

Abstract
Two complementary routes to a new integral equation theory for site-site molecular fluids are presented. First, a simple approximation to a subset of the atomic site bridge functions in the diagrammatically proper integral equation theory is presented. This in turn leads to a form analogous to the reactive fluid theory, in which the normalization of the intramolecular distribution function and the value of the off-diagonal elements in the density matrix of the proper integral equations are the means of propagating the bridge function approximation. Second, a derivation from a topological expansion of a model for the single-site activity followed by a topological reduction and low-order truncation is given. This leads to an approximate numerical value for the new density coefficient. The resulting equations give a substantial improvement over the standard construction as shown with a series of simple diatomic model calculations.
I. Introduction
A quantitative theory of the structure of molecular fluids described by atoms or sites has remained elusive. The qualitative and quantitative inconsistencies of the reference interaction site model1 (RISM) integral equation theory for predicting the structure and thermodynamic properties of site-site model molecular fluids have been understood for some time.2–4 Several means have been proposed to correct these inconsistencies, many concentrating on the closure.5–8 A second, formally distinct method for constructing a diagrammatically proper theory, due to Chandler, Silbey, and Ladanyi (CSL), generates only terms consistent with the molecular connectivity and intermolecular interactions for the model.2,9 This diagrammatically proper approach eliminates terms in the expansion of RISM which correspond to unphysical intramolecular interactions or so-called bad graphs. Unfortunately, while certain qualitative advances using the proper CSL theory have been successful,10–12 the quantitative results appear to be uniformly disappointing in comparison to simulation.9,13,14
Our work in the direct calculation of the bridge diagrams for simple fluids15,16 leads naturally to the question of whether the bridge function is perhaps the reason that the diagrammatically proper theory gives disappointing results. Other researchers 13,14,17,18 have also examined this question, using somewhat different approaches, and reached the conclusion that the bridge graphs (as opposed to heuristic bridge functions) did not appear to make a qualitative change. In reproducing the work of these groups, we have also been forced to conclude that, in the current form, the diagrammatically proper theory is not sensitive to the exact low-order bridge diagrams in the same manner as simple fluid theory. Considering the success with simple fluids, 15,16 this result was unexpected. It has led us to examine the bridge diagrams in the diagrammatically proper theory more closely and to propose a modest approximation method to these diagrams in extension to the proper interaction site model (PISM) equations in the CSL theory.
We present two complementary routes to a new integral equation theory for site-site molecular fluids. We will use analogies and insights from the theory of reactive fluids and the theory of full molecular correlations to generate a new approximate site-site theory of nonreactive fluids. First we examine a simple bridge function approximation. We consider the proper site-site expansion of the full molecular distribution functions and show that most of the resulting site-site diagrams are site (atomic) bridge diagrams. Then we distinguish two classes of atomic bridge diagrams, those which are derived from the molecular bridge diagrams and those which are derived from the simple, nonbridge molecular diagrams generated by the generalized convolution product in the molecular Ornstein-Zernike equation. The form of this second class of atomic bridge diagrams then leads us to suggest a simple approximation method. We take by analogy from the reactive fluid form of the proper interaction site model theory12,19,20 an approximate diagrammatic correction term, to use in the nonreactive fluid. That is, the diagrams which are affected by the reactive molecular density can also be written as an approximation to that second class of atomic bridge diagrams which we have distinguished in a nonreactive system. In effect, the equation system used to properly model reactive site-site fluids is also an effective approximation to a subset of the bridge diagrams in nonreactive fluids.
This approximation is similar in spirit to a topological expansion of the Percus-Yevick approximation in simple fluids,21,22 in which the “difficult” bonds in the simple fluid bridge functions are set to −1. In our case, many of the atomic site bridge functions which would be included in a full molecular treatment can be approximated by a set of included atomic diagrams multiplied by a coefficient. We show that the coefficient may be introduced into the generating equations, producing a form equivalent by analogy to certain reactive fluid theories.12,19,20 As this form is imposed upon a nonreactive fluid in this case, the bridge function series coefficient can be thought of as an effective molecular density.
For the second route, following this effective density interpretation, we examine the topological expansion of the molecular partition function with site activities and derive the site-based singlet and pair functions from a diagrammatic topological reduction. From this we show that the density coefficient in the off-diagonal elements is distinct from the density coefficient in the diagonal elements of the density matrix. We use the low-order expansion as a generating series to obtain a value for our approximation by analogy to the low-order singlet terms recently utilized23 in the study of reactive fluids.
We then show a few simple examples. For homonuclear diatomics, the effective density is a function of bond length and total density. We present encouraging numerical results for three standard cases in comparison to simulation and the standard results of RISM and the CSL theory.
II. Theory
In the usual notation,9 the total correlation function matrix H of PISM theory is generated in Fourier space from the proper site-site Ornstein-Zernike (OZ) matrix equation
| (1) |
The correlation function matrix is a species-labeled matrix whose elements are the submatrices denoted
| (2) |
where the sublabels o, l, r, and b are from the CSL notation, such that o means that no intramolecular S-bonds are attached to either root point (or labeled site), l means that one or more S-bonds are properly attached to the left root point, r means that one or more S-bonds are properly attached to the right root point, and b means that both root points have S-bonds associated with them. Here and below we use proper in the sense of CSL. For a nonreactive diatomic molecule this requires that, for a given site-site diagram or related function, a root or field point3 may be intersected by at most one S-bond.
This proper connectivity is enforced through the density matrix ρ in the OZ equation. All elements of the ρij species-labeled submatrices are zero when i ≠ j and
| (3) |
otherwise, where the nonzero ρii elements are the number densities of site i in the fluid. The construction of this matrix ensures that the normalized intramolecular distribution function,
| (4) |
and the both terms in C, connect properly with only the o, l, and r terms in C (or H).
As with the generating OZ equation, the closure system requires proper connectivity. The exact closure is taken to be9
| (5) |
with β=1/(kbT), kb the Boltzmann constant, T the temperature, and Uij(r) the pair potential between sites i and j on different molecules. The indirect correlation functions t(r) are defined t ≡ H–C, with as in H. The b(r) = bo(r) + bl(r) + br(r) + bb(r) are the bridge functions. In general, some approximation to the bridge function is required to proceed. In the usual cases, either we assume that b(r) = 0, giving the hypernetted-chain (HNC) closure, or we take b(r) = 0 and linearize with respect to to(r) to give the Percus-Yevick (PY) closure. As with the simple fluids, the bridge function approximation is central to this type of theory of liquids.3
A. A bridge function approximation
To proceed, we will make a distinction between two classes of diagrams which are subsets of the bridge function. This distinction is easily illustrated by the atomic site diagrams
 |
and
 |
where, following standard graphical notation,3 the solid lines represent the Mayer f-bond exp(−βUij(r))−1, the wiggly line represents the intramolecular S-bond, the open 1 circles are unintegrated variables, and the closed circles are integrated ρ circles. The diagrammatic distinction we make is that the first diagram belongs to that class of site-site diagrams which are generated from the site-site expansion of the molecular bridge diagrams. The second diagram, while it is an atomic site-site bridge diagram when included in the PISM theory, is not generated by the expansion of a molecular bridge diagram in site-site f-bond and S-bonds but from a normal convolution product (or t graph). The molecular diagram from which it is generated arises from a convolution product in the molecular OZ equation. That is, if we expand the first orientationally averaged molecular bridge diagram in site-site potentials as the series
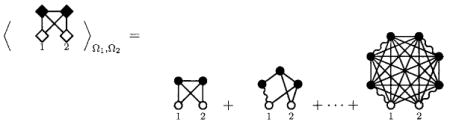 |
(6) |
with diamonds used to represent an arbitrary diatomic molecule and the bonds between them molecular F-bonds (capital F meaning from the full inter- and intramolecular potential), then we see that the first term in the series is the first diagram identified above. If we now expand the first indirect t graph generated from the molecular OZ equation, we have
 |
(7) |
In this last expansion, we note the following. Consider, for a diatomic potential, the molecular F-bond expanded in the form
 |
(8) |
with the dashed lines here representing site-site e=1+f bonds. In the full, site-site f-bond expansion of this series, there are 15 diagrams, two of which are bridge diagrams in the site-site theory. In Eq. (7), neglecting for now the homo-nuclear symmetry, there are 225 distinct diagrams, 120 of which are site-site bridge diagrams. These diagrams are included by default in a molecular series but must be addressed term by term in the site theory. Further, these diagrams are not included by means of the usual molecular bridge functions. The number of additional site-site bridge diagrams of this type will be much larger for molecules with more atomic sites, both in raw numbers and in the proportion of diagrams of a given atomic site order. We suggest that addressing the bridge diagrams term by term in the site-site theories as presently constituted is ultimately impractical.
The topological form of this second class of atomic bridge diagrams does suggest a simple approximation, however. We consider the case of a reactive system of site-site diatomic molecules,12,19,20 in which only dimers and free sites are allowed. In this case, the intermolecular site-site and atomic distributions may be calculated in the diagrammatically proper form as above by changing the density matrix and intramolecular distribution functions so that
| (9) |
and
| (10) |
with η the effective density of sites i in dimers to be determined separately, ρ the total density of sites i, both in dimers and free of molecules in the system, and Sij now normalized by η. We note that, since the normalization condition ∫drSijηj=1 remains the same as for the nonreactive case, then the only diagrams which are affected by η are those in which an S bond connects only field points,
 |
(11) |
where we have labeled the field points as η points to be clear, and this series will also contain diagrams with left, right, and both connectivity, though again only the S bonds not incident on a root point in these diagrams are affected by η due to the normalization on S. If, for each order of η, we group together the set of such η-affected diagrams, the members of each group will have the same number of points as one or more atomic bridge diagrams. These atomic bridge diagrams are members of those diagrams derived from the nonbridge molecular antecedent diagram set discussed above. That is, if we take η=ρ−α with α a constant to be determined, then we have that
 |
(12) |
where the unlabeled field points on the right-hand graphs are now 1 circles, since a factor of η may be eliminated due to the η normalization of S in Eq. (10), and we define the s bonds,
 |
(13) |
This gives in turn
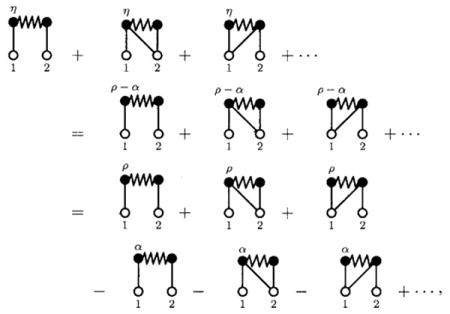 |
(14) |
and we approximate in the nonreactive system,
 |
(15) |
and it is understood that the S bond on the left-hand diagram is normalized according to Eq. (4), and the s-bonds on the right-hand diagrams are normalized according to Eq. (13). Similarly, for each order ηρ, η2, etc., the series of diagrams included using the reactive density matrix can be separated into groups of ρ-weighted diagrams and α-weighted diagrams, and the α-weighted set of diagrams compared to the analogous atomic bridge diagrams derived from the non-bridge molecular antecedent diagrams.
We may summarize the approximation as follows. If we classify the atomic bridge diagrams in the proper interaction site theory into two subclasses, the first class containing diagrams which are derived from molecular bridge diagrams and the second class containing diagrams which are derived from simple, nonbridge molecular diagrams, then a simple approximation may be made to the second class by utilizing the reactive fluid analog of the proper interaction site equations. In particular, if we take the convention that ρ is the total site density and η is the total effective molecular site density, then if we take η= ρ − α with α to be determined and expand the resulting series from the proper OZ equations, we have that the nonbridge molecular antecedent site bridge diagrams in the nonreactive system with site density ρ can be approximated as α times a set of included, nonbridge site diagrams. That is, the proper site-site reactive fluid equations are an effective bridge function approximation in the nonreactive fluid.
This approximation can be extended to most, but not all, of this class of nonbridge molecular antecedent atomic bridge diagrams. The bridge diagrams not approximated by this method are those which are functions related to the molecular F-bond, i.e., diagrams such as
 |
(16) |
in which an α substitution is made irrelevant by the normalization of the S bond, which cancels η. It is possible, in principle, to account for these diagrams. However, this would require that the normalization for the entire series be reconstructed, making it necessary to reexpress the thermodynamic integrals on the root point normalizations. We leave the details of such extensions for later work.
To continue, we require a method to assign a value for η. Since the equation form is equivalent to the propagating system used in the theory of reactive fluids, η may be thought of as an effective molecular density. As such, our second route to this approximation yields a value from a simple analogy with the theory of reactive fluids.
B. The screened density
In the case of a reactive fluid, the density elements we have labeled η are known19,24 to be topologically distinct from the ρo elements. At the most fundamental level, this is the distinction between the total site density of the fluid and the total density of sites bonded in molecules in the system. The topological distinction arises from the expansion of terms in the partition function of the system; in particular, the f-bond expansion of the interaction of sites as constrained by the S bonds leads by necessity to a different class of diagrams associated with the l and r points, as opposed to those associated with the o points. This term-by-term expansion is not restricted to reactive fluids, however. To show this, we use the methods of Morita and Hiroike,25 beginning with a partition function expansion of a nonreactive, diatomic fluid. For notational convenience, we use a homonuclear molecule, the extension to more complicated systems being straightforward.
Given a site-site intermolecular potential, we define a grand canonical partition function Ξ and its activity expansion in terms of clusters of molecules and express the resulting integrals as diagrams.3,26
Ξ = 1 + the sum of all distinct, simple, proper graphs consisting of 1 or more intramolecular e-bonds, z* circles, and intermolecular e-bonds such that there is either an intramolecular or intermolecular e-bond properly connecting each pair of z* circles,
 |
(17) |
where we use dashed lines to represent site-site intermolecular e-bonds, e=1+f for f the Mayer f-bond used above, wiggly lines here represent intramolecular e-bonds on a nondissociative intramolecular potential, and the field points are the generalized sitewise activities z*(r) = exp(−βφ(r) + βμN). φ(r) is an arbitrary field, μ is the chemical potential, and N is the number of sites. We use only sitewise properties for z*(r) and do not deal with nondecomposable pair activities and fields in this work. We expand the intermolecular e bonds in f bonds to give the series
Ξ = 1 + the sum of all distinct, simple, proper graphs consisting of one or more intramolecular e-bonds, z* circles, and some or no intermolecular f-bonds such that any pair of z* circles not properly connected by an intramolecular e-bond may be connected by an intermolecular f-bond,
 |
(18) |
We take the logarithm to eliminate the disconnected diagrams and obtain
ln Ξ = the sum of all distinct, simple, proper, connected graphs consisting of one or more intramolecular e-bonds, z* circles, and some or no intermolecular f-bonds,
 |
(19) |
The first functional derivative of ln Ξ with respect to the activity for a single site at r in the fluid yields the singlet site density at r
= the sum of all distinct, simple, proper, connected graphs consisting of one intramolecular e-bond connecting one open z* circle labeled 1 with one closed z* circle, some or no other closed z* circles, some or no other allowed intramolecular e-bonds, and some or no intermolecular f-bonds,
 |
(20) |
where, for clarity, we follow convention and replace the coordinate r with the usual coordinate labels (1, 2, etc.).
Given the single-site density ρ(1) above, the method as used below allows us to reach the topological expansion of ρ(2)(1,2) without being forced to take the logarithm of any intermediate steps beyond ln Ξ. It would be necessary to follow a different method if the question of the closure and the generating equations were to be addressed, but for now, we are interested only in the topological reductions and the diagrammatic series necessary to describe the resulting pair functions. To proceed, we recall the functional definition,
| (21) |
where δ(1,2) is the usual Kronecker delta.
Taking the second functional derivative we find
= the sum of all distinct, simple, proper, connected graphs consisting of two open z* circles labeled 1 and 2, respectively, one or more intramolecular e-bonds, some or no intermolecular f-bonds, and some or no closed z* circles such that the open z* circles are either properly connected by an intramolecular e-bond or may be connected by an intermolecular f-bond,
 |
(22) |
In order to reduce Eq. (22) further, i.e., eliminate the articulation circles, we require two distinct singlet series. The first is ρ(1)(1) as given above. The second series is given by
η(1)(1) = the sum of all distinct, simple, proper, connected graphs consisting of one open z* circle labeled 1, some or no closed z* circles, some or no intermolecular f-bonds, and some or no allowed intramolecular e-bonds such that no intramolecular e-bond intersects the open z* circle,
 |
(23) |
The distinction and necessity of η(1)(1) arises due to the fact that in Eq. (22), for any given articulation circle, the attached intramolecular e-bond connects either a z* circle which must be retained in the reduced graph or an unretained z* circle. In the latter case, the articulation series attached at that articulation circle is ρ(1)(1). For the first case, the necessary retention of the connected circle and connecting intramolecular e-bond in the resulting reduced graph requires the singlet series η(1)(1), as defined in Eq. (23), in order to make the reduction of the articulation series on both ends (open and closed). In short, we find two different kinds of “hair” on the reducible diagrams leading to two different series ρ(1)(1) and η(1)(1).
Given Eqs. (23) and (20), we may reduce Eq. (22) to
= the sum of all distinct, simple, proper, connected graphs consisting of two open circles, either or both of which may be either ρ(1)(1) circles or η(1)(1) circles, some or no closed ρ(1)(1) circles, some or no closed η(1)(1) circles, some or no allowed intramolecular e-bonds, and some or no intermolecular f-bonds, such that there are no articulation circles, ρ(1)(1) circles are intersected exclusively by f-bonds, and η(1)(1) circles are properly intersected by an intramolecular e-bond,
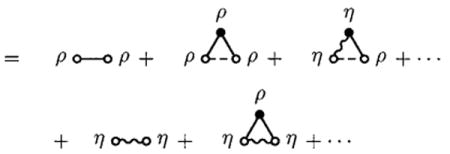 |
(24) |
= ρ(1)(1)ho(1,2)ρ(1)(2) + η(1)(1)hl(1,2) ρ(1)(2) + ρ(1)(1)hr(1,2)η(1)(2) + η(1)(1)hb(1,2)η(1)(2) + η(1)(1)S(1,2)η(1)(2),
where we recall that the dashed lines are intermolecular e=1+f bonds.
Now that we have the pair-correlation function series, we will examine the topological reduction of the site functions ρ(1) and η(1). From here, we will drop the bracketed superscript without loss of generality for these one-body properties. In most cases in the literature, for the simple fluid, ln ρ(1) is usually reduced, rather than ρ(1) directly. This is not strictly necessary,25 so that we have
ρ(1) = the sum of all distinct, simple, proper, connected graphs consisting of one intramolecular e-bond connecting one open z* circle labeled 1 with one closed η circle, some or no closed ρ circles, some or no other closed η circles, some or no intermolecular f-bonds, and some or no other allowed intramolecular e-bonds such that ρ circles are intersected exclusively by f-bonds, η circles are properly intersected by an intramolecular e-bond, and the open circle is the only articulation circle in the graph,
| (25) |
and
η(1) = the sum of all distinct, simple, proper, connected graphs consisting of one open z* circle labeled 1, some or no closed ρ circles, some or no closed η circles, some or no intermolecular f-bonds, and some or no allowed intramolecular e-bonds such that ρ circles are intersected exclusively by f-bonds, η circles are properly intersected by intramolecular e-bonds, no intramolecular e-bond intersects the open circle, and the open circle is the only articulation circle in the graph,
 |
(26) |
Given these two series, it is clear that, if we define the irreducible set,
σ(1) = the sum of all distinct, simple, proper graphs consisting of one intramolecular e-bond connecting one open 1 circle with one closed η circle, some or no closed ρ circles, some or no other closed η circles, some or no intermolecular f-bonds, and some or no other allowed intramolecular e-bonds such that ρ circles are intersected exclusively by f-bonds, η circles are properly intersected by intramolecular e-bonds, and there are no articulation circles,
 |
(27) |
then we have that ρ and η are related by
| (28) |
In a nonreactive, isotropic fluid, by construction σ(1) =1. However, in analogy to reactive fluids, this diagrammatic result gives us a reasonable numerical starting point to generate a coefficient in the l and r matrix elements in the bridge function approximation above. In particular, as in our recent work on reactive fluids,23 if we truncate the series for σ(1) at some small number of terms,
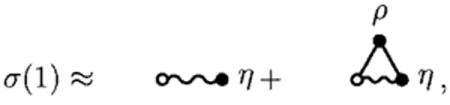 |
(29) |
then, for a hard-sphere diatomic molecule of bond length l and site diameter d, we have that (there is a simple symmetry factor of 2 different here than for the case in Ref. 23)
 |
(30) |
and
| (31) |
We present calculations below using this low-order approximation to η.
III. Numerical Results
For numerical examples using the current approximation to η given in Eq. (30), we consider three different rigid diatomic Lennard-Jones models roughly corresponding to Cl2, N2, and a slightly longer homonuclear diatomic at a dense fluid-phase point studied previously in the literature.9 The phase and model parameters are the reduced density ρ* = ρσ3 = 0.524 for σ the Lennard-Jones contact parameter, the reduced temperature T*=kbT/ε=2.2 for ε the Lennard-Jones energy parameter, and the reduced bond lengths l*= l/ σ= 0.547 (Cl2), l*=0.329 (N2), and l* =0.729. Taking these phase parameters and using σ=d in our approximation, then η* = ρ*/(1+ρ*π/3l*) = ρ*/2.003 ≈ ρ*/2 for Cl2, η* = ρ*/2.668 for N2, and η* = ρ*/1.753 for the l* = 0.729 model. These values give ρl = ρr = η* and
| (32) |
in the appropriate matrix positions. Using the HNC closure, we compare the radial distribution functions, g(r) = h(r) +1 =1+ho+hl+hr+hb, to those of simulation in Figs. 1–3. We also compare with the results of the HNC closures for the extended reference interaction site model (XRISM) and standard CSL equations. In Tables I and II, we compare the values predicted by the theories and calculated from simulation for the excess termwise internal energy3 and the Kirkwood G integral,27 respectively. All simulation results were calculated in a standard NVT ensemble with Nosé thermostat and 1000 molecules. All integral equation calculations were carried out on a grid of 2048 points with a range of 30σ.
FIG. 1.

Numerical results for a Lennard-Jones Cl2 model. The diamonds are simulation data, the solid line is the result of this work, the dashed line is the result of the standard proper interaction site model equation construction (Ref. 9), and the dotted line is the XRISM approximation. The phase point parameters are described in the text.
FIG. 3.

Numerical results for a Lennard-Jones model of N2, with l*=0.329. The diamonds are simulation data, the solid line is the result of this work, the dotted line is the XRISM approximation, and there was no stable, precise solution for the standard proper interaction site model.
TABLE I.
Termwise excess internal energy from simulation and predicted by the various integral equation theories.
| l* | η | XRISM | CSL | This work | Simulation |
|---|---|---|---|---|---|
| 0.329 | ρ/2.668 | −3.41 | … | −3.63 | −3.50 |
| 0.547 | ρ/2 | −3.11 | −3.52 | −3.31 | −2.99 |
| 0.729 | ρ/1.753 | −2.80 | −3.05 | −2.85 | −2.64 |
Table II.
Kirkwood G from simulation and predicted by the various integral equation theories.
| l* | XRISM | CSL | This work | Simulation |
|---|---|---|---|---|
| 0.547 | −1.64 | −1.77 | −1.92 | −1.79 |
| 0.729 | −1.81 | −1.78 | −2.11 | −1.88 |
In Fig. 1 we show a comparison of this work with XRISM, CSL, and simulation for a model roughly corresponding to Cl2. The current work shows a dramatic improvement over CSL with respect to the simulation. While XRISM is not bad for these types of models we note that the current work shows significant improvement in the first peak.
The longer the bond length the better all three theories become. Each of these theories has a correct separated atom limit and so this is not unexpected. As shown in Fig. 2, the current work is still a significant improvement over CSL most distinctly in the longer distance region of the outer solvation shells.
FIG. 2.

Numerical results for a Lennard-Jones model with l*=0.729. The diamonds are simulation data, the solid line is the result of this work, the dashed line is the result of the standard proper interaction site model, and the dotted line is the XRISM approximation.
At the shortest bond length studied here roughly corresponding to N2 with its triple bond, we see that RISM greatly underestimates the density waves in the system in Fig. 3. More importantly, we found no well-converged, stable solution to CSL. As is clear from the construction of the series for η our theory has the correct united atom behavior.23
The N2 model bond length (l* = 0.329) in particular deserves some comment. First, we were unable to calculate a stable numerical solution for the standard CSL construction, in that no solution for either the HNC- or PY-type closures were better converged than three digits for this model and phase point. As a result, we do not report results for the CSL equations at this model phase point. The fact that we can then get a well-converged, numerically stable solution using the current approximation to η is very encouraging. Second, as can be seen from the radial distribution function plots in Fig. 3, the phase point was such that we were unable to reliably report the results of the Kirkwood G integral from simulation for that model and phase point. This was a limitation of the box size chosen. In the longer bond lengths, the simulation box was sufficiently large to converge the Kirkwood G integrand before the cutoff distance, but for the l* =0.329 system this was not the case.
Overall, as compared to the numerical solution for the proper interaction site equations, the structural results are particularly encouraging, especially in the region of the contact peak. The results are quantitatively in better agreement with simulation, and the phase of the oscillations is a considerable improvement over CSL theory and closer to the XRISM results, which are surprisingly good. The results of the internal energy are also encouraging as the energies are more toward the XRISM result, which is more accurate than the CSL predictions. The Kirkwood-Buff integral results (zeroth moment), on the other hand, appear to overcompensate from the CSL result.
IV. Conclusions
In this work, we have presented two routes to a new integral equation for molecular fluids. In the first, we found a simple approximation to a distinct subclass of the bridge diagrams which are absent in the proper interaction site model integral equation theories. This approximation was included by way of the off-diagonal elements of the density matrix in the proper equations and the normalization of the intramolecular distribution function. The idea was introduced by analogy to the method used in some reactive fluid theories. Next, we have expanded the partition function for a nonreactive, homonuclear diatomic and have shown that the topological distinction between diagram series for η and ρ necessary in reactive fluids also arises for the single-site functions in a nonreactive fluid. In the reactive fluid case the series arises due to real dissociated atoms and in the nonreactive case it is due to the Mayer f-bond expansion in terms of individual sites. We have used the series as an analogy, and approximated the value of the introduced coefficient by employing a result recently used in examination of the reactive fluids. The numerical result for the radial distribution function of a simple model was calculated, and found to be an encouraging improvement over the standard results for CSL and RISM.
The physical analogy here is straightforward. In particular, the bridge diagrams which are approximated are those involving the indirect effect of whole molecules on single Mayer sites in the labeled intermolecular site pair functions. These terms are those which should be most affected by the angular correlations which are neglected when a site-site pair are chosen as the generating functions. The approximation used here, in which the missing diagrams are approximated by a coefficient multiplied by included diagrams, is equivalent to an effective molecular density. The effective density can be thought of as a measure of local-density anisotropy, i.e., screening of the local site density due to the presence of other, integrated, molecular sites. The topological expansions we have used here then take the next logical step.
Finally, we conclude with the following. The variability of the term we have here labeled η in the context of reactive fluids is of course well known. Given that, as indicated above, most of the diagrams in the site-site expansion of the molecular distribution function are atomic bridge diagrams and thus inaccessible in the proper site-site generating equations or by considering molecular bridge functions, we have shown that η is a reasonable variable to approximate the effect of these diagrams which would be included in a molecular OZ treatment of the problem. Furthermore, it is clear that the interpretation of the variable η and the role it plays in the site-site equations is entirely context driven. Due to the fact that η is introduced in the same way in the proper equations, regardless of the context, we have chosen in this work to approximate η using methods similar to those of the reactive fluids. To proceed further, there are two paths which could be investigated. First, the density analogy can be extended, both in terms of more complex molecules and more complete approximation equations, either drawing from variational or inhomogeneous fluid methods. Second, similar to some of the expansions used in this work, there remain unresolved questions about the place of site-site expansions in the larger theory of molecular distribution functions. The particular question raised here regarding the role that certain series expansions play in unaccounted for site-site angular dependencies may ultimately be best approximated in terms of the angular-dependent pair functions. Both ideas may ultimately lead to a better quantitative description of site-site structure and thermodynamics.
Acknowledgments
We gratefully acknowledge the support of the Robert A. Welch Foundation and NIH.
References
- 1.Chandler D, Andersen HC. J Chem Phys. 1972;57:1930. [Google Scholar]
- 2.Chandler D, Silbey R, Ladanyi BM. Mol Phys. 1982;46:1335. [Google Scholar]
- 3.Hansen JP, McDonald IR. Theory of Simple Liquids. 2nd. Academic; London: 1986. [Google Scholar]
- 4.Sullivan DE, Gray CG. Mol Phys. 1981;42:443. [Google Scholar]
- 5.Høye JS, Stell G. J Chem Phys. 1976;65:18. [Google Scholar]
- 6.Hirata F, Rossky PJ. Chem Phys Lett. 1981;83:329. [Google Scholar]
- 7.Cummings PT, Stell G. Mol Phys. 1981;44:529. [Google Scholar]
- 8.Perkyns JS, Pettitt BM. J Chem Phys. 1992;97:7656. [Google Scholar]
- 9.Rossky PJ, Chiles RA. Mol Phys. 1984;51:661. [Google Scholar]
- 10.Chandler D, Joslin CG, Deutch JM. Mol Phys. 1982;47:871. [Google Scholar]
- 11.Chandler D, Richardson DM. J Phys Chem. 1983;87:2060. [Google Scholar]
- 12.Kalyuzhnyi YV, Stell G. Mol Phys. 1993;78:1247. [Google Scholar]
- 13.Lue L, Blankschtein D. J Chem Phys. 1995;102:4203. [Google Scholar]
- 14.Lue L, Blankschtein D. J Chem Phys. 1995;102:5427. [Google Scholar]
- 15.Perkyns JS, Dyer KM, Pettitt BM. J Chem Phys. 2002;116:9404. [Google Scholar]
- 16.Dyer KM, Perkyns JS, Pettitt BM. J Chem Phys. 2002;116:9413. [Google Scholar]
- 17.Vatamanu J, Cann NM. J Chem Phys. 2004;121:6922. doi: 10.1063/1.1789131. [DOI] [PubMed] [Google Scholar]
- 18.Attard P. Mol Phys. 1994;83:273. [Google Scholar]
- 19.Wertheim MS. J Chem Phys. 1987;87:7323. [Google Scholar]
- 20.Stell G. Physica A. 1996;231:1. [Google Scholar]
- 21.Attard P, Patey GN. J Chem Phys. 1990;92:4970. [Google Scholar]
- 22.Gurikov YV, Khim ZF. Russ J Phys Chem. 1982;56:714. [Google Scholar]
- 23.Dyer KM, Perkyns JS, Pettitt BM. J Chem Phys. 2005;122:236101. doi: 10.1063/1.1893829. [DOI] [PubMed] [Google Scholar]
- 24.Wertheim MS. J Stat Phys. 1984;35:35. [Google Scholar]
- 25.Morita T, Hiroike K. Prog Theor Phys. 1961;25:537. [Google Scholar]
- 26.Chandler D. In: The Liquid State of Matter: Fluids, Simple and Complex. Montroll EW, Lebowitz JL, editors. North Holland: Amsterdam; 1982. p. 275. [Google Scholar]
- 27.Kirkwood JG, Buff FP. J Chem Phys. 1951;19:774. [Google Scholar]