

Abstract
The most ubiquitous pathway for regulated calcium (Ca2+) entry into the cells is the store-operated Ca2+ (SOC) entry pathway (also called capacitative Ca2+ entry) that is conserved from lower organisms such as yeast, worms, and flies to man. The SOC concept was proposed over two decades ago, and SOC channels are defined by their activation in response to depletion of the internal Ca2+ stores. Influx through SOC channels is necessary for the replenishment of the Ca2+ stores and is also involved in cell signaling to the nucleus. Despite intensive investigations, most of which are focusing on transient receptor potential (TRP) channels as molecular candidates for SOC channels, the mechanisms of activation and the identity of the key molecular players participating in this signaling pathway have long remained elusive. In the last 2–3 years, however, the improvements of RNA silencing protocols combined with high throughput platforms have yielded significant breakthroughs, with the identification of Stim1 as the Ca2+ store sensor and Orai1 (CRACM1) as the pore-forming subunit of the archetypical SOC channel, CRAC. This review summarizes the recent advances in the mechanisms of activation of SOC channels and their molecular composition, with emphasis on the roles of Stim, Orai, and TRP proteins.
Keywords: Calcium signaling, SOC channels, CRAC channels, Orai1, Stim1, TRPC channels
Introduction
Cytosolic calcium (Ca2+) signals control many cellular functions from short-term responses such as contraction and secretion to long-term regulation of transcription, growth, and cell division [11, 12]. The receptor-evoked Ca2+ signal entails Ca2+ release from the internal stores, namely the endoplasmic reticulum (ER) and activation of Ca2+ influx channels at the plasma membrane (PM). The action of an agonist on its specific receptor typically activates isoforms of the phosphoinositide-specific phospholipase C (PLC). PLC breaks down the phosphatidylinositol 4,5 bisphosphate (PIP2) to generate two second messengers, the inositol 1,4,5 trisphosphate (IP3) and diacylglycerol (DAG). DAG is known to activate protein kinase C (PKC) isoforms but can also regulate ion channels in a PKC-independent manner [29, 50, 70]. IP3 is responsible for Ca2+ release from the ER into the cytoplasm [9] by its action on the IP3 receptor (IP3R).
The PLC pathway and Ca2+ entry channels
The activation of the PLC pathway leads to increased Ca2+ entry from the extracellular space through two major mechanisms. The first involves direct activation of PM Ca2+ entry channels by second messengers such as DAG, IP3, arachidonic acid, and Ca2+ itself; these channels are commonly referred to as receptor-operated channels (ROC) or second messenger-operated channels (SMOC) [7, 13]. Second, as a result of the fall of the Ca2+ concentration within the lumen of the ER that follows IP3-induced Ca2+ release, a concomitant activation of another type of Ca2+ entry channels at the PM takes place. These channels are termed store-operated Ca2+ (SOC) channels and the processes which was originally proposed [56] and later refined [57] by Putney is named capacitative Ca2+ entry or store-operated Ca2+ entry (SOCE). The arrival on the scene of thapsigargin, a specific inhibitor of the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase pump that activates SOC entry by causing passive Ca2+ release without raising IP3 levels [68] along with the successful measurement of the first SOC current in mast cells [30] catapulted SOC entry from a curious phenomenon to a bona fide signaling pathway. The first and best characterized SOC channel is found in hematopoietic cells and conducts a highly Ca2+-selective, non voltage-gated, inwardly rectifying current first described by Hoth and Penner in RBL mast cells and termed the Ca2+ release activated Ca2+ current (CRAC) [30]. Ever since, however, a number of less Ca2+-selective or completely non-selective Ca2+ entry pathways seemingly activated by store depletion have been described in a variety of cells from different tissue origins [52].
Proposed mechanisms of activation for SOC entry
Two major questions have remained enigmatic in the SOC field for almost 20 years: (1) the molecular identity of the SOC channels and (2) the exact mechanisms that signal the depletion of internal Ca2+ stores to open SOC channels at the PM. These two questions are clearly intertwined; solving either would solve the other in the process. Regarding the latter question, two general hypotheses have been advanced to account for the communication between internal stores and SOC channels at the PM. The first involves the action of a diffusible messenger or Ca2+ influx factor (CIF) that is released from the ER as a result of store depletion to activate plasma membrane SOC channels. The CIF idea was first implied by Takemura et al. [68], while in recent years, this concept has been championed by Bolotina and colleagues [15]. Although CIF has yet to be purified and identified, experimental evidence supporting the existence of such messenger was first presented by Randriamampita and Tsien [59] who successfully isolated an acid-extractable factor from thapsigargin-treated lymphocytes. Csutora et al. subsequently showed that this extract can activate CRAC currents more rapidly than when IP3 was included in the patch pipette, hinting that CIF production is downstream of Ca2+ release [19]. The second major hypothesis for SOC activation is referred to as “conformational coupling”; this idea, first introduced by Irvine [31] and later refined by Berridge [10] calls for direct protein–protein interaction upon store depletion between the SOC channel at the PM and the IP3R in the ER, by analogy to excitation–contraction coupling in skeletal muscle where L-type Ca2+ channels at the PM directly couple with the ryanodine receptor in the sarcoplasmic reticulum. It makes implicit sense however that this hypothesis is difficult to test experimentally without a clear molecular candidate for SOC channels and as such studies investigating the conformational coupling hypothesis focused on transient receptor potential (TRP) channels as candidates for SOC channels [14].
Molecular candidates for SOC channels
The discovery of the product of Drosophila trp gene as a light-sensitive Ca2+ permeable channel-activated downstream of PLC during phototransduction in the eye of the fruit fly [47, 72] has led Hardie and Minke to suggest mammalian TRP homologs as molecular correlates for SOC channels [27]. For over a decade, the hunt for the molecule (s) that make up SOC channels have focused predominantly on the TRP superfamily of ion channels and particularly on the canonical TRP family (TRPC) by virtue of their activation downstream of PLC. However, the resultant literature is plagued with controversy with some laboratories providing evidence for TRPC involvement as components of SOC channels while others refuting such involvement (for a thorough review, see [52, 63]).
The last 2 years however brought about significant insights into the molecular composition and the activation mechanisms of SOC and had a remarkable impact on our understanding of the SOC pathway. Using RNA interference (RNAi)-based high throughput screens, four independent laboratories clearly identified two conserved genes that are required for thapsigargin-induced SOC entry, Stim1 and Orai1 [22, 37, 60, 75, 81]. Stim1, a single EF-hand containing protein resident in the ER, is the long-sought Ca2+ sensor capable of oligomerization and reorganization into puncta upon store depletion to somehow signal the activation of Orai1, the pore-forming subunit of the SOC channel at the PM.
Mammals have two Stim proteins, Stim 1 and 2, and three Orai proteins, Orai1, Orai2, and Orai3, which are ubiquitously expressed in different cell types [76]. This review will cover recent advances in the mechanisms of activation of SOC entry as of the spring of 2008 with emphasis on Stim and Orai proteins and will attempt to reconcile their role in SOC entry with the involvement of old players such as TRPC and CIF.
Stim1, Orai1, and their role in the SOC pathway
Stim1
Stim1 is a type I transmembrane protein residing primarily in the ER but can be found to a limited extend in the PM [28, 41]. Stim1 protein contains multiple discrete regions (Fig. 1), including an EF-hand and a sterile α-motif domain (SAM) in the N-terminus directed towards the lumen of the ER. In addition to a transmembrane region, Stim1 also contains a coiled-coil domain, a serine–proline region, and a lysine-rich region in the C-terminus facing the cytoplasmic side.
Fig. 1.
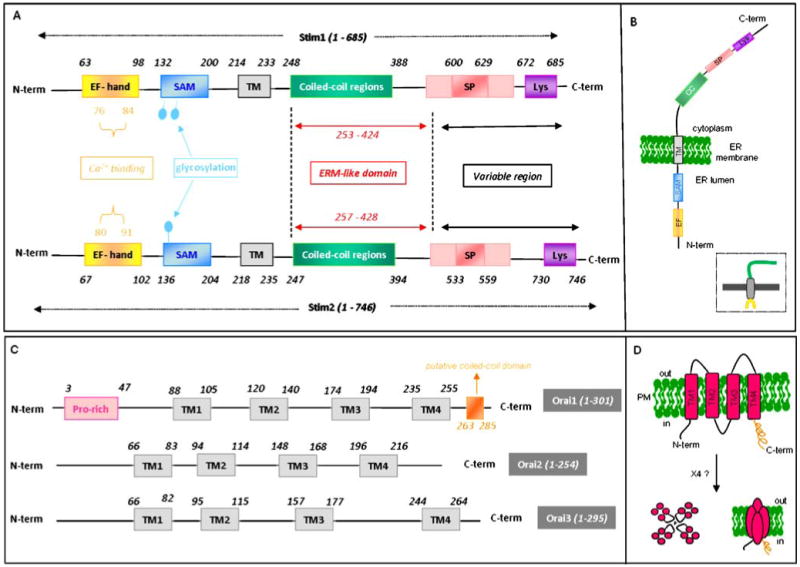
A schematic representation of the functional domains of Stim and Orai proteins. a Stim proteins contain an EF-hand Ca2+ binding motif, a sterile-alpha-motif (SAM), glycosylation sites, 1 transmembrane domain (TM), a coiled-coil region, an ezrin–radixin–myosin-like domain (ERM), a serine/proline-rich region (SP), and a polylysine rich region (Lys). b Schematic representation of a functional STIM1 protein in the ER membrane. c Functional domains of Orai include four transmembrane domains (TM), with its N- and C-termini facing the cytoplasmic side. In addition, Orai1 includes a proline-rich domain in its N-terminus and a reported coiled-coil sequence in the C-terminus [48]. d Schematic representation of a subunit protein of Orai1 in the PM. The functional Orai1 protein channel might be an association of four subunits to form tetramers
In 2005, using a high throughput RNAi-based screen in Drosophila S2 cells where 170 genes were targeted for their involvement in SOC entry, Roos et al. were the first to identify Drosophila Stim as an important player in SOC entry [60]. This was in part possible by the improvement of RNAi targeting in recent years and by the relative simplicity of RNAi in S2 cells; the uptake of double-stranded RNA by S2 cells occurs naturally and does not require transfection reagents or electroporation [17]. Patch-clamp recordings revealed nearly complete suppression of the S2 CRAC current that has biophysical characteristics similar to human CRAC currents in hematopoietic cells. Similarly, knockdown of the human homologue Stim1 significantly reduced CRAC channel activity in Jurkat T cells, as well as thapsigargin- and agonist-mediated Ca2+ entry in HEK293 cells, as measured by Fura-2 imaging. Shortly after, Liou et al. published data identifying Stim1 as an essential protein for SOC entry by using a similar high throughput RNAi knockdown strategy against 2,304 human genes in Hela cells [37]. The same authors were first to show that Ca2+ store depletion leads to translocation of Stim1 into puncta that accumulated near, but not in, the PM. Stim1 puncta vary in size and are believed to be the product of protein oligomerization, although the minimum number of oligomers necessary for SOC activation is still unknown [36]. Biochemical studies on EF-SAM domain of Stim1 shows a low affinity for binding to Ca2+, with a Kd in the 200 to 600 μM range [67], consistent with values proposed for ER Ca2+ content [21]. In fact, a point mutation of aspartate to alanine in the EF-hand Ca2+ binding domain of Stim1 (D76A) resulted in constitutive localization of Stim1 in puncta and constitutively activated SOC entry, as D76A Stim1 is “fooled” into sensing nonexistent store depletion [37]. The same year, Zhang et al. confirmed the results regarding the EF-hand mutant of Stim1 but provided evidence based on membrane biotinylation experiments that Stim1 translocates to the PM upon store depletion, while a study by Gill and coworkers concluded that PM Stim1 plays a role in SOC entry based on extracellular Stim1 antibody blockade of SOC entry [66]. However, based on evidence presented by different laboratories [5, 37, 40, 43, 77], it is fair to conclude that plasma membrane Stim1, although present, does not play an obligatory role in SOC entry and that Ca2+ store depletion leads to translocation of the Stim1 molecules resident in the ER into discrete ER areas closely associated with the PM but not to the PM per se (for review, see [58]).
In resting cells, Stim1 tagged with the yellow fluorescent protein (Stim1-YFP) appears to be in fibrillar structures reminiscent of microtubule staining. Upon store depletion, Stim1 oligomers move on average 2 μm to reach the ER–PM junctions [36], and Smyth et al. presented pharmacological evidence pointing towards a facillatory role of the microtubule cytoskeleton in SOC entry by optimizing the localization of Stim1 for Ca2+ sensing and/or communication with Orai1 [62]. Recently, Grigoriev et al. described a role of Stim1 in ER extension and remodeling through the microtubule “tip attachment complex” mechanism and showed that Stim1 directly binds to the microtubule-plus-end-tracking protein EB1 and forms EB1-dependent comet-like structures at the sites of contact between microtubule ends and ER network, suggesting that the tubulovesicular motility of Stim1 is not motor-based but rather a wave of diffusion in the ER membrane [24]. The redistribution of Stim1 into puncta upon store depletion appears to be the initiator for SOC entry, as Wu et al. showed that puncta formation precedes CRAC channel activation by 6–10 s [77]. Furthermore, another study demonstrated using live-cell imaging that Stim1 forms oligomers within 5 s after store depletion and that Stim1 oligomerization precedes its translocation [36]. Taken together, these studies have provided solid evidence clearly establishing Stim1 as the Ca2+ store sensor in the SOC entry pathway.
Orai1
Using gene mapping on lymphocyte cell lines established from a family with a severe combined immunodeficiency syndrome (SCID) due to the lack of CRAC current combined with a genome-wide RNAi screen of Drosophila S2 cells, a hypothetical protein was first identified as another essential component of the SOC pathway and termed Orai1 [22]. Orai1 is a four-transmembrane protein present in the PM with intracellular N- and C-termini and shows no homology to any known protein (Fig. 1). A single Orai1 mutation of arginine to tryptophan (R91W) in immunodeficient lymphocytes was shown to be responsible for the loss of CRAC channel function [22]. Within the same year, two separate groups used a similar genome-wide RNAi screen on S2 cells and independently identified Orai1 (called CRACM1 by Vig et al. [75]) as necessary for SOC entry and CRAC currents [75, 81]. Mutagenesis studies showed that mutation of a glutamate to alanine in position 106 in human Orai1 (E106A) generated a dead channel, while a more conservative mutation of the same residue to aspartate (E106D) yielded a functional channel that exhibited reduced selectivity to Ca2+ [55, 73, 78]. These data strongly argue that Orai1 is the pore-forming subunit of the CRAC channel. Using overexpression of preassembled tandems containing different number of Orai1 subunits co-expressed with or without a “titrating” dominant-negative Orai1 mutant, Mignen et al. published results suggesting that the functional CRAC channel pore is formed by Orai1 tetramers [46]. However, thorough biochemical and structural studies on purified Orai1 proteins are needed to unequivocally determine the exact oligomeric state of Orai1.
Functional interaction between Stim1 and Orai1
Orai1 and Stim1 interact functionally and together can recapitulate most of the electrophysiological properties of CRAC currents. Four separate laboratories have independently demonstrated that ectopic co-expression of either human Orai1 and Stim1 or Drosophila Orai and Stim generate huge (up to 100-fold of native CRAC) CRAC-like currents [43, 54, 65, 81]. These CRAC-like currents exhibit a similar time course of activation, selectivity for Ca2+, Na+ currents in divalent free bath solutions, sensitivity to 2-APB, and the same inwardly rectifying I/V relationship as the well-characterized CRAC current [54]. At first glance, these results imply that Stim1 and Orai1 are both necessary and limiting for SOC entry and that any additional interacting partners in the cell are either not functionally required or constitutively present in non-limiting quantities. However, when Ca2+ was substituted with either Sr2+ or Ba2+, the large CRAC-like currents generated by co-expression of Stim1 and Orai1 seem smaller than those described for native CRAC. This difference in divalent permeation could be explained by either the requirement of either scaffolding proteins or additional interacting partners that are limiting within the cell, such as Stim2, other Orai isoforms, or possibly TRP proteins.
Strong evidence suggests that upon store depletion, Stim1 aggregates in sub-regions of the ER that are close to the PM. Lewis and colleagues have demonstrated that Stim1 accumulates in specialized regions of junctional ER that are located within 10–25 nm of the PM [77]. An elegant study by the same group showed that open CRAC channels in T cells, as detected by hot spots of local Ca2+ increase, are tightly restricted to sub-regions of the PM that are in close proximity to Stim1-containing junctional ER [40]. While Baba et al. provided evidence supporting a role of the SAM domain of Stim1 in puncta formation [5], a study reported that a mutant Stim1 lacking the C-terminal polybasic motif, which has been shown in other proteins to function as a PM-targeting motif, formed oligomers after store depletion but failed to translocate to ER–PM junctions [36]. A subsequent report by Li et al. determined that amino acids 425–671, which correspond to the serine-proline-rich region of Stim1, are important for the correct targeting of Stim1 puncta to ER–PM junctions upon store depletion, while the polybasic C-terminal motif plays a facillatory but not essential role in Stim1 targeting [33]. The same authors reported that the C-terminal coiled-coil domain of Stim1 is necessary for Stim1 aggregation and that Orai1 C-terminus is required for functional interaction with Stim1 [33]. Work from Muik et al. identified a putative coiled-coil region in the C-terminus of Orai1 (amino acids 263–285) as necessary for the functional interaction between Stim1 and Orai1, while Orai1 N-terminus along with the arginine residue found to be mutated in SCID patients (R91W) are critical for Orai1 gating [48].
Based on the aforementioned studies, it is reasonable to conclude that Stim1 interacts functionally with Orai1 to activate SOC entry, but does Stim1 convey information to Orai1 channels via direct protein–protein interaction or through intermediary proteins? And how does the CIF and TRP fit with these new players? The answer to the former question is beginning to unravel albeit not without some controversy. Using co-immunoprecipitation, Yeromin et al. showed that Drosophila Stim and Orai associate and that store depletion enhanced their association, arguing for a direct Stim–Orai interaction as the necessary signal for SOC activation [78]. Co-immunoprecipitation of ectopically expressed human Stim1 and Orai1 was also reported by Vig et al; the effect of store depletion on this association was not reported in this study. However, studies from a different laboratory failed to detect any association between human Stim1 and Orai1 in co-immunoprecipitation and glycerol gradient centrifugation experiments [22, 26]. In vitro pull-down experiments using the C-terminus of Stim1, which constitutively activates Orai1, showed that the C-terminal domain of Stim1 can associate with Orai1 in cell-free conditions, arguing for a direct Stim1–Orai1 interaction [48]. The same authors showed a close interaction (within <10 nm range) between tagged Orai1 and Stim1 expressed in HEK293 cells using FRET microscopy and speculated about direct protein–protein interactions between the respective coiled-coil domains in the C-termini of Orai1 and Stim1 [48].
An innovative strategy by Balla and colleagues that controlled for the distance between the PM and the ER with the use of chemically inducible bridges showed that Orai1 requires a larger space than Stim1 in the ER–PM junctions; Orai1 was proposed to be part of a large macromolecular complex, with an estimated protrusion to the cytoplasm that is larger than 8–9 nm but smaller than 12–14 nm, while the cytoplasmic domain of Stim1 could fit in a space of approximately 4–6 nm [71], estimates that are in agreement with other studies [48, 77]. Although these results are not incompatible with direct Stim1–Orai1 interactions, they point towards the involvement of auxiliary proteins in SOC activation and raise the possibility that these proteins might act as intermediates in Stim1–Orai1 interaction.
These auxiliary proteins might well be formed in part by components of the CIF pathway, namely the Ca2+-independent iPLA2 and calmodulin proposed by Bolotina and colleagues [61]. For CIF to co-exist with the Stim1–Orai1 paradigm, it has to be formed and act downstream of Stim1 reorganization, as SOC activation occurs with the D76A mutant and the C-terminal fragment of Stim1 without store depletion. This is exactly what a recent report is suggesting; CIF production was reported to be tightly coupled with stim1 expression as knockdown or overexpression of Stim1 resulted in corresponding impairment or amplification of CIF production [18]. These authors showed that the glycosylation sites in ER-resident SAM domain of Stim1 were required for the initiation of CIF production [18].
It is plausible to imagine a scenario where stim1 movement and organization into puncta is followed by recruitment of a macromolecular complex (containing membrane and/or cytoplasmic proteins) in the vicinity of Orai1 channels and in situ CIF synthesis. Under this scheme, CIF would act in a similar fashion to a neurotransmitter and the necessary recycling machinery or enzymes that synthesize and hydrolyze CIF under store depletion/repletion might be either part of this protein complex or contained within the areas of close PM-ER junctions. Figure 2 summarizes the different possibilities discussed here as to how ER Stim1 might convey store depletion to Orai1 at the PM.
Fig. 2.
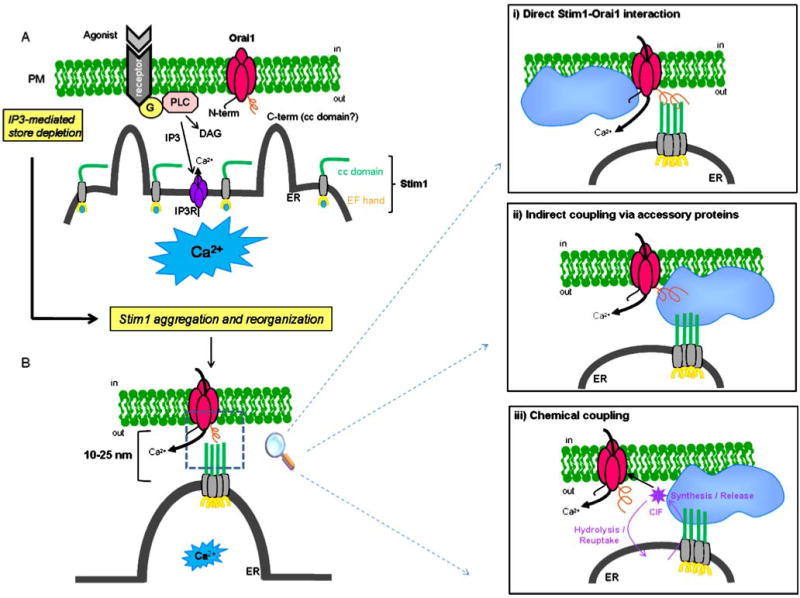
Possible scenarios for coupling between Stim1 and Orai1 in SOC activation. a Activation of membrane receptors causes IP3-mediated store depletion followed by Stim1 reorganization into puncta. The decrease in luminal ER Ca2+ causes dissociation of bound Ca2+ from the low affinity EF-hand in the N-terminus of Stim1. This Ca2+ dissociation causes Stim1 molecules to aggregate (puncta formation) and to diffuse into regions of ER in close proximity with the plasma membrane (PM) within 10–25 nm. b Stim1 oligomers then interact functionally with Orai1 channels in the PM. Three coupling models are proposed: (i) a model involving direct protein–protein interaction between the C-terminal domain of Stim1 in the ER and the C-terminal region of Orai1 in the PM. (ii) An alternative physical coupling model in which the Stim1–Orai1 interaction is mediated via accessory membrane and/or cytoplasmic proteins, either constitutively present or recruited to the tight spaces of ER–PM junctions upon store depletion. (iii) A coupling model involving a diffusible calcium influx factor (CIF) produced or released within the ER–PM junctions to activate Orai1
Role of TRP channels in SOC entry
The mammalian TRP superfamily of proteins comprise cation channel members with varying permeabilities to Ca2+ ions and, over the past 13 years, have been the focus of intensive investigations aimed at implicating them in the SOC pathway. Many of these studies have focused on the canonical TRP members (TRPC), which comprises seven channel proteins (TRPC1–7), by virtue of their activation by mechanisms downstream of the PLC pathway. The evidence for a role of TRPC in the SOC pathway have been discussed in detail elsewhere [63, 69], and as of this writing, the involvement of TRPC proteins in SOC remains a highly controversial topic. It is quite revealing to contrast the failure to reach a consensus in over a decade of research on the role of TRPC proteins in SOC entry, with the clear establishment in less than 2 years of Orai1 and Stim1 as central players in the SOC pathway. Namely, ectopic TRPC expression consistently failed to recapitulate the electrophysiological and pharmacological characteristics of the archetypical CRAC current. While several laboratories reported that knockdown protocols targeting TRPC channels in several cell types inhibited SOC entry, others laboratories failed to reproduce these results; this is true for T cells, B cells, and particularly for the widely studied HEK293 cells (see [52] and references therein). However, as we argued elsewhere [63, 69], while CRAC is the first discovered SOC current and the most intensely studied, SOC currents are quite heterogeneous in terms of pharmacological profile and ion selectivity patterns in different cell types (for extensive review, see [52], and references therein). Thus, it is conceivable that these SOC channels are made up of heteromultimers from a heterogeneous group of ion channel with different ion selectivities. Under these conditions, the native SOC channel could be formed by a combination of different TRPC isoforms and additional accessory molecules, conditions that are not easily recapitulated during ectopic expression. The SOC entry mediated by TRPC1 either in human salivary gland cells [3, 4] or in vascular smooth muscle cells [1, 8] is an example of such pathway.
Interestingly, a recent report suggested that the association of Stim1 with TRPC1 favors the insertion of TRPC1 into lipid rafts and converts it from a ROC to a SOC channel [2], and an increasing number of studies from different laboratories are reporting interactions between Stim1 and TRPC isoforms [79] or ternary complex formation between Stim1, Orai1, and TRPC [51], as revealed by co-immunoprecipitation experiments. A role of TRPC proteins as a subunit of SOC channels and the possibility that Orai1 could mediate Stim1-dependent activation of non-selective SOC channels containing TRPC molecules as their pore-forming subunits cannot be ruled out at this stage. Similarly, the involvement of Stim and Orai proteins in SOC in all cell types remains to be demonstrated.
Based on co-expression studies of unusually small cDNA quantities (60 ng as opposed to microgram) of Orai1 in HEK293 cells stably expressing TRPC proteins, the Birnbaumer group have proposed an alternative model whereby TRPC are the pore-forming unit of the CRAC channel and Orai1proteins regulatory subunits that interact with TRPC to mediate their Stim1-dependent activation [34, 35]. To reconcile their model with published evidence from several laboratories showing mutations of residue E106 in the putative pore region of Orai1 either yields a dead channel or changes the ion selectivity of the CRAC channel [55, 73, 78], Birnbaumer and coworkers [35] have drawn an analogy between Orai1 and KCNE1 (minK), the regulatory β-subunit of the K+ channel KCNQ1. KCNE1 neither lines the pore of the KCNQ1 channel nor forms part of its voltage-regulated gate yet point mutations in KCNE1 transmembrane domain-altered KCNQ1 current voltage dependency and ion selectivity [42]. However, it is worth mentioning that in all the RNAi screens discussed herein, which clearly implicated Stim1 and Orai1 in SOC entry, not a single TRP channel gave a hit. The Birnbaumer model is also difficult to reconcile with established data demonstrating that co-expression of Stim1 and Orai1 in HEK293 cells generates huge CRAC-like currents [43, 54, 65, 81]; it assumes that under native conditions, TRPC proteins are not limiting. Furthermore, it is not clear how Orai1 interaction with a non-selective TRPC channel would endow the latter sub-unit with high Ca2+ selectivity. Clearly, additional studies are needed to understand the dynamics of Stim/Orai/TRPC interactions in mammalian cells and their role in shaping native Ca2+ entry pathways.
Stim2 and Orai2/3
Stim2
In vertebrates, Stim1 has a closely related homologue, Stim2. Stim2 is also a type I transmembrane protein and contains 833 amino acids but is only found in the ER (Fig. 1). The two molecules share similar domain architecture, with the N-terminal EF-hand and SAM domains in the ER lumen and the C-terminal domains in the cytoplasm [28, 82]. In their original screen mentioned above, Liou et al. reported a slight reduction in SOC entry after knock-down of Stim2 [37]. Soboloff et al. showed by using immunoprecipitations that Stim1 and Stim2 interact and provided evidence, suggesting an inhibitory role of Stim2 on Stim1-mediated SOC activation when expressed in various cell lines such as HEK293, PC12, A7r5, and Jurkat T cells [64]. Interestingly, when Stim2 is co-expressed with Orai1, it resulted in a substantial increase in constitutive SOC entry [64]. The ability of Stim2 to increase the SOC entry implies that under certain conditions, Stim2 function might overlap with that of Stim1 in regulating the SOC pathway. In fact, an elegant study by Brandman et al. has shed light into the role of Stim2; using an RNAi screen, these authors provided compelling evidence implicating Stim2 as a powerful regulator of basal cytoplasmic Ca2+ concentrations. Unlike Stim1, Stim2 is active at basal ER Ca2+ concentrations and can further activate Ca2+ influx through Orai1 upon smaller decrease in ER Ca2+ content [16]. In addition to this store-operated mode, another store-independent mode of CRAC channel activation by Stim2 have been described; in this case, Stim2 action on CRAC channels was shown to be counteracted by calmodulin [53]. In HEK29 cells, Stim2 exists in at least two functional states, with most of the Stim2 molecules coupling to CRAC channels in a store-independent manner (constitutively coupled) and a smaller population of stim2 molecules remaining available to activate CRAC channels after store depletion (store coupled). The store-operated Stim2 population can be suppressed by maintaining the stores filled with Ca2+, while the store-independent population can be revealed by loss of cytosolic calmodulin that appears to inhibit the constitutive activity of Stim2 and its activation of Orai1 [53].
Studies on knockout animals are beginning to emerge, confirming the RNAi results obtained earlier with cultured cells. Not surprisingly, Stim1-deficient fetal liver-derived mast cells had impaired FcεRI-mediated mast cell activation and anaphylaxis; Ca2+ influx mediated by the high-affinity IgE receptor FcεRI and activation of the transcription factors NF-κB and NFAT were impaired [6]. Studies on knockout mice with conditionally targeted alleles of Stim1 and Stim2 showed that while T cells and fibroblasts lacking Stim2 had a smaller impairment of SOC entry compared to Stim1 mice; T cells lacking either Stim1 or Stim2 showed impaired nuclear translocation of the transcription factor NFAT and cytokine production. T-cell-specific knockout of both Stim1 and Stim2 resulted in a lymphoproliferative phenotype and a selective decrease in regulatory T cell numbers [49]. This data suggest that Stim1 and Stim2 have a non-redundant role in cell function and are consistent with previous data, suggesting that Stim2 is active at higher ER Ca2+ concentrations compared to Stim1, thus maintaining SOC entry and NFAT nuclear localization when partial store replete conditions have inactivated Stim1 [16].
Orai2/3
In addition to Orai1, Feske et al. described two homologous genes, which were designated Orai2 and Orai3 [22]. Mercer et al. have demonstrated in HEK293 ectopic expression experiments that when co-expressed with Stim1, all Orai proteins augmented SOC entry with efficacies in the order: Orai1>Orai2>Orai3 [43]. Orai3 failed to produce any detectable increase in Ca2+-selective currents, while Na+ currents (in divalent free bath solutions) were significantly larger in Orai3-expressing cells than in control cells, perhaps pointing towards low levels of Orai3 expression [20]. However, Orai3 was capable of rescuing the knock-down of Orai1 in HEK293 cells [43]. De Haven et al. showed that Orai1, 2, and 3 channels are similarly inhibited by extracellular Ca2+, but Orai3 channels appeared to differ from Orai1 and 2 by being to some extent resistant to the process of Ca2+ depotentiation [20]. Lis et al. also described distinct patterns of ion selectivity, pharmacological inhibition, and regulation by intracellular Ca2+ between Orai1, 2, and 3 (referred to as CRACM1-3) [38]. While Orai1 displayed fast and slow Ca2+-dependent inactivation, Orai2 and Orai3 showed only fast inactivation; Orai2 appeared fairly resistant to Ca2+-induced inactivation in general, with only a small component of fast inactivation, whereas Orai3 displayed a much greater degree of fast inactivation. All Orai isoforms share high Ca2+ selectivity and can discriminate against Na+ ions as long as Mg2+ ions (2 mM) are present. However, when Ca2+ is substituted by Ba2+ as the charge carrier, Orai1 currents are greatly reduced, while Orai2 and Orai3 maintained significant inward currents; it appears that in the presence of Ba2+, mainly Na+ ions or a mixture of Na+ and Ba2+ may be carrying Orai currents. Regarding pharmacological differences between different Orai homologs, the drug 2-APB was shown to increase Orai3 and inhibit Orai1 currents [38, 80]. Knock-down of endogenous Stim1 or Orai1 in cells overexpressing Orai3 did not influence the 2-APB-evoked Ca2+ influx, suggesting that 2-APB could sensitize or activate Orai3 in a Stim1- and store-depletion-independent manner [80]. Parvez et al. showed rapid store-independent gating of CRAC channel by 2-APB that appeared to be mediated by some close interaction between Stim2 and Orai1; this rapid gating was not observed in cells overexpressing Orai1 either with or without Stim1 [53]. These subtle differences in the biophysical properties of different Orai proteins could contribute to the heterogeneity of the SOC currents observed in different cell types. Additionally, Lis et al. suggested that Orai proteins can form heteromultimeric complexes based on experiments, showing that the dead channel mutation glutamate 106 to glutamine (E106Q) in orai1 acts as a dominant negative for all Orai homologs [38].
Orai2 and Orai3 may be responsible for SOC entry in different cell types of non-lymphoid origin; this is consistent with the fact that SCID patients defective for Orai1 displayed only minor alterations in other cell types. Interestingly, recent studies on Orai1 knockout mice showed alteration of CRAC currents in mast cells but not in T cells [74]. This implies that unlike human T cells where Orai1 is involved in SOC entry, in mice T cells, this pathway appears to be fulfilled by Orai2 [74]. In fact, Vig et al. reported undetectable levels of Orai1 in the thymic and spleenic lymphoid areas of newborn mice, and wild-type mouse thymocytes and T cells had much higher expression of Orai2 mRNA than Orai1 and Orai3 mRNA [74]. The heteromultimerization of the three Orai homologs might provide further flexibility for subtle control of Ca2+ signals in different cell types and tissues. Along these lines, co-expression of mouse Orai2 variants (S and L, for short and long N-terminus, respectively) with Stim1 showed substantial CRAC current enhancement in HEK 293 but not in RBL 2H3 cells, while Orai1 enhanced CRAC currents in both cell lines, suggesting that the capability of Orai2 to form CRAC channels depends on the cell background and the expression levels of native Orai and Stim homologs in these cells [25]. Further work is needed to understand the molecular interactions between different Orai and Stim proteins and their role in shaping regulated Ca2+ signals in mammalian cells from different tissues.
In addition, the involvement of Stim and Orai proteins seems to extend beyond the SOC pathway to store-independent Ca2+ entry pathways. Thus, while the levels of Orai1 alone determine the magnitude of the CRAC channel currents, both Orai1 and Orai3 are critical for the corresponding currents through the store-independent arachidonic acid-regulated Ca2+ (ARC) channels. Mignen et al. showed that in HEK293 cells stably expressing STIM1, overexpression of Orai1 increases CRAC and ARC channels currents. While overexpression of Orai3 alone had no effect on ARC currents, increased ARC currents were observed by co-expression of Orai3 in cells stably expressing Orai1. Expression of a dominant-negative mutant of Orai3 (E81Q), alone or in cells expressing wild-type Orai1, reduces currents through ARC channels without affecting CRAC currents [45]. The same group reported earlier that ARC channels are also regulated by Stim1, but unlike SOC channels, this regulation depends exclusively on Stim1 proteins constitutively present in the plasma membrane [44].
Conclusions
The SOC pathway is one of the most ubiquitous means of regulated Ca2+ entry in mammalian cells found not only in non-excitable cells but also increasingly appreciated in electrically excitable cells. SOC entry is not only vital for the replenishment of the ER Ca2+ stores but also plays a key role in driving important physiological processes such as lymphocyte activation and cell proliferation [23, 32, 39, 52]. The identification of Stim1 and Orai1 as key components of this pathway have provided much needed molecular “starting points” for future investigations on the signaling mechanisms for SOC entry and constitute a major advancement in the SOC field. As discussed above, insights into the communication between Stim1 and Orai1 are starting to emerge. Some of the obvious unanswered questions that come to mind are the following: (1) what are the accessory proteins that are involved in this molecular communication? (2) What is the exact in vivo oligomeric state of the CRAC channel and the molecular make up of other non-selective SOC channels? (3) What is the contribution of Orai isoform homo- and heteromultimerization to regulated Ca2+ entry in different cell types? (4) Does Stim2 communicate with Orai homologs in a fashion similar to that of Stim1? The answer to these questions and many others will enhance our understanding of the signaling mechanisms for SOC entry in different cell types, so we might in the future specifically target components of this important pathway in therapeutic strategies of human disease.
Acknowledgments
Work in the authors' laboratory is supported by start up funds from the Albany Medical College, NY and by an early career NIH grant to M.T. (K22ES014729).
References
- 1.Albert AP, Saleh SN, Peppiatt-Wildman CM, Large WA. Multiple activation mechanisms of store-operated TRPC channels in smooth muscle cells. J Physiol. 2007;583:25–36. doi: 10.1113/jphysiol.2007.137802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alicia S, Angelica Z, Carlos S, Alfonso S, Luis V. STIM1 converts TRPC1 from a receptor-operated to a store-operated channel: moving TRPC1 in and out of lipid rafts. Cell Calcium. 2008 doi: 10.1016/j.ceca.2008.03.001. in press. [DOI] [PubMed] [Google Scholar]
- 3.Ambudkar IS. TRPC1: a core component of store-operated calcium channels. Biochem Soc Trans. 2007;35:96–100. doi: 10.1042/BST0350096. [DOI] [PubMed] [Google Scholar]
- 4.Ambudkar IS, Ong HL, Liu X, Bandyopadhyay B, Cheng KT. TRPC1: the link between functionally distinct store-operated calcium channels. Cell calcium. 2007;42:213–223. doi: 10.1016/j.ceca.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 5.Baba Y, Hayashi K, Fujii Y, Mizushima A, Watarai H, Wakamori M, Numaga T, Mori Y, Iino M, Hikida M, Kurosaki T. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc Natl Acad Sci USA. 2006;103:16704–16709. doi: 10.1073/pnas.0608358103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baba Y, Nishida K, Fujii Y, Hirano T, Hikida M, Kurosaki T. Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat Immunol. 2008;9:81–88. doi: 10.1038/ni1546. [DOI] [PubMed] [Google Scholar]
- 7.Barritt GJ. Receptor-activated Ca2+ inflow in animal cells: a variety of pathways tailored to meet different intracellular Ca2+ signalling requirements. Biochem J. 1999;337(Pt 2):153–169. [PMC free article] [PubMed] [Google Scholar]
- 8.Beech DJ. TRPC1: store-operated channel and more. Pflugers Arch. 2005;451:53–60. doi: 10.1007/s00424-005-1441-3. [DOI] [PubMed] [Google Scholar]
- 9.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 10.Berridge MJ. Capacitative calcium entry. Biochem J. 1995;312(Pt 1):1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 12.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 13.Bird GS, Aziz O, Lievremont JP, Wedel BJ, Trebak M, Vazquez G, Putney JW., Jr Mechanisms of phospholipase C-regulated calcium entry. Curr Mol Med. 2004;4:291–301. doi: 10.2174/1566524043360681. [DOI] [PubMed] [Google Scholar]
- 14.Birnbaumer L, Boulay G, Brown D, Jiang M, Dietrich A, Mikoshiba K, Zhu X, Qin N. Mechanism of capacitative Ca2+ entry (CCE): interaction between IP3 receptor and TRP links the internal calcium storage compartment to plasma membrane CCE channels. Recent Prog Horm Res. 2000;55:127–161. discussion 161–122. [PubMed] [Google Scholar]
- 15.Bolotina VM, Csutora P. CIF and other mysteries of the store-operated Ca2+-entry pathway. Trends Biochem Sci. 2005;30:378–387. doi: 10.1016/j.tibs.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 16.Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131:1327–1339. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cahalan MD, Zhang SL, Yeromin AV, Ohlsen K, Roos J, Stauderman KA. Molecular basis of the CRAC channel. Cell calcium. 2007;42:133–144. doi: 10.1016/j.ceca.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Csutora P, Peter K, Kilic H, Park KM, Zarayskiy V, Gwozdz T, Bolotina VM. Novel role of STIM1 as a trigger for calcium influx factor (CIF) production. J Biol Chem. 2008 doi: 10.1074/jbc.M709575200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Csutora P, Su Z, Kim HY, Bugrim A, Cunningham KW, Nuccitelli R, Keizer JE, Hanley MR, Blalock JE, Marchase RB. Calcium influx factor is synthesized by yeast and mammalian cells depleted of organellar calcium stores. Proc Natl Acad Sci USA. 1999;96:121–126. doi: 10.1073/pnas.96.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeHaven WI, Smyth JT, Boyles RR, Putney JW., Jr Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J Biol Chem. 2007;282:17548–17556. doi: 10.1074/jbc.M611374200. [DOI] [PubMed] [Google Scholar]
- 21.Demaurex N, Frieden M. Measurements of the free luminal ER Ca(2+) concentration with targeted “cameleon” fluorescent proteins. Cell calcium. 2003;34:109–119. doi: 10.1016/s0143-4160(03)00081-2. [DOI] [PubMed] [Google Scholar]
- 22.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 23.Feske S, Okamura H, Hogan PG, Rao A. Ca2+/calcineurin signalling in cells of the immune system. Biochem Biophys Res Commun. 2003;311:1117–1132. doi: 10.1016/j.bbrc.2003.09.174. [DOI] [PubMed] [Google Scholar]
- 24.Grigoriev I, Gouveia SM, van der Vaart B, Demmers J, Smyth JT, Honnappa S, Splinter D, Steinmetz MO, Putney JW, Jr, Hoogenraad CC, Akhmanova A. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr Biol. 2008;18:177–182. doi: 10.1016/j.cub.2007.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gross SA, Wissenbach U, Philipp SE, Freichel M, Cavalie A, Flockerzi V. Murine ORAI2 splice variants form functional Ca2+ release-activated Ca2+ (CRAC) channels. J Biol Chem. 2007;282:19375–19384. doi: 10.1074/jbc.M701962200. [DOI] [PubMed] [Google Scholar]
- 26.Gwack Y, Srikanth S, Feske S, Cruz-Guilloty F, Oh-hora M, Neems DS, Hogan PG, Rao A. Biochemical and functional characterization of Orai proteins. J Biol Chem. 2007;282:16232–16243. doi: 10.1074/jbc.M609630200. [DOI] [PubMed] [Google Scholar]
- 27.Hardie RC, Minke B. Novel Ca2+ channels underlying transduction in Drosophila photoreceptors: implications for phosphoinositide-mediated Ca2+ mobilization. Trends Neurosci. 1993;16:371–376. doi: 10.1016/0166-2236(93)90095-4. [DOI] [PubMed] [Google Scholar]
- 28.Hewavitharana T, Deng X, Soboloff J, Gill DL. Role of STIM and Orai proteins in the store-operated calcium signaling pathway. Cell calcium. 2007;42:173–182. doi: 10.1016/j.ceca.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 29.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 30.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 31.Irvine RF. ‘Quantal’ Ca2+ release and the control of Ca2+ entry by inositol phosphates—a possible mechanism. FEBS lett. 1990;263:5–9. doi: 10.1016/0014-5793(90)80692-c. [DOI] [PubMed] [Google Scholar]
- 32.Lewis RS. Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol. 2001;19:497–521. doi: 10.1146/annurev.immunol.19.1.497. [DOI] [PubMed] [Google Scholar]
- 33.Li Z, Lu J, Xu P, Xie X, Chen L, Xu T. Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J Biol Chem. 2007;282:29448–29456. doi: 10.1074/jbc.M703573200. [DOI] [PubMed] [Google Scholar]
- 34.Liao Y, Erxleben C, Abramowitz J, Flockerzi V, Zhu MX, Armstrong DL, Birnbaumer L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci USA. 2008;105:2895–2900. doi: 10.1073/pnas.0712288105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liao Y, Erxleben C, Yildirim E, Abramowitz J, Armstrong DL, Birnbaumer L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc Natl Acad Sci USA. 2007;104:4682–4687. doi: 10.1073/pnas.0611692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA. 2007;104:9301–9306. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lis A, Peinelt C, Beck A, Parvez S, Monteilh-Zoller M, Fleig A, Penner R. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr Biol. 2007;17:794–800. doi: 10.1016/j.cub.2007.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luik RM, Lewis RS. New insights into the molecular mechanisms of store-operated Ca2+ signaling in T cells. Trends Mol Med. 2007;13:103–107. doi: 10.1016/j.molmed.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 40.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER–plasma membrane junctions. J Cell Biol. 2006;174:815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manji SS, Parker NJ, Williams RT, van Stekelenburg L, Pearson RB, Dziadek M, Smith PJ. STIM1: a novel phosphoprotein located at the cell surface. Biochim Biophys Acta. 2000;1481:147–155. doi: 10.1016/s0167-4838(00)00105-9. [DOI] [PubMed] [Google Scholar]
- 42.Melman YF, Krummerman A, McDonald TV. KCNE regulation of KvLQT1 channels: structure-function correlates. Trends Cardiovasc Med. 2002;12:182–187. doi: 10.1016/s1050-1738(02)00158-5. [DOI] [PubMed] [Google Scholar]
- 43.Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW., Jr Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem. 2006;281:24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mignen O, Thompson JL, Shuttleworth TJ. STIM1 regulates Ca2+ entry via arachidonate-regulated Ca2+-selective (ARC) channels without store depletion or translocation to the plasma membrane. J Physiol. 2007;579:703–715. doi: 10.1113/jphysiol.2006.122432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mignen O, Thompson JL, Shuttleworth TJ. Both Orai1 and Orai3 are essential components of the arachidonate-regulated Ca2+-selective (ARC) channels. J Physiol. 2008;586:185–195. doi: 10.1113/jphysiol.2007.146258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mignen O, Thompson JL, Shuttleworth TJ. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J Physiol. 2008;586:419–425. doi: 10.1113/jphysiol.2007.147249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Montell C, Rubin GM. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron. 1989;2:1313–1323. doi: 10.1016/0896-6273(89)90069-x. [DOI] [PubMed] [Google Scholar]
- 48.Muik M, Frischauf I, Derler I, Fahrner M, Bergsmann J, Eder P, Schindl R, Hesch C, Polzinger B, Fritsch R, Kahr H, Madl J, Gruber H, Groschner K, Romanin C. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem. 2008 doi: 10.1074/jbc.M708898200. [DOI] [PubMed] [Google Scholar]
- 49.Oh-Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S, Rao A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol. 2008;9:432–443. doi: 10.1038/ni1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Okada T, Inoue R, Yamazaki K, Maeda A, Kurosaki T, Yamakuni T, Tanaka I, Shimizu S, Ikenaka K, Imoto K, Mori Y. Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca(2+)-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. J Biol Chem. 1999;274:27359–27370. doi: 10.1074/jbc.274.39.27359. [DOI] [PubMed] [Google Scholar]
- 51.Ong HL, Cheng KT, Liu X, Bandyopadhyay BC, Paria BC, Soboloff J, Pani B, Gwack Y, Srikanth S, Singh BB, Gill D, Ambudkar IS. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J Biol Chem. 2007;282:9105–9116. doi: 10.1074/jbc.M608942200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 53.Parvez S, Beck A, Peinelt C, Soboloff J, Lis A, Monteilh-Zoller M, Gill DL, Fleig A, Penner R. STIM2 protein mediates distinct store-dependent and store-independent modes of CRAC channel activation. FASEB J. 2008;22:752–761. doi: 10.1096/fj.07-9449com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, Koblan-Huberson M, Lis A, Fleig A, Penner R, Kinet JP. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8:771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 56.Putney JW., Jr A model for receptor-regulated calcium entry. Cell calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 57.Putney JW., Jr Capacitative calcium entry revisited. Cell calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- 58.Putney JW., Jr Recent breakthroughs in the molecular mechanism of capacitative calcium entry (with thoughts on how we got here) Cell calcium. 2007;42:103–110. doi: 10.1016/j.ceca.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Randriamampita C, Tsien RY. Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature. 1993;364:809–814. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- 60.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. A novel mechanism for the store-operated calcium influx pathway. Nat Cell Biol. 2004;6:113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- 62.Smyth JT, DeHaven WI, Bird GS, Putney JW., Jr Role of the microtubule cytoskeleton in the function of the store-operated Ca2+ channel activator STIM1. J Cell Sci. 2007;120:3762–3771. doi: 10.1242/jcs.015735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smyth JT, Dehaven WI, Jones BF, Mercer JC, Trebak M, Vazquez G, Putney JW., Jr Emerging perspectives in store-operated Ca(2+) entry: roles of Orai, Stim and TRP. Biochim Biophys Acta. 2006;1763:1147–1160. doi: 10.1016/j.bbamcr.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 64.Soboloff J, Spassova MA, Hewavitharana T, He LP, Xu W, Johnstone LS, Dziadek MA, Gill DL. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ entry. Curr Biol. 2006;16:1465–1470. doi: 10.1016/j.cub.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 65.Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 66.Spassova MA, Soboloff J, He LP, Xu W, Dziadek MA, Gill DL. STIM1 has a plasma membrane role in the activation of store-operated Ca(2+) channels. Proc Natl Acad Sci USA. 2006;103:4040–4045. doi: 10.1073/pnas.0510050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stathopulos PB, Li GY, Plevin MJ, Ames JB, Ikura M. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: an initiation mechanism for capacitive Ca2+ entry. J Biol Chem. 2006;281:35855–35862. doi: 10.1074/jbc.M608247200. [DOI] [PubMed] [Google Scholar]
- 68.Takemura H, Hughes AR, Thastrup O, Putney JW., Jr Activation of calcium entry by the tumor promoter thapsigargin in parotid acinar cells. Evidence that an intracellular calcium pool and not an inositol phosphate regulates calcium fluxes at the plasma membrane. J Biol Chem. 1989;264:12266–12271. [PubMed] [Google Scholar]
- 69.Trebak M, Lemonnier L, Smyth JT, Vazquez G, Putney JW., Jr Phospholipase C-coupled receptors and activation of TRPC channels. Handb Exp Pharmacol. 2007;179:593–614. doi: 10.1007/978-3-540-34891-7_35. [DOI] [PubMed] [Google Scholar]
- 70.Trebak M, St JBG, McKay RR, Birnbaumer L, Putney JW., Jr Signaling mechanism for receptor-activated canonical transient receptor potential 3 (TRPC3) channels. J Biol Chem. 2003;278:16244–16252. doi: 10.1074/jbc.M300544200. [DOI] [PubMed] [Google Scholar]
- 71.Varnai P, Toth B, Toth DJ, Hunyady L, Balla T. Visualization and manipulation of plasma membrane-endoplasmic reticulum contact sites indicates the presence of additional molecular components within the STIM1–Orai1 complex. J Biol Chem. 2007;282:29678–29690. doi: 10.1074/jbc.M704339200. [DOI] [PubMed] [Google Scholar]
- 72.Venkatachalam K, Montell C. TRP channels. Ann Rev Biochem. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP, Penner R. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol. 2006;16:2073–2079. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vig M, DeHaven WI, Bird GS, Billingsley JM, Wang H, Rao PE, Hutchings AB, Jouvin MH, Putney JW, Kinet JP. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol. 2008;9:89–96. doi: 10.1038/ni1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wissenbach U, Philipp SE, Gross SA, Cavalie A, Flockerzi V. Primary structure, chromosomal localization and expression in immune cells of the murine ORAI and STIM genes. Cell calcium. 2007;42:439–446. doi: 10.1016/j.ceca.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 77.Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol. 2006;174:803–813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol. 2007;9:636–645. doi: 10.1038/ncb1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang SL, Kozak JA, Jiang W, Yeromin AV, Chen J, Yu Y, Penna A, Shen W, Chi V, Cahalan MD. Store-dependent and -independent modes regulating CRAC channel activity of human Orai1 and Orai3. J Biol Chem. 2008 doi: 10.1074/jbc.M801536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci USA. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zheng L, Stathopulos PB, Li GY, Ikura M. Biophysical characterization of the EF-hand and SAM domain containing Ca(2+) sensory region of STIM1 and STIM2. Biochem Biophys Res Commun. 2007 doi: 10.1016/j.bbrc.2007.12.129. [DOI] [PubMed] [Google Scholar]