). Such autoreactive T cells apparently escape thymic deletion and reach the periphery, where they can be activated to induce effective antitumor immune responses (4, 5). Among melanoma-associated antigens recognized by T cells are gp100 and tyrosinase-related proteins TRP-1 and TRP-2, which are lineage-specific differentiation antigens expressed by both melanocytes and melanoma cells of humans and mice (6–10). In an attempt to determine the requirements for breaking peripheral T cell tolerance to such naturally expressed antigens, cytotoxic T lymphocytes (CTLs) against gp100 were elicited by immunization of mice with a recombinant vaccinia virus encoding human gp100, which fortuitously generated a heteroclitic response against the corresponding murine gp100 epitope. These T cells, once cloned, substantially reduced established pulmonary B16WT melanoma metastases in C57BL/6J mice. Importantly, an autologous vaccine encoding murine gp100 failed to generate tumor-protective immunity in this model system. The mechanism responsible for the greater immunogenicity of the human version of gp100 became apparent when mice were immunized with recombinant vaccinia virus containing minigenes encoding either human gp10025–33 or murine gp10025–33 peptide epitopes. Although the human and murine gp10025–33 epitopes were homologous and conformed to the predicted H-2Db antigen binding motif, differences in three NH2-terminal amino acids resulted in a 2-log increase in the ability of the human peptide to stabilize “empty” H-2Db antigens on RMA-S cells and a 3-log increase in triggering IFN-γ release by T cells compared with the murine peptide (10).
TRP-2181–188, a peptide that shares the same amino acid sequence in humans and mice and conforms to a predicted H-2Kb antigen binding motif, 5(F) and 8(L), was identified as the major reactive epitope within the TRP-2 antigen recognized by anti-B16 CTLs. A CTL line raised from normal mouse splenocytes by repeated stimulation with the TRP-2181–188 peptide was therapeutic against 3-day-old established murine B16 melanoma lung metastases when low-dose systemic IL-2 was given with adoptive cell transfer (9). Recently, a recombinant vaccinia virus encoding the murine melanoma self-antigen TRP-1 was reported to induce autoimmune vitiligo and tumor cell destruction in mice that required CD4+ T lymphocytes to protect C57BL/6J mice completely against s.c. B16 murine melanoma cell challenge (11). However, immunizations with DNA vaccines containing minigenes encoding mouse gp10025–33 combined with TRP-2181–188 epitopes have not reported thus far to induce antitumor immunity capable of rejecting s.c. challenges of wild-type B16 murine melanoma cells.
The induction of a most effective antigen-specific immunity by DNA vaccines involves optimization of the vaccine design, including different approaches for vaccine delivery and effective antigen processing. This modality includes the use of an oral delivery system with an attenuated strain of Salmonella typhimurium (12). Oral immunization with S. typhimurium harboring plasmid DNA vaccines proved effective in DNA delivery and subsequent induction of immunity against antigens encoded by the plasmid (12). DNA immunization can be enhanced significantly by exploiting the natural pathways of antigen presentation. Thus, most peptides presented by MHC class I antigens that induce CTL are derived from cytosolic proteins degraded by the proteasome. Proteins are targeted to this organelle by ubiquitination, a process in which many copies of the cellular protein ubiquitin are covalently attached to the target protein. Reasoning that covalent attachment of ubiquitin to the target protein might enhance its degradation by the proteasome, a DNA vaccine was prepared in which the lymphocytic choriomeningitis virus nucleoprotein was cotranslationally expressed with a modified ubiquitin (13). The ubiquitinated nucleoprotein was more rapidly degraded by the proteasome, and this DNA vaccine conferred significantly better antiviral immunity than did a vaccine lacking ubiquitin. These findings were confirmed and extended by analyses of ubiquitin attached to minigenes encoding lymphocytic choriomeningitis virus CTL epitopes; again, protective immunity was greatly enhanced, and a 6-fold increase in CTL precursor frequency was observed (14).
Herein, we applied an alternative mode of DNA delivery in an attempt to break peripheral T cell tolerance to murine melanoma self-antigens with an attenuated strain of S. typhimurium serving as a carrier for eukaryotic expression vectors encoding for ubiquitinated gp10025–33 and TRP-2181–188 peptide epitopes. An autologous, oral vaccine encoding these ubiquitinated murine melanoma peptide epitopes induced an effective tumor-protective immunity that was mediated by CD8+ T cells in an MHC class I-restricted manner. This immunity was achieved in a prophylactic setting against an s.c. challenge with highly tumorigenic but poorly immunogenic B16G3.26 melanoma cells.
Materials and Methods
Animals, Bacterial Strains, and Cell Lines.
C57BL/6J mice and C57BL/6J scid/scid mice 6–8 weeks of age were purchased from The Jackson Laboratory or from the breeding colony at the Scripps Research Institute. The attenuated S. typhimurium AroA− strain SL7207 {S. typhimurium 2337-65 derivative his G46.DEL407 [aroA∷Tn10 (Tc-S)]} was kindly provided by B. A. D. Stocker (Stanford University, Stanford, CA). Bacterial strains DH5α and JM101 were purchased from Invitrogen. Bacteria were grown routinely at 37°C in LB broth or on agar plates (Sigma) supplemented, when required, with 50 μg/ml ampicillin. The B16G3.26 melanoma cell line was obtained originally from C. Stackpole (New York Medical College, Valhalla, NY) and cultured in RPMI medium 1640 supplemented with 10% (vol/vol) FBS as reported (15). The TRAMP-C1 (pTC1) prostate tumor cell line was kindly provided by N. M. Greenberg (Baylor College of Medicine, Houston, TX). This line is an early passage murine prostate cancer cell line derived from TRAMP mice that spontaneously developed prostate cancer because of prostate-specific simian virus 40 large tumor antigen (Tag) expression (16). The murine macrophage cell line RAW was obtained from the American Type Culture Collection.
Construction of Expression Vectors.
The following three expression vectors were constructed: pUb-M and pUb-H containing ubiquitinated murine and human melanoma gp10025–33 and TRP-2181–188 peptide epitopes, respectively. p-Mini is an expression vector including the nonubiquitinated murine peptide epitopes, and pUb-V contains ubiquitin only (13) and serves as an empty vector control. Specifically, mouse gp10025–33 (EGSRNQDWL) and TRP-2 181–188 (VYDFFVWL) peptide epitopes containing dominant H-2Db 5(N), 9(L), H-2Kb 5(F), and 8(L) anchor residues were amplified by PCR with oligonucleotides with a 20-bp overlap. The PCR-amplified DNA fragments were then cloned into the TOPO-TA cloning vector (Invitrogen). To generate the pUb-M expression vector, the fragment containing the ubiquitinated murine peptide epitope was excised by BglII and cloned into the same site of the pCMV-Ub vector (13). The pUb-H expression vector was constructed by using the same strategy, except that the epitopes were changed to human gp10025–33 (KVPRNQDWL) and TRP-2 181–188 (VYDFFVWL) CTL peptide epitopes with dominant H-2Db 5(N), 9(L), H-2Kb 5(F), and 8(L) anchor residues, respectively. The third expression vector, p-Mini, was generated by removing the ubiquitin sequence from pUb-M or pUb-H. Vector construction is illustrated schematically in Fig. 1.
Figure 1.

Schematic map of vector construction. Minigenes encoding for human or mouse gp10025–33 and TRP-2181–188 were assembled by PCR with overlapping oligonucleotides as templates. The PCR fragments generated were cloned into pCMV-Ub (13) by using a unique BglII restriction site downstream of the coding sequence for mouse ubiquitin. SV40, simian virus 40.
Transduction of S. typhimurium with DNA Vaccine Plasmids.
Attenuated S. typhimurium bacteria were transduced with DNA vaccine plasmids by electroporation according to the following protocol. A single colony of bacteria was inoculated into 3 ml of LB medium, and the bacteria were harvested during mid-log phase after washing them twice with ice-cold water. Freshly prepared bacteria (1 × 108) were then admixed with 10 pg to 0.1 μg of plasmid DNA on ice in a 0.2-cm cuvette and electroporated with a Bio-Rad Gene Pulser at 2.5 kV, 25 μF, and 200 Ω. The bacteria were removed subsequently from the cuvette into a sterile culture tube containing 1 ml of LB broth medium and incubated with moderate shaking for 30 min at 37°C. The transformed culture (100 μl) was then plated onto LB plates with 50 μg/ml ampicillin. Resistant colonies harboring the DNA vaccine minigene were cultured and stored after confirmation of the coding sequence. For mammalian expression, 5 μg of enhanced green fluorescent protein (EGFP)-N plasmid was added to 1 × 105 RAW cells in a 0.4-cm cuvette and electroporated at 300 V, 200 Ω, and 25 μF.
Assessment of Gene Transfer from S. typhimurium to Phagocytes.
Gene transfer from S. typhimurium to murine peritoneal macrophages and the RAW macrophage cell line was determined by using plasmid DNA encoding the EGFP as a tracer. Briefly, 1 ml of 3% (vol/vol) Brewer's thioglycolate medium was injected into the peritoneal cavities of C57BL/6J mice to produce inflammatory exudate-derived macrophages, which were harvested 7 days after this initial stimulation. Isolated macrophages were allowed to adhere for 2 h at 37°C, at which time the nonadherent cells were removed by two washes with antibiotic-free medium. An attenuated strain of S. typhimurium bearing the EGFP-N plasmid was added and incubated for 30 min. Cells were washed again with a medium containing 50 μg/ml of gentamycin to kill extracellular bacteria. After another 4 h incubation, 10 μg/ml tetracycline was added to the cell culture to block intracellular bacterial multiplication, and thereafter incubation was continued for another 30–48 h in DMEM [10% (vol/vol) FCS/2 mM l-glutamine]. The medium was then removed, and the cells were washed three times with PBS (pH 7.4) before evaluation of EGFP by fluorescence microscopy.
Oral Immunization and Tumor Cell Challenge.
Groups of eight female C57BL/6J mice were fed by oral gavage with 100 μl of PBS containing 1 × 108 recombinant S. typhimurium harboring one of the expression plasmids (pUb-V, pUb-M, pUb-H, or p-Mini) at 2-week intervals (three times). At 1 week after the last oral immunization, mice were challenged with 1 × 105 B16G3.26 melanoma cells by s.c. injection in the right front flank. Animals were examined daily until the tumor became palpable, and thereafter, its diameter was measured every other day with microcalipers in two dimensions.
Cytokine Release Assay.
CD8+ T cells isolated from splenocytes of C57BL/6J mice that were immunized three times with a DNA vaccine and that had been challenged 1 week after the last oral immunization were collected from the pUb-M, pUb-H, and p-Mini experimental groups of mice, as well as from control groups injected with PBS or immunized with S. typhimurium carrying only the empty ubiquitin vector. After splenectomy, lymphocytes were collected on Ficoll/Hypaque (Amersham Pharmacia), and CD8+ T cells were isolated by using the CD8+ T cells Subset Enrichment Column according to the manufacturer's directions (R & D Systems). To determine cytokine release, purified CD8+ T cells were cultured in T cell medium [DMEM/10% (vol/vol) FCS/2 mM l-glutamine/5 × 10−4 M β-mercaptoethanol/100 units/ml penicillin/100 μg/ml streptomycin/1:25 T-STIM (BDB Bioscience, Bedford, MA)] with 5 × 104 irradiated B16G3.26 syngeneic melanoma cells. Supernatants were harvested from each group of mice after 48 h and assayed for IFN-γ release with a commercially available cytokine detection kit by using a solid-phase sandwich ELISA (BioSource International, Camarillo, CA).
Cytotoxicity Assay.
Cytotoxicity was measured by a standard 51Cr-release assay (17). CD8+ T cells were isolated from spleens of syngeneic C57BL/6J mice 1 week after challenge with 1 × 105 B16G3.26 melanoma cells and subsequently cultured for 3 days at 37°C in T cell medium. B16G3.26 melanoma target cells (3 × 106) were labeled with 0.5 mCi of 51Cr for 2 h at 37°C, washed three times, and suspended in the T cell medium. Target cells were added to each well of a U-bottomed microtiter plate and incubated with effector cells at various effector-to-target cell ratios at 37°C for 4 h. The percentage of specific target cell lysis was calculated with the formula [(E − S)/(T − S)] × 100, where E is the average experimental release, S is the average spontaneous release, and T is the average total release.
Adoptive Transfer of CD8+ CTLs.
Mice that were previously immunized three times with either the pUb-V empty vector control or the pUb-M vaccine served as donors of tumor-specific CD8+ T cells for adoptive transfer experiments. These animals were killed 1 week after challenge with B16G3.26 melanoma cells, and CD8+ T cells were isolated as described above. Thereafter, syngeneic C57BL/6J scid/scid mice were injected i.v. with a total of 4.5 × 107 CD8+ T cells in three separate injections of 1.5 × 107 T cells each on days −3, −1, and +3. On day 0, mice were challenged with an s.c. injection of 1 × 105 wild-type B16G3.26 melanoma cells. Animals were examined daily, and two diameters of each tumor were measured with microcalipers until day 35.
Statistical Analysis.
The statistical significance of differential findings between experimental groups was determined by Student's t test. Findings were regarded as significant, if two-tailed P values were <0.05.
Results
DNA Is Transferred from S. typhimurium to Mammalian Host Cells in Vitro.
To test the hypothesis that the attenuated S. typhimurium is a suitable carrier for DNA vaccines, we analyzed the effective DNA transfer from S. typhimurium AroA to mammalian host cells. The transfer was accomplished by demonstrating direct transfer of DNA from the S. typhimurium carrier to mouse primary peritoneal macrophages by infecting such cells with Salmonellae harboring the EGFP expression vector. The data depicted in Fig. 2 A and B show that approximately one-fourth of the adherent macrophage/dendritic-like cells expressed EGFP, indicating the attenuated bacteria to be a suitable carrier for the oral DNA vaccine. The mouse macrophage cell line RAW transduced with the eukaryotic expression vector EGFP by electroporation was used as a positive control. At least half of the RAW cells expressed green fluorescence after 36 h, indicating functional integrity of the EGFP vector (Fig. 2 C and D).
Figure 2.
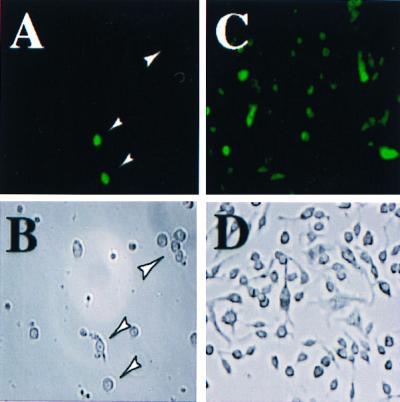
Gene transfer from S. typhimurium into mouse macrophages: Effective gene transfer from S. typhimurium into primary mouse macrophages was determined by using an EGFP-encoding plasmid. For this purpose, inflammatory exudate macrophages were cultured with S. typhimurium harboring the EGFP plasmid, and green fluorescence was analyzed after 36–48 h by UV microscopy at 488 nm (A and C). Arrowheads indicate location of macrophages. A macrophage cell line (RAW) transfected with the same EGFP plasmid by electroporation and green fluorescence was analyzed after 36 h (C and D). Identical microscopic fields were analyzed in the presence and absence of UV light (A corresponds to B; C corresponds to D). Photographs were taken at ×100 magnification.
Protection Against Lethal Challenge with B16 Melanoma Cells.
The working hypothesis was tested that an oral DNA vaccine could induce tumor-protective immunity against a lethal challenge of C57BL/6J mice with wild-type B16G3.26 melanoma cells. Thus, C57BL/6J mice (n = 8) were immunized by oral gavage three times at 2-week intervals with 1 × 108 S. typhimurium AroA, harboring the expression plasmid pUb-H and encoding ubiquitin and the human gp10025–33 and TRP-2181–188 peptide epitopes; the mice were challenged 1 week thereafter by s.c. injection of 1 × 105 B16G3.26 murine melanoma cells. These mice showed tumor-protective immunity; two of them rejected the tumor cell challenge completely, and the remaining six animals had a marked suppression in tumor growth (Fig. 3). An even better tumor-protective immunity (P < 0.05) was achieved by vaccination with the ubiquitinated expression plasmid pUb-M encoding the autologous murine gp10025–33 and TRP-2181–188 peptide epitopes (Fig. 3). These vaccination effects were in contrast to control animals that received either PBS or the empty ubiquitin vector (pUb-V) instead of the vaccine, because all mice in these experimental groups had tumors that were three times as large. Ubiquitination seemed to be absolutely essential to achieve tumor-protective immunity, because immunization with the autologous minigene (p-Mini) lacking the coding sequence for murine ubiquitin resulted in large tumor growth in all mice equal to that of controls (Fig. 3). Similar results were obtained with p-Mini encoding the human peptide epitopes (data not shown).
Figure 3.

Induction of tumor-protective immunity. C57BL/6J mice (n = 8) received three vaccinations of 108 attenuated S. typhimurium (SL7207) harboring DNA vaccines by oral gavage at 2-week intervals. Experimental groups were immunized with vaccines encoding ubiquitinated murine gp10025–33 and TRP-2181–188 peptide epitopes (pUb-M), ubiquitinated human gp10025–33 and TRP-2181–188 epitopes (pUb-H), or the murine peptide epitopes without ubiquitin (p-Mini). Control groups of mice (n = 8) included those treated with PBS or a vaccine containing only the empty vector (pUb-V). Mice were challenged with 1 × 105 B16G3.26 murine melanoma cells 1 week after the final vaccination. Bars represent means and standard deviations of eight mice per group. Differences in tumor volume between experimental groups treated with either pUb-M or pUb-H and all control groups were statistically significant (P < 0.01).
Adoptive Transfer of CD8+ T Cells from Immunized Mice Results in Suppression of Tumor Growth.
To test whether the DNA vaccines would elicit a CD8+ T cell response that could limit tumor growth, CD8+ T cells were isolated from splenocytes of mice immunized three times with either the control vaccine containing the empty ubiquitin vector (pUb-V) or the pUb-M vector encoding the murine gp10025–33 and TRP-2181–188 T cell epitopes. These CD8+ T cells were then adoptively transferred to syngeneic scid/scid mice. After subsequent challenge of these mice with 1 × 105 wild-type B16G3.26 melanoma cells, all animals that received CD8+ T cells isolated from mice vaccinated with either pUb-M or pUb-H showed >60% reduction in tumor growth compared with control animals that received CD8+ T cells from mice vaccinated with only the empty ubiquitin vector (Fig. 4). These data imply that the autologous pUb-M DNA vaccine can induce a functional CD8+ T cell-mediated immune response in vivo, because such T cells, when adoptively transferred to scid/scid mice, retained sufficient memory to recognize tumor-specific T cell epitopes initially presented to them by antigen-presenting cells.
Figure 4.

Horizontal transfer of tumor immunity by adoptive transfer of T lymphocytes. CD8+ T cells were isolated from C57BL/6J mice successfully immunized with pUb-M DNA vaccine or pUb-V empty vector control. All donor animals were killed 1 week after tumor cell challenge, and their CD8+ T cells were isolated from their pooled splenocyte suspensions. At 48 h before i.v. challenge with 1 × 105 wild-type B16G3.26 cells, naïve syngeneic SCID mice were reconstituted with a total of 4.5 × 107 CD8+ T cells by i.v. injection of 1.5 × 107 T cells on days −3, −1, and +3. Tumor growth was analyzed in mice receiving CD8+ T cells either from animals vaccinated with pUb-M (┌) or pUb-H (▴) or from mice vaccinated with the empty vector control (●). Tumor growth was determined by microcaliper measurements, and tumor volume was calculated according to 0.5 × width2 × length.
Immunization with DNA Vaccines Induces MHC Class I-Restricted CTL-Mediated Killing of B16G3.26 Melanoma Cells in Vitro.
To demonstrate that immunization with DNA vaccines induces tumor-specific CTLs capable of killing B16 melanoma cells in vitro in an MHC class I antigen-restricted manner, we isolated CD8+ T cells from splenocytes of various groups of C57BL/6J mice, each of which had been immunized with the autologous pUb-M vaccine, the xenogeneic pUb-H vaccine, or the pUb-V control vaccine. The data depicted in Fig. 5A indicate that CD8+ T cells isolated from splenocytes of mice immunized with either the pUb-M or pUb-H DNA vaccines were equally effective in killing B16G3.26 melanoma cells in vitro at different effector-to-target cell ratios. In contrast, controls such as CD8+ T cells isolated from naïve mice or from mice immunized with a vaccine carrying the empty vector produced only background levels of tumor cell lysis (Fig. 5A). The CD8+ T cell-mediated killing of B16G3.26 melanoma cells was specific, because syngeneic but unrelated prostate carcinoma target cells (TRAMP-C1) were not lysed (Fig. 5B). Importantly, the CD8+ T cell-mediated tumor cell lysis was MHC class I antigen-restricted, because addition of 10 μg/ml anti-H2Kb/H-2Db antibody (clone 28-8-6; PharMingen) specifically inhibited melanoma cell lysis (Fig. 5B).
Figure 5.

Cytotoxicity induced by CD8+ T cells of C57BL/6J mice against murine B16G3.26 melanoma cells after immunization with DNA vaccines encoding ubiquitinated melanoma-specific epitopes. Splenocytes were isolated 1 week after the third vaccination, and CD8+ T cells were analyzed for their lytic activity in a 4-h 51Cr-release assay at different effector-to-target cell ratios (E:T) ratios. (A) Specific lysis mediated by CD8+ T cells from different experimental groups of mice: pUb-H (○), pUb-M (♦), empty vector pUb-V (▵), and PBS (*). (B) Lysis mediated by CD8+ T cells was blocked by anti-MHC class I antibody (H-2Kb/Db) after immunization with the pUb-M (♦) and pUb-H (○) DNA vaccines. Nonspecific syngeneic prostate cancer target cells (TRAMP-C1) were not lysed by pUb-M-specific CD8+ T cells (▿).
TH1 Cytokine Release by CD8+ T Cells Isolated from Mice Immunized with DNA Vaccines.
To determine whether T cells activated by DNA vaccines will secrete inflammatory TH1 cytokines, CD8+ T cells were isolated from splenocytes of mice that were successfully immunized three times with either the pUb-M or pUb-H DNA vaccines and then challenged 1 week thereafter with 1 × 105 B16 melanoma cells. When purified CD8+ T cells were isolated from splenocytes of these mice 1 week after tumor cell challenge and cultured in T cell medium with 5 × 104 irradiated B16G3.26 melanoma cells, increasing amounts of IFN-γ (Fig. 6) were detected in the supernatants of these cultures when compared with controls. There was no release of IL-4 (data not shown). These data suggest that the DNA vaccine-activated CD8+ T cells secreted the TH1 inflammatory cytokine IFN-γ.
Figure 6.

Cytokine release by CD8+ T cells isolated from splenocytes of C57BL/6J mice treated with DNA vaccines. Release of IFN-γ by CD8+ T cells isolated from splenocytes of C57BL/6J mice was measured by solid-phase sandwich ELISA. Splenocytes were obtained from mice immunized with either pUb-H or pUb-M DNA vaccines and from control mice treated with either PBS or the empty ubiquitin vector (pUb-V). Bars represent means and standard deviations of eight mice per group. Differences in IFN-γ release observed between experimental groups treated with either pUb-M or pUb-H and all control groups were statistically significant (P < 0.01).
Discussion
We tested the hypothesis that peripheral T cell tolerance to melanoma self-antigens can be broken by oral DNA vaccines that were optimized for antigen processing and presentation of encoded peptide epitopes derived from these antigens. Proof for this hypothesis was established by the induction of a tumor-protective immune response after a lethal challenge of vaccinated C57BL/6J mice with murine B16G3.26 melanoma cells resulting in rejection of tumors and suppression of growth. Three lines of evidence indicated that the cells responsible for this tumor-protective immunity were primarily activated CD8+ T cells. First, CD8+ T cells isolated from splenocytes of successfully vaccinated mice specifically killed B16G3.26 melanoma target cells in an in vitro cytotoxicity assay in an MHC class I-restricted manner. Second, CD8+ T cells were clearly responsible for this protective immunity, because such cells isolated from splenocytes of successfully vaccinated C57BL/6J mice could be adoptively transferred to syngeneic scid/scid mice and had sufficient memory to suppress markedly the growth of a lethal challenge of wild-type B16G3.26 melanoma cells. Third, the DNA vaccines did indeed activate CD8+ T cells, because such cells isolated from splenocytes of successfully vaccinated mice secreted the TH1 inflammatory cytokine IFN-γ.
The most important consequence of the immunization achieved with the DNA vaccines in a prophylactic setting is the protective immunity achieved against a lethal challenge with B16G3.26 murine melanoma cells. Immunity was accomplished by using an attenuated strain of S. typhimurium as carrier for eukaryotic expression vectors encoding minigenes consisting of ubiquitinated gp10025–33 (H-2Db) and TRP-2181–188 (H-2Kb) peptide epitopes. Importantly, a completely autologous DNA vaccine encoding the ubiquitinated murine gp10025–33 and TRP-2181–188 peptide epitopes elicited an MHC class I-restricted CD8+ T cell response capable of inducing tumor-protective immunity against a challenge of B16G3.26 murine melanoma cells. Interestingly, the extent of this tumor-protective immunity was better (P value was <0.05) than that achieved by xenoimmunization (P value was <0.05), i.e., by replacing the murine gp10025–33 epitope with the human gp10025–33 peptide epitope. There was no difference in the MHC class I-restricted CD8+ T cell responses induced by these two DNA vaccines or in the release of TH1 cytokine IFN-γ from these T cells when stimulated by irradiated B16 melanoma cells. The CD8+ T cell responses were directed specifically against these melanoma cells, because syngeneic prostate carcinoma cells TRAMP-C1 were not lysed by the CD8+ effector T cells in vitro.
Our success in eliciting an effective CD8+ T cell-mediated tumor-protective immunity with a completely autologous oral DNA vaccine is most likely due to our efforts in optimizing antigen processing and presentation by ubiquitination. Support for this contention comes from our findings indicating that the DNA vaccines (p-Mini) encoding either the murine gp10025–33 and TRP-2181–188 peptide epitopes or their human counterparts and lacking in ubiquitination were equally ineffective in inducing tumor-protective immunity as the controls, i.e., mice immunized with only PBS or the empty vector (pUb-V). In contrast, both ubiquitinated DNA vaccines, pUb-H and pUb-M, were clearly capable of inducing tumor-protective immunity against a lethal challenge of B16G3.26 murine melanoma cells.
Our findings clearly show the beneficial effects of ubiquitination in inducing stronger CD8+ T cell responses and protective immunity. These results are in agreement with previous studies that used a modified ubiquitin in a viral model system (13, 14). Although ubiquitination does not invariably enhance CD8+ T cell responses (18), the present work and studies by other investigators (13, 14, 19–22) confirm the important role of ubiquitination in the MHC class I antigen presentation pathway.
In summary, we demonstrated that an autologous, oral DNA vaccine encoding ubiquitinated murine gp10025–33 and TRP-2181–188 peptide epitopes carried by an attenuated strain of S. typhimurium protected C57BL/6J mice against a lethal challenge of murine B16G3.26 melanoma cells and that optimization of antigen processing and presentation by ubiquitination played a major role in achieving this successful vaccination.
Acknowledgments
This work was supported by an Outstanding Investigator Award CA42508 (to R.A.R.) and by Grant AI-37186 (to J.L.W.) from the National Institutes of Health. J.M.R. is a fellow of the Deutsche Forschungsgemeinschaft. This article is the Scripps Research Institute's manuscript no. 12935-IMM.
Abbreviations
- CTL
cytotoxic T lymphocyte
- EGFP
enhanced green fluorescent protein
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.090097697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.090097697
References
- 1.Boon T, Cerottini J C, Van Der Eynde B, Van der Bruggen P, Van Pel A. Annu Rev Immunol. 1994;12:337–365. doi: 10.1146/annurev.iy.12.040194.002005. [DOI] [PubMed] [Google Scholar]
- 2.Van Pel A, Van der Bruggen P, Coulie P G, Brichard V G, Lethe B, Van Der Eynde B, Uyttenhove C, Renauld J-C, Boon T. Immunol Rev. 1995;154:229–250. doi: 10.1111/j.1600-065x.1995.tb00084.x. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg S A. J Natl Cancer Inst. 1996;88:1635–1644. doi: 10.1093/jnci/88.22.1635. [DOI] [PubMed] [Google Scholar]
- 4.Sprent J, Schaefer M. Immunol Rev. 1990;117:213–234. doi: 10.1111/j.1600-065x.1990.tb00574.x. [DOI] [PubMed] [Google Scholar]
- 5.Wang R-F, Rosenberg S A. Immunol Rev. 1999;170:85–100. doi: 10.1111/j.1600-065x.1999.tb01331.x. [DOI] [PubMed] [Google Scholar]
- 6.Wang R F, Appella E, Kawakami Y, Kang X, Rosenberg S A. J Exp Med. 1996;184:2207–2216. doi: 10.1084/jem.184.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawakami Y, Eliyahu S, Delgado C H, Robbins P F, Sakaguchi K, Appella E, Yannelli J R, Adema G J, Miki T, Rosenberg S A. Proc Natl Acad Sci USA. 1994;91:6458–6462. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bakker A B H, Schreurs M W J, de Boer A J, Kawakami Y, Rosenberg S A, Adema G J, Figdor C G. J Exp Med. 1994;179:1005–1009. doi: 10.1084/jem.179.3.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bloom M B, Perry-Lalley D, Robbins P F, Li Y, El-Gamil M, Rosenberg S A, Yang J C. J Exp Med. 1997;185:453–459. doi: 10.1084/jem.185.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Overwijk W W, Tsung A, Irvine K R, Parkhurst M R, Goletz T J, Tsung K, Carroll M W, Liu C, Moss B, Rosenberg S A, et al. J Exp Med. 1998;188:277–286. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Overwijk W W, Lee S D, Surinam D R, Irvine K R, Touloukian C E, Chan C-C, Carroll M W, Moss B, Rosenberg S A, Restifo N P. Proc Natl Acad Sci USA. 1999;96:2982–2987. doi: 10.1073/pnas.96.6.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darji A, Guzman C A, Gerstel B, Wachholz P, Timmis K N, Wehland T, Chakraborty T, Weiss S. Cell. 1997;91:765–775. doi: 10.1016/s0092-8674(00)80465-1. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez F, Zhang J, Whitton J L. J Virol. 1997;71:8497–8503. doi: 10.1128/jvi.71.11.8497-8503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez F, Ling L A, Harkins S, Zhang J, Yokoyama M, Widera G, Fuller J T, Kinkaid C, Campbell I L, Whitton J L. J Virol. 1998;72:5174–5181. doi: 10.1128/jvi.72.6.5174-5181.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stackpole C W, Alterman A L, Fornabaio D M. Invasion Metastasis. 1985;5:125–143. [PubMed] [Google Scholar]
- 16.Foster B A, Gingrich J R, Kwon E D, Madias C, Greenberg N M. Cancer Res. 1997;57:3325–3330. [PubMed] [Google Scholar]
- 17.Xiang R, Lode H N, Dreier T, Gillies S D, Reisfeld R A. Cancer Res. 1998;58:3918–3925. [PubMed] [Google Scholar]
- 18.Fu T-M, Guan L, Friedman A, Ulmer J B, Liu M A, Donnelly J J. Vaccine. 1998;16:1711–1717. doi: 10.1016/s0264-410x(98)00134-0. [DOI] [PubMed] [Google Scholar]
- 19.Townsend A, Bastin J, Gould K, Brownlee G, Andrew M, Coupar B, Boyle D, Chan S, Smith G. J Exp Med. 1988;168:1211–1224. doi: 10.1084/jem.168.4.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Y, Kipps T J. J Immunol. 1997;159:6037–6043. [PubMed] [Google Scholar]
- 21.Tobery T W, Siliciano R F. J Exp Med. 1997;185:909–920. doi: 10.1084/jem.185.5.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michalek M T, Grant E P, Gramm C, Goldberg A L, Rock K L. Nature (London) 1993;363:552–555. doi: 10.1038/363552a0. [DOI] [PubMed] [Google Scholar]