

Abstract
Inflammatory bowel disease is a chronic inflammatory response of the gastrointestinal tract mediated in part by an aberrant response to intestinal microflora. Expression of IL-23 subunits p40 and p19 within cells of the innate immune system plays a central role in the development of lower bowel inflammation in response inflammatory challenge. The NF-κB subunit c-Rel can regulate expression of IL-12/23 subunits suggesting that it could have a critical role in mediating the development of chronic inflammation within the lower bowel. Here we have analyzed the role of c-Rel within the innate immune system in the development of lower bowel inflammation, in two well-studied models of murine colitis. We have found that the absence of c-Rel significantly impaired the ability of H. hepaticus to induce colitis upon infection of RAG-2-deficient mice, and ameliorated the ability of CD4+CD45RBhigh T cells to induce disease upon adoptive transfer into RAG-deficient mice. The absence of c-Rel interfered with the expression of IL-12/23 subunits both in cultured primary macrophages and within the colon. Thus, c-Rel plays a critical role in regulating the innate inflammatory response to microflora within the lower bowel, likely through its ability to modulate expression of IL-12/23 family members.
Keywords: Rodent, Transcription Factors, Inflammation, Macrophages, Mucosa
Introduction
Crohn's disease is characterized by a Th1-like inflammatory response associated with elevated expression of IFN-γ and IL-12 p40 (1, 2). Consistent with this, treatment of patients with α-IL-12 p40 antibody has been reported to reduce disease severity (3). Many mouse models of colitis also exhibit an apparent Th1 like bias (4, 5), and α-IL-12 p40 antibody has been shown to reduce colitis observed in IL-10-deficient mice, and in lymphocyte-deficient mice that received CD4+CD45RBhigh T cells (6, 7). Interestingly, elevated expression of IL-12 p40 can also be observed in lymphocyte-deficient mice that develop chronic lower bowel inflammation in response to H. hepaticus (Hh3) infection (8, 9), and this innate inflammatory response can likewise be inhibited by antibody directed at p40. Several recent publications demonstrate that while mice lacking p40 are resistant to both T cell-mediated and innate lower bowel inflammation in response to Hh, p35 appears to be dispensable. Rather, the absence of p19 significantly reduces the development of inflammation (10-13). This strongly suggests that IL-23 rather than IL-12 p70 is essential for the development of colitis. While IL-12 p70 induces expression of IFN-γ from differentiated T cells, it has recently been demonstrated that IL-23 strongly enhances expression of IL-17 from previously primed T cells (14). Expression of IL-17 has been reported to closely correlate with the development of colitis in both T cell-mediated and innate models of colitis (10-13), although in several studies the absence of p40 and p19 interfered with the expression of the effector cytokine IFN-γ, as well as IL-17 (12, 13). These results strongly suggest that regulation of the IL-12/IL-23 family of cytokines within the innate immune system has a central role in mediating the development of colitis.
It has been reported that members of the NF-κB family of transcription factors have critical roles in the regulation of IL-12 and IL-23 expression (15-19). The NF-κB family consists of 5 members; p65, c-Rel, RelB, p50, and p52 (20). These members form homodimers and heterodimers with other family members, and influence transcription by interacting with κB sites present in the promoter/enhancer region of many pro-inflammatory genes. NF-κB activity is induced in response to a wide range of inflammatory stimuli that induce degradation of the IκB inhibitor family, which allows nuclear translocation of NF-κB subunits. IκB degradation and nuclear accumulation of NF-κB subunits is a central feature of human inflammatory bowel disease as well as mouse models of colitis (2, 21, 22), and treatment of mice with general inhibitors of NF-κB function can ameliorate disease severity (22, 23). Despite the near universal association of lower bowel inflammation and activation of NF-κB, the role of individual subunits in the development of colitis has been somewhat surprising. Mice lacking the p50 subunit of NF-κB are susceptible to colitis induced by H. hepaticus and this is exacerbated by heterozygosity at the p65 locus (24). Colitis in mice lacking p50 is caused at least in part by a defect within the hematopoietic compartment of the innate immune system that results in a defect in responding to the inhibitory effects of IL-10 (9, 25). Consistent with this, suppression of LPS-induced IL-12 p40 expression by IL-10 is significantly less efficient in macrophages lacking p50 (25). In contrast to p50, it has been reported that the NF-κB subunit c-Rel is essential for expression of p40 in macrophages (15), as well as p35 and p19 in dendritic cells (18, 19). There is a defect in the development of encephalitic Th1 effector cells in c-Rel-deficient mice immunized with MOG in a mouse EAE model (26), and c-Rel-deficient APCs are unable to induce the differentiation of Th1 effector cells after stimulation of WT CD4+ T cells in an in vitro culture system (27). However, whether c-Rel plays a physiologic role in regulating IL-12/IL-23 family members within the bowel in response to inflammatory challenge, and whether c-Rel might be essential for development of colitis has to our knowledge not been determined.
To address these issues, we have used well-described models of innate and T cell-mediated colitis to examine the role of c-Rel in the development of lower bowel inflammation. We have found that c-Rel is required within the innate immune system for the development of H. hepaticus-induced inflammation in RAG-deficient mice, and further that the ability of CD4+CD45RBhigh T cells to induce colitis is markedly compromised after transfer into RAG-deficient hosts that also lack c-Rel.
Materials and Methods
Experimental Animals
All mice were housed in facilities approved by the Association for the Assessment and Accreditation of Laboratory Animal Care. All experiments were approved by the Massachusetts Institute of Technology Committee on Animal Care and the Harvard Medical Area Standing Committee on Animals. c-Rel-/-;IL-10-/- (c-Rel/IL-10) mice, c-Rel-/-;RAG-2-/- (c-Rel/RAG) mice, p50-/-;c-Rel-/-;RAG-2-/- (p50/c-Rel/ RAG) mice, IL-10-/-,RAG-2-/- (IL-10/RAG) mice, and IL-10-/-,c-Rel-/-,RAG-2-/- (IL-10/c-Rel/RAG) mice were generated by crossing the c-Rel deficient allele (28), p50-deficient allele (29), or IL-10-deficient allele (30) onto either the 129S6/SvEvTac or 129S6/SvEvTac-RAG2tm1 background for 6-10 generations and then intercrossing to generate compound strains. Mice were maintained under conditions free of known Helicobacter species prior to targeted infection.
In vitro stimulation of bone marrow-derived macrophages (BMDM)
BMDM were grown as previously described (9) and re-plated in 6 well dishes at concentrations of 3×106/well. BMDM were challenged with LPS from E. coli 0127:B8 (Sigma, St Louis, MO) at the final concentration of 1 ng/mL.
ELISA
Elisa for IL-12 p40 (Caltag, Burlingame, CA) and IL-12 p70 (Endogen, Wuburn, MA) were performed per the manufactures instructions.
Analysis of gene expression
RNase protection analyses were performed using RiboQuant Multi Probe Template sets (BD Biosciences, San Diego, CA). Intensities of the protected fragments were quantified by phosphorimager analysis. Real-time PCR analysis was performed using probes from Applied Biosystems (Foster City, CA), as per the manufacturer's instructions.
Isolation of CD4+CD45RBhigh T cells
CD4+ cells were isolated from spleens and mesenteric lymph nodes (MLN) of WT 129SvEv mice as previously described (31). Purified CD4+ cells were stained with fluorescent antibodies to CD4, C45RB, and CD25 (BD Biosciences) and the 40% of CD4+ cells that were stained highest for CD45RB and negative for CD25 were purified by fluorescence-activated cell sorting.
Induction of colitis
RAG and c-Rel/RAG mice were infected with H. hepaticus bacteria by gastric gavage as previously described (24), received 4×105 CD4+CD45RBhigh T cells by retroorbital injection, or received T cells and then on the following day infected with Hh. Colitis score (0-8) was determined as previously described (25).
Production of protein homogenates
Ascending colon was snap frozen in liquid nitrogen and then homogenized in Tris-HCl 20mM, NaCl 125mM, EDTA-Na2 10mM and protease inhibitors (Roche Diagnostics, Manheim, Germany). Protein content was normalized using the Bradford assay (Sigma).
Intracellular flow cytometry
Single cell suspensions of MLN and spleen were stimulated with PMA 50 ng/ml (Sigma) and Ionomycin 500 ng/ml (Sigma) for 4 hours. Brefeldin A (BD Biosciences) was added for the last 2 hours of incubation. Intracellular staining was performed using the Cytofix/Cytoperm Kit (BD Biosciences) and α-IFN-γ and α-IL-17 (BD Bioscience) per the manufacture's instructions. The percentage of cells within the live cell gate that expressed either CD4 and IFN-γ, or CD4 and IL-17 was determined by flow cytometry.
Statistical evaluation
All data analysis was performed using GraphPad Prism software (GraphPad Software, inc., San Diego, CA). Mann-Whitney t test for non-parametric data were used to compare colitis scores. t-test for parametric data was used to compare inflammatory gene expression, cytokine levels, and flow cytometry data. Welch's correction was applied when the F test indicated that the variances between groups were significantly different. Differences between groups were considered statistically significant when P < 0.05.
Results
Defective induction of IL-12/IL-23 family members in the absence of c-Rel is independent of dysregulated expression of IL-10
Previous results have demonstrated that the ability of LPS and IFN-γ to induce IL-12 p40 in bone marrow or fetal liver-derived macrophages is severely compromised in the absence of c-Rel (15). However, published data and data from our lab (data not shown) suggest that LPS induces higher levels of IL-10 in c-Rel-deficient macrophages than in WT macrophages (15). Because IL-10 is a potent inhibitor of IL-12 p40, we wondered whether increased expression of IL-10 could be responsible for the lower levels of IL-12 p40 expression observed in c-Rel-deficient macrophages. To definitively address this issue, we crossed c-Rel-deficient mice onto the IL-10-deficient background (c-Rel/IL-10). Bone marrow derived macrophages (BMDM) were harvested from these c-Rel/IL-10 mice as well as IL-10-deficient control mice and stimulated with LPS. 12 hours after LPS-challenge, c-Rel/IL-10 BMDM exhibited a marked defect in the accumulation of IL-12 p40 and IL-12 p70 within the culture supernatants compared to IL-10-deficient BMDM (Fig. 1A). In addition, while LPS induced similar or higher levels of mRNA for the LPS-induced genes IL-6, TNF, and IP-10 in c-Rel/IL-10 BMDM than in IL-10-deficient BMDM (Fig. 1B), expression of IL-12 p40 was severely compromised (Fig. 1C). Interestingly, there was also marked reduction of LPS-induced IL-12 p35 and IL-23 p19 expression in c-Rel/IL-10 BMDM compare to IL-10-deficient BMDM (Fig. 1C). These data confirm that c-Rel has a focused and critical role in regulating the induction of IL-12/IL-23 family members, and that reduced expression observed in the absence of c-Rel is independent of increased secretion of IL-10.
Figure 1.

A) Levels of IL-12 p40 and p70 in the culture medium of IL-10-deficient (filled bars) and c-Rel/IL-10 (open bars) BMDM 12h after stimulation with LPS (1ng/ml), as determined by ELISA. B) Relative mRNA expression compared to GAPDH of IL-6, TNF, IP-10, as determined by RT-PCR in BMDM derived from IL-10-deficient (filled bars) and c-Rel/IL-10 (open bars) mice either prior to (0h) or 4 hours after (4h) stimulation with LPS. C) Relative mRNA expression of IL-12 p40, IL-12 p35, and IL-12 p19 in groups described in B. Macrophages from 4 independent mice were included in each group, and experiments were repeated twice. Bars represent mean value per group with SEM as shown. *, P<0.05 comparing IL-10-deficient to c-Rel/IL-10 BMDM
The absence of c-Rel interferes with H. hepaticus induced innate colitis
Infection of RAG-deficient (RAG) mice with H. hepaticus has become an important model for studying the contribution of the innate immune system to the development of chronic lower bowel inflammation (8). Therefore, to probe the role of c-Rel in regulating the innate inflammatory response within the lower bowel, we crossed c-Rel-deficient mice onto the RAG-2-deficient background (c-Rel/RAG). RAG and c-Rel/RAG mice were left uninfected or infected with Hh, and the development of bowel inflammation assessed 6 weeks later. To quantify differences between genotypes, histological sections from the colons of individual animals were evaluated by a veterinary pathologist, and severity of colitis scored on a previously validated ascending scale from 0-8. As expected, there was little inflammation in any of the uninfected groups (data not shown), while Hh infection induced disease within the colon of RAG mice (Fig. 2A). Remarkably, inflammation was markedly less severe within the colons of Hh infected c-Rel/RAG mice compared to the infected RAG mice (Fig. 2A), indicating the absence of c-Rel interferes with the ability of Hh to induce innate inflammation within the colon.
Figure 2.

A) Colitis scores in the colon of RAG, c-Rel/RAG, p50/RAG, p50/c-Rel/RAG, IL-10/RAG, and IL-10/c-Rel/RAG mice were determined 6 weeks after Hh-infection. Each symbol represents the score of an individual animal. Horizontal lines represent the median colitis score within each group. One of two experiments with similar results. **, P<0.01; ***, P<0.001. B) Expression of IL-12 p40 and IL-23 p19 within the colons of RAG, c-Rel/RAG, p50/RAG, p50/c-Rel/RAG, IL-10/RAG, and IL-10/c-Rel/RAG mice prior to or after infection with Hh. 4-10 mice per group. Relative expression was determined by quantitative RT-PCR. Bars represent mean value per group with SEM as shown. **, P<0.01; *, P<0.05. C) The concentration of p40 within colon homogenate of RAG and c-Rel/RAG mice prior to (uninfected) or 6 weeks after infection with Hh (Hh) was measured by ELISA. 8-10 animals were included in each group. Bars represent mean with SEM as shown. *, P<0.05.
The absence of c-Rel interferes with the development of Hh-induced colitis in p50/RAG mice
We have previously shown that there is a defect in the ability of IL-10 to inhibit Hh-induced innate colitis in RAG mice that lack the p50 subunit of NF-κB (25). We have proposed that this defect may be the result of excessive expression of IL-12 p40 in the absence of p50. To determine whether colitis that is observed in p50/RAG mice also depends upon c-Rel, we compared the ability of Hh to induce colitis in RAG mice that lack p50 (p50/RAG) and those that lack p50 and c-Rel (p50/c-Rel/RAG). Notably, colitis scores were significantly lower within the colons of Hh-infected p50/c-Rel/RAG mice than within the colons of p50/RAG mice (Fig. 2A). This suggests that c-Rel plays an important role in the induction of Hh-induced inflammation in both RAG and p50/RAG mice.
Reduced innate colitis in the absence of c-Rel is not the result of increased expression of IL-10
We had hypothesized that decreased expression of IL-12 p40 in c-Rel-deficient BMDM might be the results of elevated expression of IL-10 that had previously been observed. As shown above, we found that c-Rel is necessary for LPS-induced expression of IL-12/23 subunits even in the absence of endogenous IL-10, suggesting that increased expression of IL-10 observed in c-Rel deficient BMDM is not solely responsible for the defect in the induction of IL-12 p40. However, as we, and others, have shown that IL-10 can inhibit Hh-induced colitis in RAG mice (25, 32), we wondered whether altered expression of IL-10 after Hh-challenge of c-Rel/RAG mice might play a role in the reduced severity of disease observed in these animals. To address this issue, we compared the severity of Hh-induced innate colitis in RAG mice that lack IL-10 alone (IL-10/RAG) and those that lack both c-Rel and IL-10 (IL-10/c-Rel/RAG). Colitis scores were significantly lower in IL-10/c-Rel/RAG mice than in IL-10/RAG mice (Fig. 2A), indicating that altered expression of IL-10 is not responsible for the decreased severity of Hh-induced inflammation observed in the absence of c-Rel. Taken together with results from the previous paragraph, these observations indicate that absence of c-Rel has a powerful inhibitory effect on the development of innate colitis in several colitis-prone mouse models.
The absence of c-Rel interferes with Hh-induced expression of p40 and p19 within the colon
Previous studies have demonstrated that both IL-12 p40 and p19 are essential for Hh-induced innate colitis (13). To determine whether the presence of c-Rel is required within the innate immune system for the induction of these subunits in response to Hh infection, we examined expression of p40 and p19 mRNA within the colons of the strains of RAG mice described above. Hh infection induced expression of p40 within the colons of RAG, p50/RAG, and IL-10/RAG mice, while induction was significantly compromised in Hh-infected c-Rel/RAG mice, p50/c-Rel/RAG mice, and IL-10/c-Rel/RAG mice (Fig. 2B). Interestingly, while Hh infection induced only a modest increase of p19 expression within the colon of c-Rel/RAG mice, Hh induced considerable higher expression of p19 in both p50/RAG mice and IL10/RAG mice (Fig. 2B). Remarkably, the absence of c-Rel significantly reduced Hh-induced expression of p19 in both of these strains (Fig. 2B). In concordance with mRNA levels for p40, we found that Hh-induced the production of p40 protein within the colon of Hh-infected RAG mice, as judged by ELISA of colon homogenates, and this was abrogated in Hh-infected c-Rel/RAG mice (Fig. 2C). Thus, the absence of c-Rel interferes with the ability of Hh to induce both subunits of IL-23, even in the absence of p50 or IL-10.
The absence of c-Rel interferes with Hh-induced expression of pro-inflammatory cytokines
Previous work has demonstrated that depletion of IL-23 with α-p19 antibodies inhibits the ability of Hh to induce expression of inflammatory cytokines including IL-17 within the colon of RAG mice (13). Consistent with a defect in the ability to express IL-23 in the absence of c-Rel, there was significantly lower expression of the mRNA for inflammatory cytokines associated with the development of colitis, including TNF, IP-10, IL-6, and IL-17 within the colons of Hh-infected c-Rel/RAG mice than within the colon of infected RAG mice (Fig. 3). As we did not detect a defect in the ability of LPS to induce several of these cytokines in macrophages lacking c-Rel (see Fig. 1), we suggest that lower expression within the colon of Hh-infected c-Rel/RAG mice is likely secondary to reduced expression of IL-12/23 subunits and overall reduced levels of inflammation observed in these mice, rather than a primary defect in the ability of Hh infection to induce these cytokines. Taken together, we believe that these results strongly support the hypothesis that c-Rel plays a key role in the ability of Hh-infection to induce expression of IL-12/IL-23 subunits and the development of innate colitis.
Figure 3.

Relative expression of indicated cytokine mRNA within the colons of RAG (filled bars) and c-Rel/RAG (open bars) mice left uninfected (uninfected) or six weeks after infection with Hh (Hh). Expression of TNF, IP-10, and IL-6 determined by RPA. Expression of IL-17 determined by real-time quantitative PCR. Each group included eight to ten animals. Bars represent mean with SEM as shown. *, P<0.05; ***, P<0.001.
c-Rel-independent pathways within the cecum
In addition to examining inflammation in the colon, we also evaluated inflammation within the cecum after Hh infection. While median colitis scores were significantly lower within the cecum of both c-Rel/RAG and p50/c-Rel/RAG mice compared to RAG and p50/RAG mice respectively, residual inflammation was clearly present (Fig. 4A). This is in contrast to the colon where there was little residual inflammation in the absence of c-Rel (see Figure 2A). Further, while expression of p40 was reduced within the cecum of Hh-infected c-Rel/RAG mice compared to levels observed within the cecum of infected RAG mice, the level of p40 observed in the cecum of Hh-infected c-Rel/RAG mice was considerably higher than levels observed within the colon (Fig. 4B). These results indicate that while c-Rel plays an essential role in the ability of Hh to induce innate inflammation within the colon, the absence of c-Rel only partially interferes with the ability of Hh to induce inflammation within the cecum.
Figure 4.

A) Colitis scores within the cecum of mice described in Figure 2. Symbols represent individual animals. Horizontal lines represent the median colitis score within each group. **, P<0.01. B) Expression of IL-12 p40 within the cecum and colons of RAG and c-Rel/RAG mice prior to (Hh-) or 6 weeks after Hh infection (Hh+) was determined by RT-PCR. 4-10 animals per group. Bars represent mean with SEM as shown. **, P<0.01; ***, p<0.001.
The ability of CD4+CD45RBhigh T cells to induce colitis is compromised in c-Rel/RAG mice
In addition to the innate colitis induced by infection with Hh, the ability of adoptively transferred CD4+CD45RBhigh T cells to induce colitis in RAG mice has been studied in detail (4, 33). To determine if the absence of c-Rel within cells of the innate immune system interfered with the ability of T cells to induce colitis, RAG and c-Rel/RAG mice received 4×105 purified CD4+CD45RBhigh T cells. 6-8 weeks after transfer, the median colitis score within the colon was significantly lower in c-Rel/RAG mice than in RAG mice (Fig. 5A). While median colitis scores were also lower within the cecum of c-Rel/RAG mice than RAG mice after transfer of CD4+CD45RBhigh T cells (Fig. 5A), this difference did not reach statistical significance (P=0.13). These results demonstrate that c-Rel is required within cells of the innate immune system for the activation of T cell-dependent as well as innate mechanisms of mucosal inflammation.
Figure 5.

A) RAG or c-Rel/RAG mice as indicated were injected with 4×105 purified CD4+CD45RBhigh T cells (Teff) or injected with 4×105 purified CD4+CD45RBhigh T cells and then infected with Hh (Teff+Hh). Colitis scores in the colon and cecum were determined 6-8 weeks later. Circles represent individual animals. Horizontal lines represent the median colitis score within each group. *, P<0.05; ***, P<0.001. B) Relative expression of IL-12 p40, IL-23 p19, IL-12 p35, and IL-17a mRNA within the colons of RAG (filled bars) and c-Rel/RAG (open bars) mice left uninfected (uninfected) or six weeks after adoptive transfer of CD4+CD45RBhigh T cells and infection with Hh (Teff+Hh), as determined by RPA (p40, and p35) or RT-PCR (p19 and IL-17a). Bars represent mean with SEM as shown. **, P<0.01.
Previous results have suggested that the severity of colitis induced by adoptive transfer of CD4+CD45RBhigh T cells into Helicobacter-free lymphocyte-deficient mice is enhanced by concomitant infection with Hh (34). As all hosts in experiments described here were maintained in conditions free of Helicobacter species (prior to targeted infection), we wondered whether the observed defect in the ability of CD4+CD45RBhigh T cells to induce colitis after adoptive transfer into c-Rel/RAG mice could be overcome by Hh infection. To address this possibility, RAG and c-Rel/RAG mice received CD4+CD45RBhigh T cells and then were infected with Hh on the following day. When these groups were analyzed 6-8 weeks later, median colitis scores in both the colon and the cecum were significantly lower in c-Rel/RAG mice than in RAG mice (Fig. 5A). These results demonstrate that the absence of c-Rel within cells of the innate immune interferes with the ability of CD4+CD45RBhigh T cells to induce colitis even in the presence of Hh-infection.
Induction of IL-12/23 subunits within the bowel following adoptive transfer of CD4+CD45RBhigh T cells is compromised in c-Rel/RAG mice
As in the innate model of colitis, a role for IL-23 in T cell-mediated colitis has recently been documented (10, 12, 13). Mice that both received CD4+CD45RBhigh T cells and were then infected with Hh exhibited higher expression of p40, p19, and p35 with the colon when compared to control mice (Fig. 5B). The mean expression of all three of these subunits was lower within the colon of c-Rel/RAG mice that received CD4+CD45RBhigh T cells and were then infected with Hh than similarly treated RAG mice, although this decrease only reached statistical significance for p40 and p35 (Fig. 5B). Consistent with lower levels of IL-12/23 subunit expression, we also observed lower levels IL-17a mRNA within the colons of c-Rel/RAG mice that received CD4+CD45RBhigh T cells (Fig. 5B).
Altered T cell differentiation after transfer of CD4+CD45RBhigh T cells into c-Rel/RAG mice
It has been shown that the absence of IL-23 leads to altered differentiation of T cells within the mesenteric lymph node (MLN) of RAG mice that received CD4+CD45RBhigh T cells (13). In this previous study, the absence of either p40 or p19 appears to lead to decreased percentages CD4+ T cells capable of expressing IFN-γ and increased percentages of T cells capable of expressing IL-17, although it is important to note that this did not lead to increased expression of IL-17 message within the bowel. Therefore, we wondered whether the absence of c-Rel might also impact differentiation of Th17 cells. To address this, we compared the differentiation of CD4+CD45RBhigh T cells after adoptive transfer into RAG and c-Rel/RAG mice. MLN and spleen cells were isolated from RAG and c-Rel/RAG mice 6-8 weeks after the adoptive transfer of CD4+CD45RBhigh T cells and infection with Hh. These cells were then stimulated with PMA and ionomycin and the percentage of CD4+ T cells that expressed IFN-γ and IL-17 was determined by intracellular flow cytometry. We observed that the ratio of cells expressing IL-17 to cells expressing IFN-γ within each individual animal was significantly higher in c-Rel/RAG hosts compared to RAG hosts (Fig. 6, A and B). We observed parallel results in RAG and c-Rel/RAG mice that received CD4+CD45RBhigh T cells in the absence of Hh infection (data not shown). This suggests that c-Rel limits the differentiation of Th17 cells, although as in previous results with p40/RAG and p19/RAG mice, this was not associated with increased expression of IL-17a mRNA within the bowel itself (See Figure 5B).
Figure 6.
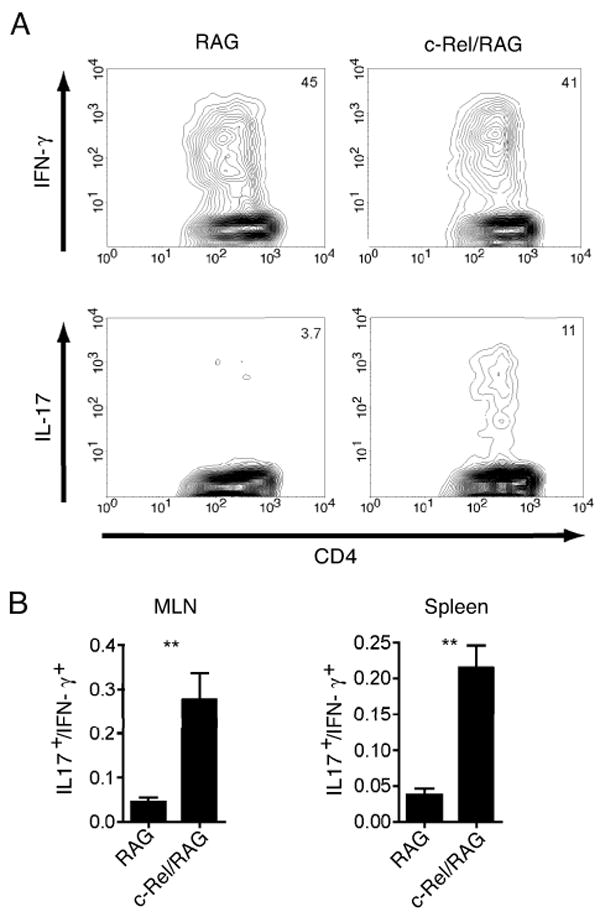
(A) Percentages of IFN-γ and IL-17 expressing CD4+ T cells within the mesenteric lymph node of representative RAG and c-Rel/RAG mice that received CD4+CD45RBhigh T cells and were infected with Hh 6 weeks earlier. (B) Ratio of CD4+ T cells expressing IL-17 to those expressing IFN-γ within the MLN and spleen of mice that received CD4+CD45RBhigh T cells and were infected with Hh. Each group represents 15-17 mice. Bars represent mean with SEM as shown. **, P<0.01.
Discussion
c-Rel is a widely expressed member of the NF-κB family. However, there are few genes that appear to absolutely require c-Rel (35). Interestingly, subunits of the IL-12 family appear to represent clear exceptions to this general observation, as LPS has virtually no ability to induce p40, p35, or p19 within bone marrow-derived macrophages in the absence of c-Rel. This observation extends previous results evaluating IL-12/23 subunit expression in c-Rel-deficient bone marrow-derived macrophages (15). It has been suggested that expression of p35 but not p40 requires c-Rel in CD8+ splenic dendritic cells (18), and c-Rel is required for LPS-mediated induction of p19 in myeloid dendritic cells as well (19). We have now shown that the failure of LPS to induce IL-12 subunits in c-Rel-deficient macrophages is not due to elevated IL-10 expression, as there is defective expression of these IL-12 subunits in macrophages that lack both c-Rel and IL-10.
It has previously been shown that Hh-infection of RAG mice induces robust p40 expression within the colon (9, 13) and that p40 is necessary for the development of Hh-induced innate colitis in these mice (8). Consistent with a critical role for c-Rel in the regulation of p40 within the colon, the severity of Hh-induced colitis was markedly compromised in c-Rel/RAG mice, and this was associated with defective induction of IL-12 p40. It has also been demonstrated that Hh-infection can induce expression of p19 mRNA within the colon, although induction was considerably less robust than for p40 (13). Consistent with this previous study, we found that Hh infection induced only modest increases in p19 mRNA expression within the colon of RAG mice. However, we found that induction of p19 mRNA was considerably more robust within the colons of both p50/RAG and IL-10/RAG mice following Hh infection, and in both cases this induction was abrogated in the absence of c-Rel. Thus, Hh clearly has the ability to induce both p40 and p19 mRNA expression, and this requires the presence of c-Rel. It therefore seems plausible that that the defect in the ability of Hh to induce innate colitis in the absence of c-Rel is due to a defect in expression of IL-23 subunits.
While the absence of c-Rel markedly interfered with the development of colonic inflammation after Hh-challenge, inflammation was less severely affected within the cecum. Consistent with the levels of inflammation, we found that in the absence of c-Rel, Hh retains partial ability to induce p40 within the cecum but not the colon. Interestingly, is has previously been documented that Hh colonizes the cecum at higher levels than the colon (36). Therefore, we hypothesize that the most plausible explanation for this observation is that at higher colonization levels Hh is able to mobilize p40 through c-Rel independent pathways. We speculate that these c-Rel independent pathways may be the result of activation of additional APC populations at higher Hh colonization levels observed in the cecum, consistent with previous observations that the ability of some APC populations to express IL-12 p40 is not inhibited by the absence of c-Rel. We believe that this explanation for residual inflammation within the cecum of c-Rel-deficient mice is more likely than the possibility that Hh-induces IL-23 independent pathways within the cecum, as it has been demonstrated that the depletion of p19 effects inflammation within the cecum as well as the colon (12, 13).
We were somewhat surprised that despite reductions in the expression of IL-12 p40 in c-Rel/RAG mice that received CD4+CD45RBhigh T cells, we observed elevated percentages of IL-17 positive cells after restimulation of splenic and mesenteric lymph node cells isolated from c-Rel/RAG mice. However, this alteration in the differentiation of adoptively transferred T cells is highly similar to alterations previously reported in RAG-deficient mice that also lack either p19 or p40 (13). Although this initially may seem incongruous with a role for IL-23 in IL-17 cell function, recent studies suggest that differentiation of Th17 cells is independent of IL-23 (37). Further it has been shown that continued stimulation of differentiated Th17 cells with IL-6 and TGF-β in the absence of IL-23 leads to the development of cell populations that express high amounts of IL-10 and exhibit regulatory properties (37). While we have not yet evaluated expression of IL-10 in T cell populations that differentiate in c-Rel/RAG mice, it seems likely that the Th17 cells differentiating in this environment have reduced pathogenic potential, as we have observed lower levels of IL-17 mRNA within the colon of c-Rel/RAG mice that received CD4+CD45RBhigh T cells. This is consistent with our hypothesis that c-Rel-dependent IL-23 expression is necessary for T cell-mediated pathology within the colon. Evaluating the pathogenicity and regulatory properties of T cells that differentiate in c-Rel/RAG mice is an important future goal in our laboratory.
Our results demonstrate that the presence of c-Rel within the innate immune system is essential for the development of both innate and T cell-dependent colitis, likely based on its role in the regulation of IL-12/IL-23 family members. Interestingly, as the absence of c-Rel profoundly influences IL-23 mediated inflammatory responses but appears to have very limited effects on the expression of other genes induced by innate inflammatory stimuli, inhibition of c-Rel may could prove a highly attractive and specific approach for the treatment of inflammatory bowel disease.
Footnotes
This work was supported by NIH AI52267 (BHH and SEE), Crohn's and Colitis Foundation of America and the William and Shelby Modell Family Foundation Senior Research Grant (BHH), NIH CA108854 (SEE), and NIH CA67529 (JGF).
Abbreviations Used: BMDM, bone marrow-derived macrophages; Hh, Helicobacter hepaticus; RPA, RNase protection analyses; MLN, mesenteric lymph node.
Literature Cited
- 1.Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol. 2003;3:521–533. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- 2.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 3.Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, Dolin B, Goodman N, Groden C, Hornung RL, Quezado M, Yang Z, Neurath MF, Salfeld J, Veldman GM, Schwertschlag U, Strober W. Anti-interleukin-12 antibody for active Crohn's disease. N Engl J Med. 2004;351:2069–2079. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- 4.Powrie F, Leach MW, Mauze S, Menon S, Caddle LB, Coffman RL. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1:553–562. doi: 10.1016/1074-7613(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 5.Strober W, Fuss IJ, Ehrhardt RO, Neurath M, Boirivant M, Ludviksson BR. Mucosal immunoregulation and inflammatory bowel disease: new insights from murine models of inflammation. Scand J Immunol. 1998;48:453–458. [PubMed] [Google Scholar]
- 6.Simpson SJ, Shah S, Comiskey M, de Jong YP, Wang B, Mizoguchi E, Bhan AK, Terhorst C. T cell-mediated pathology in two models of experimental colitis depends predominantly on the interleukin 12/Signal transducer and activator of transcription (Stat)-4 pathway, but is not conditional on interferon gamma expression by T cells. J Exp Med. 1998;187:1225–1234. doi: 10.1084/jem.187.8.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kullberg MC, Ward JM, Gorelick PL, Caspar P, Hieny S, Cheever A, Jankovic D, Sher A. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect Immun. 1998;66:5157–5166. doi: 10.1128/iai.66.11.5157-5166.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maloy KJ, Salaun L, Cahill R, Dougan G, Saunders NJ, Powrie F. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J Exp Med. 2003;197:111–119. doi: 10.1084/jem.20021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomczak MF, Erdman SE, Poutahidis T, Rogers AB, Holcombe H, Plank B, Fox JG, Horwitz BH. NF-kappa B is required within the innate immune system to inhibit microflora-induced colitis and expression of IL-12 p40. J Immunol. 2003;171:1484–1492. doi: 10.4049/jimmunol.171.3.1484. [DOI] [PubMed] [Google Scholar]
- 10.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, Sathe M, Cua DJ, Kastelein RA, Rennick D. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, Robinson N, Buonocore S, Tlaskalova-Hogenova H, Cua DJ, Powrie F. Differential Activity of IL-12 and IL-23 in Mucosal and Systemic Innate Immune Pathology. Immunity. 2006;25:309–318. doi: 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 12.Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, Cua DJ, Powrie F, Cheever AW, Maloy KJ, Sher A. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med. 2006;203:2485–2494. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, Maloy KJ. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–2483. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 15.Sanjabi S, Hoffmann A, Liou HC, Baltimore D, Smale ST. Selective requirement for c-Rel during IL-12 P40 gene induction in macrophages. Proc Natl Acad Sci U S A. 2000;97:12705–12710. doi: 10.1073/pnas.230436397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murphy TL, Cleveland MG, Kulesza P, Magram J, Murphy KM. Regulation of interleukin 12 p40 expression through an NF-kappa B half-site. Mol Cell Biol. 1995;15:5258–5267. doi: 10.1128/mcb.15.10.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mason N, Aliberti J, Caamano JC, Liou HC, Hunter CA. Cutting edge: identification of c-Rel-dependent and -independent pathways of IL-12 production during infectious and inflammatory stimuli. J Immunol. 2002;168:2590–2594. doi: 10.4049/jimmunol.168.6.2590. [DOI] [PubMed] [Google Scholar]
- 18.Grumont R, Hochrein H, O'Keeffe M, Gugasyan R, White C, Caminschi I, Cook W, Gerondakis S. c-Rel regulates interleukin 12 p70 expression in CD8(+) dendritic cells by specifically inducing p35 gene transcription. J Exp Med. 2001;194:1021–1032. doi: 10.1084/jem.194.8.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carmody RJ, Ruan Q, Liou HC, Chen YH. Essential roles of c-Rel in TLR-induced IL-23 p19 gene expression in dendritic cells. J Immunol. 2007;178:186–191. doi: 10.4049/jimmunol.178.1.186. [DOI] [PubMed] [Google Scholar]
- 20.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 21.Schreiber S, Nikolaus S, Hampe J. Activation of nuclear factor kappa B in inflammatory bowel disease. Gut. 1998;42:477–484. doi: 10.1136/gut.42.4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neurath MF, Pettersson S, Meyer zum Buschenfelde KH, Strober W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in mice. Nat Med. 1996;2:998–1004. doi: 10.1038/nm0996-998. [DOI] [PubMed] [Google Scholar]
- 23.Fichtner-Feigl S, Fuss IJ, Preiss JC, Strober W, Kitani A. Treatment of murine Th1- and Th2-mediated inflammatory bowel disease with NF-kappa B decoy oligonucleotides. J Clin Invest. 2005;115:3057–3071. doi: 10.1172/JCI24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erdman S, Fox JG, Dangler CA, Feldman D, Horwitz BH. Typhlocolitis in NF-kappa B-deficient mice. J Immunol. 2001;166:1443–1447. doi: 10.4049/jimmunol.166.3.1443. [DOI] [PubMed] [Google Scholar]
- 25.Tomczak MF, Erdman SE, Davidson A, Wang YY, Nambiar PR, Rogers AB, Rickman B, Luchetti D, Fox JG, Horwitz BH. Inhibition of Helicobacter hepaticus-Induced Colitis by IL-10 Requires the p50/p105 Subunit of NF-{kappa}B. J Immunol. 2006;177:7332–7339. doi: 10.4049/jimmunol.177.10.7332. [DOI] [PubMed] [Google Scholar]
- 26.Hilliard BA, Mason N, Xu L, Sun J, Lamhamedi-Cherradi SE, Liou HC, Hunter C, Chen YH. Critical roles of c-Rel in autoimmune inflammation and helper T cell differentiation. J Clin Invest. 2002;110:843–850. doi: 10.1172/JCI15254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boffa DJ, Feng B, Sharma V, Dematteo R, Miller G, Suthanthiran M, Nunez R, Liou HC. Selective loss of c-Rel compromises dendritic cell activation of T lymphocytes. Cell Immunol. 2003;222:105–115. doi: 10.1016/s0008-8749(03)00114-x. [DOI] [PubMed] [Google Scholar]
- 28.Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, Gerondakis S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- 29.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 30.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 31.Erdman SE, Poutahidis T, Tomczak M, Rogers AB, Cormier K, Plank B, Horwitz BH, Fox JG. CD4+ CD25+ regulatory T lymphocytes inhibit microbially induced colon cancer in Rag2-deficient mice. Am J Pathol. 2003;162:691–702. doi: 10.1016/S0002-9440(10)63863-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.von Freeden-Jeffry U, Davidson N, Wiler R, Fort M, Burdach S, Murray R. IL-7 deficiency prevents development of a non-T cell non-B cell-mediated colitis. J Immunol. 1998;161:5673–5680. [PubMed] [Google Scholar]
- 33.Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int Immunol. 1993;5:1461–1471. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- 34.Cahill RJ, Foltz CJ, Fox JG, Dangler CA, Powrie F, Schauer DB. Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect Immun. 1997;65:3126–3131. doi: 10.1128/iai.65.8.3126-3131.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bunting K, Rao S, Hardy K, Woltring D, Denyer GS, Wang J, Gerondakis S, Shannon MF. Genome-wide analysis of gene expression in T cells to identify targets of the NF-kappa B transcription factor c-Rel. J Immunol. 2007;178:7097–7109. doi: 10.4049/jimmunol.178.11.7097. [DOI] [PubMed] [Google Scholar]
- 36.Ge Z, Feng Y, Whary MT, Nambiar PR, Xu S, Ng V, Taylor NS, Fox JG. Cytolethal distending toxin is essential for Helicobacter hepaticus colonization in outbred Swiss Webster mice. Infect Immun. 2005;73:3559–3567. doi: 10.1128/IAI.73.6.3559-3567.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]