

Abstract
In Fas/FasL deficient mice anti-chromatin Ab production is T cell dependent and is not apparent until after 10 weeks of age. Early control of anti-chromatin antibodies may be due to the counterbalancing influence of Treg cells. Here we show that Treg cells block lpr/lpr gld/gld Th cells from providing help to anti-chromatin B cells in an in vivo transfer system. Interestingly, the percentage and absolute numbers of Foxp3+ Treg cells is elevated in BALB/c-lpr/lpr gld/gld mice and increases with age compared to BALB/c mice. The majority of Foxp3 expression is found in the B220− CD4+ T cell population, and Foxp3-expressing cells are localized in the splenic PALS (periarteriolar lymphocyte sheath). Strikingly, although the lack of functional Fas/FasL does not affect the ability of Treg cells to block Th cell proliferation, Treg cells can block the IFN-γ differentiation of Th cells from BALB/c or young BALB-lpr/lpr gld/gld mice but not of pre-existing Th1 cells from older BALB/c-lpr/lpr gld/gld mice. Thus, we suggest autoantibody production is not caused by the lack of Treg cells but by a defect in activation-induced cell death that leads to the accumulation of T effector cells that are resistant to regulatory T cell activity.
Keywords: autoantibody, Fas/FasL, IFN-γ, T regulatory
Introduction
VH3H9/λ1 anti-chromatin B cells are present in the periphery of non-autoimmune BALB/c mice and young Fas (lpr/lpr) or FasL (gld/gld) deficient mice but in both cases autoAbs are not secreted; in the Fas/FasL deficient mice autoAbs are not detected until after 10 weeks of age [1–3]. Although anti-chromatin B cells appear to be held in check in both scenarios, the localization and cell surface phenotype of anti-chromatin B cells in non-autoimmune and autoimmune mice are distinct [1–3], suggesting different mechanisms of tolerance are in play. Anti-chromatin B cells in BALB/c mice are primarily excluded from B cell follicles but migrate into the B cell follicle upon receipt of T cell help [1]. In contrast, in autoimmune lpr/lpr or gld/gld mice these cells are found in the B cell follicle in a CD4 dependent manner [2–4]. One possibility to account for this data is that B cells from young Fas/FasL-deficient mice are partially defective in their response to T cell help, responding by migrating into the follicles but not differentiating into AFCs. Alternatively, CD4 T cell help may be partially lacking in young lpr/lpr gld/gld mice, leading to the abortive phenotype of the anti-chromatin B cells. We have hypothesized that Treg cells maintain B cell tolerance and consistent with this have demonstrated that Treg cells inhibit autoreactive B cell response to Th cells in an in vivo transfer model [5].
The status of Treg cells in lupus is controversial. Lupus patients appear to have a reduced frequency of Treg cells [6, 7]. Likewise, in some murine models of SLE Treg cells are diminished and/or have reduced Foxp3 protein expression [8–10]. In contrast, 16–20 week old C3H gld/gld mice have an increased frequency of Foxp3+ T cells with Foxp3 protein expressed at levels similar to normal mice [11]. The functional capacity of Treg cells to block T cell proliferation in vitro also varies according to strain. In the C3H gld/gld model, Treg cells inhibited Th cell proliferation while in NZM2410 mice the Treg cells were less effective [10, 11]. The ability of Treg cells to block lupus in vivo has also depended upon the symptoms examined. We have demonstrated that Treg cells can block the production of anti-chromatin Abs induced by the provision of T cell help [4, 5]. Likewise, in the NZM2328 or SNF1 lupus models, the presence of Treg cells decreases anti-dsDNA Abs; however, glomerulonephritis is not improved [12, 13].
Here, we show that Fas/FasL-deficient anti-chromatin B cells readily respond to T helper cell infusion in vivo by differentiating into AFCs. Thus, the delay in autoantibody production until 10–12 weeks of age is not due to an intrinsic defect in the anti-chromatin B cells in young lpr/lpr gld/gld mice. To account for this delay, we have analyzed the phenotype and functional abilities of Foxp3+ Treg cells before (< 8 week old) and after (> 12 week old) autoAbs are detected in lpr/lpr gld/gld mice. Immunofluorescence analyses reveal that the majority of Foxp3+ cells are located in the PALS of both BALB/c, young (serum VH3H9/λ1 autoAb−) lpr/lpr gld/gld, and old lpr/lpr gld/gld (serum VH3H9/λ1 autoAb+) mice. However, as we have previously reported [2, 14], the splenic architecture (separation of B and T cell areas) becomes progressively disrupted in older lpr/lpr gld/gld mice.
We further tested the responsiveness of lpr/lpr gld/gld T cells to suppression by Treg cells in terms of proliferation and cytokine production. Here we report a deviance in the dampening of IFN-γ potential by Treg cells upon young versus old lpr/lpr gld/gld T cells. This phenomenon, along with other factors such as the disruption of splenic architecture, may account for the breakdown of tolerance in older lpr/lpr gld/gld mice.
Materials and Methods
Mice
Male and female mice between 6–24 weeks of age were maintained in specific-pathogen-free conditions at the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited Wistar Institute under the supervision of the Institutional Animal Care and Use Committee (IACUC). All mice are on the BALB/c background. TS1 TCR (anti-hemagglutinin) Tg, TS1xHA28 Tg, lpr/lpr gld/gld, VH3H9 HACII lpr/lpr gld/gld, and VH3H9/HACII/Ig κ−/− mice were bred at the Wistar Animal Facility. BALB/c and CB17 [a congenic strain carrying the Ig heavy-chain allele (Igh-1b) from a C57BL/Ka on a BALB/c background] mice were purchased from the National Cancer Institute and Charles River Laboratory, respectively.
T and B cell injections into CB17 mice
2 million anti-HA Th (CD4+ CD25−) cells were sorted from TS1 TCR Tg BALB/c or TS1 TCR Tg BALB/c-lpr/lpr gld/gld mice (the latter were <8 weeks old), and injected i.v. with 1000 hemagglutinating units of purified PR8 influenza virus. Splenocytes from VH3H9/HACII/Ig κ−/− mice were depleted of RBC, and an aliquot was stained by flow cytometry to determine the frequency of anti-chromatin B cells. CB17 recipient mice were injected i.v. with splenocytes containing 5 × 106 anti-chromatin B cells. Mice were killed eight days later.
Flow cytometry
1–4 × 106 cells were prepared and surface stained as per standard protocol [15]. The following Abs were used: anti-B220-FITC, -PerCP-Cy5.5, or -APCCy7 (RA3-6B2), anti-CD3-FITC or -PE-Cy7 (145-2C11), anti-CD25-biotin (7D4), anti-CD95-biotin (Jo2), anti-CXCR5-biotin (2G8), anti-CD4-PerCP-Cy5.5 (RM4-5), streptavidin-PE, -PerCP-Cy5.5, or -PerCP (BD Biosciences), anti-CD4-APC (RM4-5), anti-Thy1.2-APC (53-2.1), anti-Foxp3-FITC or -PE (FJK-16s), isotype control Rat IgG2a-FITC or -PE (EBR2a), anti-IFN-γ-PE (XMG1.2), and anti-IL-4-PE (11B11) (Ebioscience). Intracellular cytokine and Foxp3 staining were performed as previously described [5]. Stained cells were run on a FACScan, FACSCalibur, LSRII (Becton Dickinson), or Cyan ADP machine (Dako) and analyzed using FlowJo software (Treestar). All plots show log10 fluorescence.
Localization of CD4+, CD4+ B220+, and DN T cells
Lymph nodes from lpr/lpr gld/gld mice aged >12 weeks of age were stained with CD19-biotin (1D3) (BD Biosciences)/ streptavidin-PE, anti-CD4-APC and anti-B220-PerCP-Cy5.5. Cells were sorted into three groups, CD19− CD4+ B220− (CD4+), CD19− CD4+ B220+, and CD19− CD4− B220+ (DN), and were subsequently CFSE labeled and transferred i.v. into BALB/c mice. The following day mice were killed and spleens were taken for Immunofluorescence analysis.
Immunostaining
Spleens were frozen, sectioned, and stained [1]. Immunohistochemistry protocols used anti-CD22-FITC (Cy34.1) and anti-IgMa-biotin (DS-1) (BD Biosciences). Secondary reagents were anti-FITC-horseradish peroxidase (HRP) and streptavidin-AP (Southern Biotech). Immunofluorescence protocols used anti-B220-FITC and anti-Foxp3-PE.
ANAs
The presence of anti-nuclear Abs (ANAs) in serum was detected as previously described using permeabilized HEp-2 cells as the substrate [2]. Serum was considered ANA+ if homogeneous nuclear staining was observed [16].
Chromatin ELISAs
ELISA plates (ThermoLabSystems) were coated with 2 µg/ml of chromatin (a generous gift of M. Monestier, Temple University, Philadelphia, PA) overnight at 4°C. The remaining steps were performed at room temperature. All washes were conducted at least eight times in 1X PBS/0.05% Tween 20. Following the coating step, plates were washed, blocked with 1% BSA/PBS/azide for at least 1h, and washed again. Sera were then added at increasing dilutions, as indicated in figure, and incubated for a minimum of 1h. Plates were washed and incubated with developing Ab (anti-IgMa biotin; BD Biosciences), for at least 1h. Finally plates were washed, incubated with streptavidin-AP (Southern Biotechnology Associates) for at least 1h, washed and developed for 14–18 h. The plates were developed with Immunopure p-nitro-phenyl phosphate (Pierce) as the substrate. Absorbances were read at dual wavelength, 405/650 nm using a microplate reader.
Treg inhibition of proliferation or cytokine assay
CD4+ CD25− (BALB/c) and B220− CD4+ CD25− (BALB-lpr/lpr gld/gld) Th cells were sorted from lymph nodes. Treg cells were sorted from lymph nodes of BALB/c or TS1xHA28 mice (CD4+ CD25+), and lpr/lpr gld/gld mice (B220− CD4+ CD25+). Sorted cells were plated at a starting ratio of 2.5 Th cells : 1 Treg cell to a final ratio of 20 Th : 1 Treg cell with anti-CD3 (2C11) (33 ng/ml) (BD Biosciences) and irradiated BALB/c spleen cells as APCs. After three days of stimulation, cells were pulsed with 3H thymidine and 16–18 hours later cpm were recorded. The percent inhibition is determined by the following formula: ((cpm of Th alone culture – cpm of Th+Treg culture) / cpm of Th alone culture) × 100 = % inhibition.
Responder T cells for in vitro proliferation or cytokine assays were either obtained by sorting CD4+ CD25− cells from BALB/c, TS1 TCR Tg mice, or B220− CD4+ CD25− cells from lpr/lpr gld/gld or mice. The Th1 and Th2 lines were established as described previously [5]. 5×104 CD4+ CD25− cells were stimulated with 0.125 µg/ml of anti-CD3 (2C11), 5×105 CD3 depleted BALB/c splenocytes and with 5×104 Treg cells where indicated. After three days cells were stimulated with PMA, Ionomycin, and Brefeldin A as previously described [5]. The cells were then stained for CD4, Thy1.2 and IFN-γ or IL-4.
CD25 depletion protocol
>BALB/c-gld/gld or lpr/lpr gld/gld mice were injected i.p. weekly from two to four weeks of age with 1 mg of PC61 (bio-express.com) dissolved in 0.5 ml of PBS. Control mice were given 0.5 ml of PBS or 1 mg of Rat IgG, respectively. Beginning at week 5 of age, mice were bled weekly and anti-chromatin ELISAs were performed to detect anti-chromatin IgM, IgG1, and IgG2a Abs in sera. The level of CD25 depletion was determined weekly starting at week 4 by staining peripheral lymphocytes for CD4 and CD25. The frequency of CD25+ cells within the CD4+ population was: week 4, PC61 treated = 0.76% +/− 0.12%, control = 6.8% +/− 0.5%; week 5, PC61 treated = 1.12% +/− 0.18%, control = 6.68% +/− 0.51%; week 6, PC61 treated = 2.97% +/− 0.68%, control = 8.03% +/− 2.17%; week 7, PC61 treated = 2.63% +/− 0.71%, control = 8.28% +/− 1.96% (n=5). All mice in the PC61 group had a significantly lower frequency of CD4+ CD25+ cells at all time points compared to control mice.
Statistical Analysis
Statistical significance was determined via the Student’s t test provided by Microsoft Excel software unless otherwise noted. Significance was ascribed when p < 0.05.
Results
In single mutant lpr/lpr mice, FasL mRNA is upregulated [17, 18]. Consistent with this finding, a high level of non-specific cell death and/or graft versus host disease is observed when lpr/lpr T cells are adoptively transferred. These outcomes are avoided when lpr/lpr gld/gld T cells are used [19, 20]. Therefore, to bypass these complications as well as to prevent the transferred cells from being killed by host cells bearing FasL, we used double mutant lpr/lpr gld/gld mice in our studies.
Anti-chromatin B cells from young VH3H9 Tg lpr/lpr gld/gld mice are responsive to T cell help
In order to address whether the abortive response of anti-chromatin B cells in young lpr/lpr gld/gld mice is due to a defect in their response to T cell help, we engineered a means to provide them with defined T cell help. This system previously allowed us to dissect the effects of Th and Treg cells on anti-chromatin B cells in Fas-sufficient BALB/c mice. TS1 transgenic T cells specific for the site 1 peptide from HA of the PR8 influenza virus were injected into VH3H9 Tg lpr/lpr gld/gld mice that expressed HA on all class II+ cells (Fig 1A). Ten days after transferring lymph node cells from TS1 mice into VH3H9/HACII Tg lpr/lpr gld/gld mice, serum was collected and assayed for anti-nuclear antibodies (ANAs). Mice that received no T cells were ANA−, however when mice received T cell help they were ANA+ (Fig 1B). Age-matched uninjected mice remained ANA−. Darkly staining extrafollicular anti-chromatin B cells were observed in the spleens of mice that had received T cell help; in contrast, these B cells were found in the B cell follicles in mice that did not receive T cells (Fig 1C). The extrafollicular localization of anti-chromatin cells is remarkably similar to that previously observed with VH3H9/HACII Tg Fas-sufficient BALB/c mice as recipients [4]. Therefore, anti-chromatin B cells from lpr/lpr gld/gld mice are capable of terminal ASC (antibody secreting cell) differentiation in the presence of sufficient T cell help. The abortive response seen in young lpr/lpr gld/gld mice must therefore be due to another factor – perhaps the quantity or nature of available T cell help.
Figure 1. lpr/lpr gld/gld anti-chromatin B cells do produce autoAbs in the presence of Th cells.

A) 20 ×106 TS1 lymph node cells were transferred into 4–5 week old VH3H9/HACII lpr/lpr gld/gld mice for 10 days. At day 10, B) serum or C) spleen was taken and assayed for ANAs and splenic localization, respectively (n=3).
Treg cells can suppress lpr/lpr gld/gld T cell help for B cells
In order to test the ability of Fas/FasL-deficient T cells to help anti-chromatin B cells, we made use of an in vivo transfer model in which anti-chromatin B cells engineered to express HA are transferred along with anti-HA TS1 TCR Tg T cells, and mice monitored for development of anti-chromatin Abs. This system allowed us to demonstrate previously that Th cells can induce the differentiation of anti-chromatin B cells into plasma cells, and that Treg cells can block this maturation [5]. We bred the TS1 Tg onto BALB/c-lpr/lpr gld/gld mice in order to test the ability of Fas/FasL-deficient T cells to help anti-chromatin B cells and the susceptibility of these cells to suppression by Treg cells. Anti-HA CD4+ CD25− cells, with or without Fas and FasL, were injected into CB17 mice along with PR8 influenza virus. In some cases, purified Treg cells were coinjected with the Th cells (Fig 2A). The following day, HA-expressing anti-chromatin B cells (Iga) were injected into the Igb-expressing CB17 mice. This system allows anti-chromatin antibody production to be tracked using anti-Iga reagents. On day 8 anti-chromatin antibodies in the sera and plasma cell localization in the spleen was analyzed (Fig. 2A).
Figure 2. Treg cells block in vivo autoAb production promoted by lpr/lpr gld/gld Th cells.

A) In vivo protocol for determining whether young lpr/lpr gld/gld Th cells can be blocked from inducing autoreactive anti-chromatin antibody production. B) Anti-chromatin Abs from the serum of day 8 CB17 mice that received B cells alone (black circles), TS1 Th cells (black diamonds), TS1 Th + Treg cells (white diamonds), TS1 lpr/lpr gld/gld Th (black squares), and TS1 lpr/lpr gld/gld Th + Treg cells (white squares). Data are the average of three experiments. (*) indicates p < 0.05 and is different compared to all groups. C) Localization of IgMa cells in the spleen of the indicated groups by immunohistochemistry (n=3).
As described previously, anti-HA CD4+ CD25− T cells induced autoAbs which were inhibited by the co-administration of Treg cells (Fig. 2B, C)[5]. When lpr/lpr gld/gld anti-HA T cells from young (≤8 week old) mice were transferred with anti-chromatin B cells, anti-chromatin Abs were likewise produced, but at lower titers than recipients of Fas/FasL-sufficient anti-HA CD4+ CD25− cells (Fig. 2B). This difference may be due to the fact that Fas and FasL can also act as costimulators for CD4+ T cells [21]. Nevertheless, when Treg cells were added in addition to the young anti-HA lpr/lpr gld/gld T cells, antibody production was dramatically curtailed and anti-chromatin B cells were not detected in the spleen by immunohistochemistry (Fig. 2B, C). Thus, T cells from young Fas/FasL-deficient mice can provide help to anti-chromatin B cells, and this help can be suppressed by Treg cells.
Characterization of T cell subsets in lpr/lpr gld/gld mice
Multiple studies of lpr and gld mice on various strains (including MRL, C3H, C57BL/6, and BALB/c) have documented an increased number of CD4+ T cells in the periphery, as well as an increase in the unusual B220+ CD4+ and B220+ CD4− CD8− (double negative, DN) T cells compared to Fas/FasL-wildtype mice [22–26]. Indeed, in older Fas/FasL-deficient mice, the DN cells account for much of the lymphoproliferation [22–26]. The BALB/c-lpr/lpr gld/gld double mutant mice studied here duplicated this trend, with increased numbers of CD4+, B220+ CD4+, and DN T cells (Fig. 3A, B).
Figure 3. T cell subsets in the lymph node and spleen.
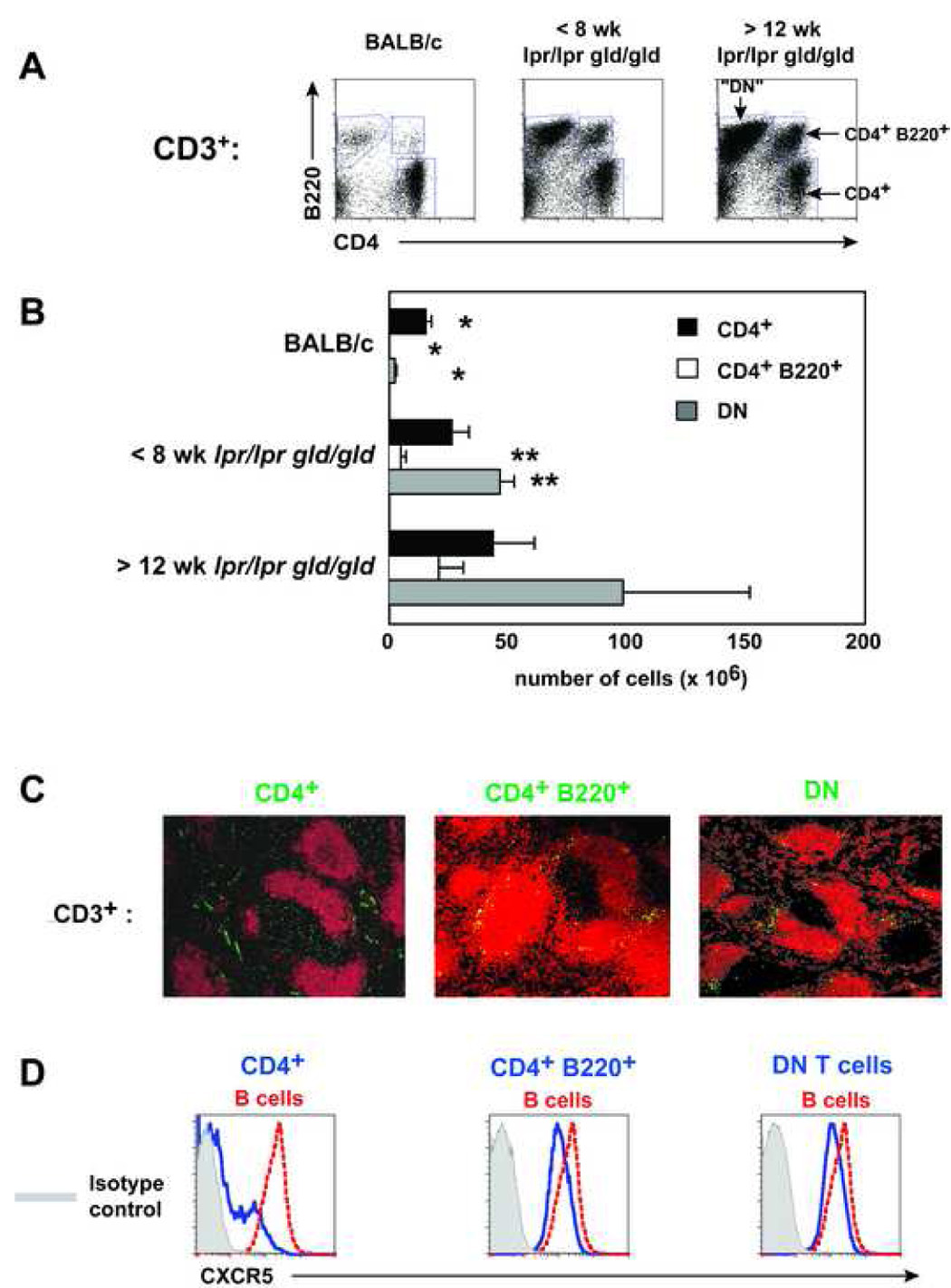
A) Representative plots of CD3+ cells from the spleens of BALB/c and young and old lpr/lpr gld/gld mice (n=4). B) Total cell numbers from the spleen of BALB/c and lpr/lpr gld/gld mice (n=4). (*) indicates significantly different compared to all group of mice except for those labeled. (**) indicateds are significantly different than old lpr/lpr gld/gld mice only. C) The indicated T cell populations were sorted from >12 week old lpr/lpr gld/gld spleens, CFSE labeled, and transferred into BALB/c mice overnight. Recipient mice were stained with anti-B220 (red) and transferred cells were visualized by immunofluorescence (n=3). D) Histograms of CXCR5 expression on the T cell population indicated in C) (n=3).
In addition to rising T cell numbers, multiple publications have described an increase in the activation level of T cells in Fas/FasL-deficient mice [27–30]. We have previously documented a rise in the frequency of CD4+ T cells found in splenic B cell follicles in lpr/lpr or gld/gld mice [2, 3]. However, because as lpr/lpr gld/gld mice age, their splenic architecture becomes increasingly disrupted [14], it is difficult to ascertain where the CD4+, B220+ CD4+, and DN T cells localize. To determine where these T cells home in the spleen, CFSE labeled T cell subsets from lpr/lpr gld/gld mice were transferred into BALB/c recipients. Sixteen hours after transfer, the majority of CD4+ T cells localized in the splenic PALS, but with a minority clearly present in the follicle (Fig 3C, left). DN T cells were located near the marginal zones, confirming a previous publication (Fig 3C, right) [31]. Like the DN T cells, the CD4+ B220+ T cells were also found on the outer edges of the B cell follicle (Fig 3C, center).
Flow cytometry showed that the CD4+ T cell population was predominantly negative for CXCR5 expression but did include a small CXCR5+ population consistent with the presence of CD4+ T cells in the follicle (Fig 3C, D, left). The DN and CD4+ B220+ cells were uniformly CXCR5+, but with a slightly lower mean fluorescence than follicular B cells (Fig 3D, right and center). The vast majority of CD4+ T cells from the recipient mouse were CXCR5− (data not shown) [5]. Therefore, CXCR5 expression on lpr/lpr gld/gld T cell subsets correlated with splenic localization in recipient BALB/c mice.
Foxp3 expression in T cells from lpr/lpr gld/gld mice is primarily in the CD4+ T cell subset
An important factor in the potential for T cells to help B cells is the counterbalancing influence of Treg cells. To characterize the resident Treg cell population in lpr/lpr gld/gld mice, Foxp3 expression was evaluated in the CD4+, B220+ CD4+, and DN T cell populations (Fig. 4A, B). As described previously, Foxp3+ cells comprise about 10% of CD4 T cells in BALB/c mice [32]. Strikingly, the frequency and absolute number of CD4+ Foxp3+ cells in the spleen and lymph nodes of both the young and old lpr/lpr gld/gld mice was significantly higher than in the BALB/c mice (Fig. 4A,B).
Figure 4. Foxp3 expression in lpr/lpr gld/gld mice.

A) Representative plots of Foxp3 in the spleens of the indicated T cell population. The average percentage of Foxp3+ cells in BALB/c mice: CD4+, inguinal LN = 10.9% +/− 2.3%, spleen = 6.8% +/− 2.6% (n=4); CD4+ B220+, inguinal LN = 12.0% +/− 3.0%, spleen = 12.1% +/− 1.7% (n=3). The average percentage of Foxp3+ cells in young lpr/lpr gld/gld mice: CD4+, inguinal LN = 11.9% +/− 6.4%, spleen = 18.0% +/− 5.0% (n=3–4); CD4+ B220+, inguinal LN = 4.0% +/− 1.6%, spleen = 3.9% +/− 0.8% (n=3). The average percentage of Foxp3+ cells in old lpr/lpr gld/gld mice: CD4+, inguinal LN = 14.7% +/− 3.7%, spleen = 25.8% +/− 5.0% (n=3–4); CD4+ B220+, inguinal LN = 1.1% +/− 0.5%, spleen = 2.5% +/− 0.7% (n=2–3). B) Absolute cell number of Foxp3+ and Foxp3− cells in the CD4+, CD4+ B220+, and DN T cell subsets (n=3). C) Foxp3 localization in the spleens of BALB/c and young and old lpr/lpr gld/gld mice (n≥3).
In the lpr/lpr gld/gld mice, a decrease in the frequency of Foxp3 staining was observed in the B220+ CD4+ compartment compared to B220− CD4+ cells, with virtually no Foxp3 staining in the DN compartment (Fig. 4A). The rare B220+ CD4+ cells in BALB/c mice contain a similar fraction of Foxp3+ cells as the B220− CD4+ cells (~12% vs. ~11%, respectively), whereas no Foxp3 staining in the miniscule DN compartment was observed (Fig. 4A). Analysis of absolute numbers of Foxp3+ cells shows that the vast majority of Foxp3+ cells reside in the B220− CD4+ population in lpr/lpr gld/gld mice, as in the Fas-sufficient BALB/c mice (Fig. 4B).
Finally, immunofluorescence was performed to determine where the Foxp3+ population is located in the spleen. In the white pulp of BALB/c and young lpr/lpr gld/gld mice the Foxp3+ population is located in the PALS and so is co-localized with CD4+ T cells (Fig. 4C). While the lymphoid architecture gets progressively disrupted in lpr/lpr, gld/gld, or lpr/lpr gld/gld mice as they age [2, 14], the Foxp3+ cells were still predominately found in the PALS of the old lpr/lpr gld/gld mice (Fig 4C).
Fas/FasL deficiency does not diminish the suppressive effects of Treg cells nor does it lessen the susceptibility of Th proliferation to suppression by Treg cells
Although there is an increase in the frequency and number of Foxp3+ cells in the CD4 compartment in Fas/FasL-deficient mice, it was possible that these cells could become functionally unable to suppress the proliferation of target T cells. If Treg cells in lpr/lpr gld/gld mice were to lose their ability to suppress proliferation as the mice aged, this could account for the onset of autoantibody production. Likewise, if Th cells developed a resistance to suppression by Treg cells, this could trigger autoAb production.
We investigated whether B220− CD4+ CD25− cells from young or old lpr/lpr gld/gld mice were susceptible to suppression by Treg cells. TS1xHA28 BALB/c mice were used as donors for the Treg cells because greater numbers of cells could be obtained [33]. Consistent with previous data, Treg cells inhibited CD4+ CD25− cell proliferation from BALB/c mice by > 80% (Fig. 5A). Likewise, the proliferation of B220− CD4+ CD25− cells isolated from either young or old lpr/lpr gld/gld mice was equally inhibited by the Treg cells (Fig. 5A).
Figure 5. Treg and Th cells from lpr/lpr gld/gld mice can inhibit proliferation and be inhibited in vitro.

A) 5×104 Treg cells (CD4+ CD25+) from TS1xHA28 BALB/c mice were cultured with 5×104 Th cells from young or old lpr/lpr gld/gld mice and stimulated as in B). The data is the average of three separate experiments. A) 5×104 Treg (B220− CD4+ CD25+) cells from young and old lpr/lpr gld/gld mice were cultured with 5×104 Th cells from BALB/c mice, 5×105 irradiated BALB/c spleen cells and stimulated with anti-CD3. After 72h stimulation the cultures were pulsed with 3H thymidine and then harvested 16h later. The data is the average of three separate experiments.
In similar experiments, Treg cells were isolated from both young and old lpr/lpr gld/gld mice and tested for their ability to inhibit CD4+ CD25− cell proliferation in vitro. Treg cells from both young and old lpr/lpr gld/gld mice were equally capable of suppressing BALB/c CD4+ CD25− cell proliferation at a 2.5 or 5 to 1 ratio of CD4+ CD25− cells to Treg cells (Fig 5B and data not shown).
IFN-γpotential is not reduced in lpr/lpr gld/gld B220− CD4+ CD25− T cells in the presence of Treg cells
As another function of CD4+ CD25− T cells is their ability to produce cytokines that direct other cells in the immune system, we investigated whether the production of cytokines by lpr/lpr gld/gld T cells was affected by the presence of Treg cells. Previously, it was shown that CD4+ T cells from 4–6 month old C3H gld/gld mice stimulated in vitro had secreted high levels of IFN-γ and low amounts of IL-4 [27]. We confirmed this observation after ex vivo stimulation with PMA and ionomycin from BALB/c-lpr/lpr gld/gld mice (Fig. 6A). Thus we focused on whether Treg cells can inhibit IFN-γ production from BALB/c, young, and old lpr/lpr gld/gld B220− CD4+ CD25− T cells after stimulation in vitro.
Figure 6. The effect of Treg cells on the IFN-γ potential of Th cells.
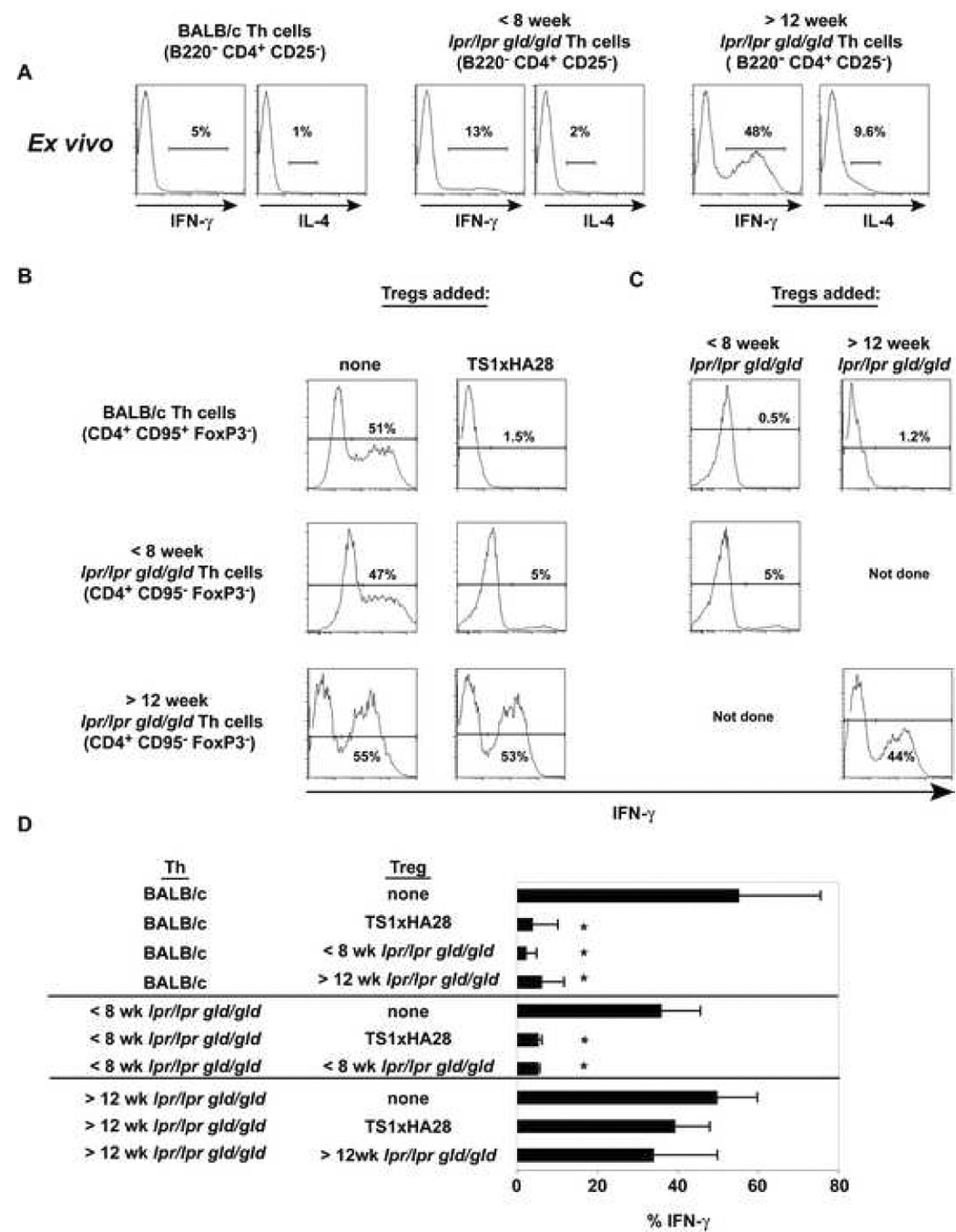
A) Th cells from the indicated mice were stimulated with PMA and Ionomycin ex vivo and 5 hours later were stained for IFN-γ and IL-4 (n=2). B) and C) Th cells and Treg cells were sorted from BALB/c and lpr/lpr gld/gld mice and stimulated with anti-CD3 for three days in the presence or absence of different Treg cell populations. On day 3 the cultures were stimulated with PMA and Ionomycin and stained for IFN-γ. The representative histograms are gated on CD4+ Foxp3− cells and the percentage of IFN-γ+ cells are shown (n=3). D) The bar graph shows the average frequency of cells making IFN-γ from all three experiments. * denotes p < 0.05 between Th cells with and without Treg cells.
When CD4+ CD25− T cells from BALB/c mice or B220− CD4+ CD25− T cells from lpr/lpr gld/gld mice were stimulated with soluble anti-CD3 and CD3 depleted BALB/c splenocytes for three days, in the absence of Treg cells, all populations produced a similar frequency of IFN-γ+ cells upon restimulation with PMA and Ionomycin (Fig 6B). In contrast, when CD4+ CD25− T cells from BALB/c mice were initially stimulated with Treg cells from one of three different sources (BALB/c, young, or old lpr/lpr gld/gld mice) the frequency of IFN-γ producing cells was reduced to nearly baseline (Fig 6B, C, top row). B220− CD4+ CD25− T cells from young lpr/lpr gld/gld mice stimulated with Treg cells from either TS1xHA28 BALB/c mice or young lpr/lpr gld/gld mice decreased their IFN-γ production, although a small IFN-γ+population remained (Fig. 6B, C, middle row). In contrast, when the B220− CD4+ CD25− T cells from old lpr/lpr gld/gld mice were stimulated with Treg cells from either TS1xHA28 BALB/c or old lpr/lpr gld/gld mice, there was no significant reduction in the percentage of IFN-γ cells observed upon restimulation (Fig 6B, C, bottom row, and D). These data suggest that Treg cells, regardless of their source, can not diminish the potential IFN-γ production that is already present in B220− CD4+ CD25− T cells from old lpr/lpr gld/gld mice.
Treg cells do not diminish the potential of IFN-γ production from in vitro generated Th1 cells but modestly affect IL-4 levels from Th2 cells
Treg cells have been shown to block the in vitro proliferation of naïve T cells, which is required for Th1/Th2 differentiation [34, 35]. However, their ability to alter the status of effector T cells is less clear. It has been previously demonstrated that lpr/lpr or gld/gld mice have a greater percentage of effector T cells [27]. In order to test whether Treg cells can influence the potential of Th1 or Th2 cells to make their signature cytokine, Th1 and Th2 cells were generated in vitro from CD4+ CD25− BALB/c TS1 TCR Tg cells, and IFN-γ or IL-4 production after culture with or without Treg cells was measured. Naïve TS1 cells stimulated with anti-CD3 had a substantial number of cells producing IFN-γ after PMA and Ionomycin stimulation that was inhibited when Treg cells were added to the initial anti-CD3 cultures (Fig. 7). As expected, the Th1 cultures also had a higher number of cells that produced IFN-γ and had a higher MFI compared to naïve T cells (Fig. 7). However unlike naïve T cells, the number of IFN-γ cells did not decrease when Treg cells were present in the same culture (Fig. 7). The Th2 cells in the absence of Treg cells had a majority of cells producing IL-4 and this frequency was modestly decreased in the presence of Treg cells (31%) (Fig 7). This data suggests that regardless of the Fas or FasL status on the CD4+ CD25− T cell, Treg cells have only a limited effect, at best, on the potential of effector T cells to make cytokines.
Figure 7. The response of Th1 and Th2 cells to Treg cells.

Using BALB/c TS1 TCR Tg mice, naïve, Th1, or Th2 cells were stimulated as in Figure 6 and stained for the number of cells producing IFN-γ or IL-4 (n=2).
CD25 depletion in gld/gld or lpr/lpr gld/gld deficient mice augments anti-chromatin IgG1 antibodies
In order to further address the role of Treg cells in delaying autoimmunity in autoimmune-prone mice, CD25+ cells were depleted from gld/gld or lpr/lpr gld/gld mice, beginning at a young age, and anti-chromatin IgM, and IgG1 and IgG2a responses assayed. It has been previously shown that IgG1 and IgG2a are associated with IL-4 (Th2) and IFN-γ (Th1) production, respectively [36]. Anti-CD25 depletion compared to PBS treatment had no effect on anti-chromatin IgM or IgG2a production at weeks 5 and 6 in gld/gld mice (Fig. 8A). However, CD25 depletion did result in significantly greater anti-chromatin IgG1 responses compared to the control mice at weeks 5 and 6 (Fig. 8A). This affect on IgG1 was even more prominent in anti-CD25 treated lpr/lpr gld/gld mice (Fig. 8B). The anti-CD25 treated lpr/lpr gld/gld mice also exhibited slightly higher levels of anti-chromatin IgM compared to control treated mice which may correspond to a slight increase in total IgM (Fig. 8B).
Figure 8. Anti-chromatin Ab responses from anti-CD25 depleted gld/gld or lpr/lpr gld/gld mice.

Weekly at 2–4 weeks of life 1 mg of PC61 (anti-CD25) was administered i.p. to A) gld/gld (n=3) or B) lpr/lpr gld/gld (n=2). Simultaneously control gld/gld or lpr/lpr gld/gld mice were treated with A) PBS (n=5) or B) Rat IgG (n=4), respectively. At weeks A) 5 and 6 or B) week 6, mice were bled and the isotype of anti-chromatin antibodies were determined. (*) denotes p<0.05 between PC61-treated and control treated mice.
Discussion
Fas/FasL-deficient mice have proven to be a useful model for studying the breakdown of B and T cell tolerance en route to the development of anti-chromatin antibodies [14]. We have tracked the phenotype and fate of VH3H9/λ1 anti-chromatin B cells in these mice before and after anti-chromatin autoantibodies become detectable [2, 3]. Although anti-chromatin B cells show certain phenotypic changes even in young mice, their differentiation into ASCs is not observed until 10 weeks of age, coinciding with the detection of serum autoantibodies [2, 3]. We have hypothesized that the apparent incomplete activation of the anti-chromatin B cells in young lpr/lpr or gld/gld mice may be due to one or more factors, including an intrinsic defect in the B cells themselves at the younger ages, a limiting quantity or quality of available T cell help at early time points, or the suppressive effects of T regulatory cells [4, 14].
Here, we show that the absence of autoAbs in young lpr/lpr or gld/gld mice is not due to an inherent defect in the B cells themselves, as anti-chromatin autoAbs are quickly produced when a defined, plentiful source of T cell help is provided. We further show that Th cells from young lpr/lpr gld/gld mice, when transferred into a third party mouse, are capable of inducing the production of anti-chromatin Abs. Why, then, do the Th cells in the young lpr/lpr gld/gld mouse not induce autoAbs until at least 10 weeks of age? One possibility is that the process of transferring the lpr/lpr gld/gld Th cells into a third-party recipient mouse may improve the apparent helper ability of the Th cells by concentrating their numbers (this transfer system uses a TCR Tg and purifies a defined number of Th cells for injection). Alternatively, the process of purification and transfer may remove the Th cells from a non-permissive environment in the young lpr/lpr gld/gld mouse, perhaps including the dampening effects of Treg cells. Indeed, we show here that the co-transfer of an equal number of Treg and Th cells effectively aborts autoantibody production.
To better understand the T cell environment in the young versus old lpr/lpr gld/gld mice, we studied CD4+ Th and Treg cells as well as further characterizing the B220+ CD4+ and DN T cells. CD4 cell numbers increase as lpr/lpr gld/gld mice age compared to BALB/c mice. Although the majority of CD4+ T cells expresses baseline levels of CXCR5 and localizes in the PALS, a small but distinct minority expresses CXCR5 and appears to co-mingle with B cells in the follicle. This may be relevant as T cells with a follicular localization (TFH) have been shown to be important in promoting germinal center development and Ab production [37].
We found that the frequency and number of Foxp3+ CD4+ Treg cells is also elevated in lpr/lpr gld/gld mice compared to BALB/c mice, and this number increases with age. Thus, even as the CD4 Th population is expanded in Fas/FasL-deficient mice, the number of Treg cells appears to keep pace with this expansion. Foxp3 expression was observed in both the typical B220− CD4+ population [38] as well as in the B220+ CD4+ subset in all mice examined. It is worth noting however that while the Foxp3 population increases in the B220− CD4+ compartment, the population of Foxp3+ B220+ CD4+ cells appears to decline as the lpr/lpr gld/gld mice age, coinciding with the onset of autoAb production. As this population comprises only a small population in total number, however, the significance of these cells is unclear.
To examine the functionality of Treg cells in lpr/lpr gld/gld mice, we tested their ability to suppress proliferation and IFN-γ production in vitro. We also examined the susceptibility of lpr/lpr gld/gld Th cells to Treg cells in similar assays. We found that Treg cells derived from both young and old lpr/lpr gld/gld mice ably suppress T cell proliferation in vitro. These data are consistent with other studies that have shown that blocking with an anti-FasL antibody has no effect on Treg cell suppression [39, 40], and that Treg cells from gld/gld mice can inhibit gld/gld Th cell proliferation [11]. Conversely, we found that Treg cells suppress the proliferation of Th cells derived from either young or old lpr/lpr gld/gld mice.
Many studies have shown that Treg cells can inhibit naïve T cell proliferation in vitro and hence this would block potential Th1 and Th2 cell development [34, 35, 41]. However, we have shown in vivo that naïve T cell proliferation at the beginning of an immune response is minimally impaired, yet the autoantibody response is inhibited [5]. Furthermore, Treg cells can suppress different ongoing autoimmune responses including gastritis and colitis [42, 43], but the mechanism for stopping autoimmunity in these models is still unknown. These data suggest that blocking of Th cell proliferation may not be the only, or even the main, mechanism of inhibition in vivo.
As one possible mechanism for the suppression of an ongoing autoimmune response would be to alter the cytokine potential of established effector T cells, we tested whether cytokine production from lpr/lpr gld/gld Th cells or in vitro-generated Th1 and Th2 cell lines was altered by Treg cells. We found that Treg cells effectively blocked IFN-γ production by Th cells from BALB/c mice and young lpr/lpr mice. This data suggests that Treg cells can block naïve T cells from differentiating into effector Th1 cells. However, if the Th cells were derived from older lpr/lpr gld/gld mice, co-culture with Treg cells did not diminish the frequency of IFN-γ-producing cells upon restimulation in vitro. Thus, although Treg cells suppress the proliferation of a majority of Th cells from old lpr/lpr gld/gld mice, a pre-existing population of cells appears resistant to this suppression and maintains the potential to produce IFN-γ. This supports the hypothesis that as lpr/lpr gld/gld mice age, they accumulate dedicated effector T cells that produce IFN-γ and are resistant to suppression by Treg cells. The assays we present here demonstrating the resistance of an established Th1 cell line to suppression of cytokine production by Treg cells bolster this hypothesis. Established Th2 cells appear less resistant than Th1 cells to the effects of Treg cells on their cytokine potential. We and others have shown that IFN-γ-producing Th cells are effective at promoting Abs in vivo, including anti-chromatin Abs [44, 45]. Furthermore, many studies have shown a preponderance of a Th1-type environment in Fas or FasL-deficient mice [27, 46, 47].
To test the ability of Treg cells to suppress Th1 or Th2-type T cells in vivo, we used the anti-CD25 Ab (PC61) to deplete Treg cells from young gld/gld or lpr/lpr gld/gld mice and tracked the appearance of Th1-and Th2-associated Ab isotypes. Young lpr/lpr gld/gld mice were used to avoid the complications of the dramatic serum hypergammaglobulinemia and other disease-associated problems (such as scabby skin, lymphoproliferation, and enlarged lymph nodes and spleens) in older lpr/lpr gld/gld mice. We demonstrated in figure 6 that even young lpr/lpr gld/gld mice have a small population of pre-existing IFN-γ cells and postulated that the IFN-γ potential of these cells would be resistant to Treg cells in vivo as in vitro. In an environment depleted of Treg cells, IgG1 (a Th2-associated isotype) but not IgG2a (a Th1-associated isotype) was significantly elevated. Thus, as in vitro, Treg cells in vivo do not affect IFN-γ secretion, thereby leaving IgG2a production unchanged.
We have shown here and in previous publications that Treg cells can block anti-chromatin Ab production in an in vivo transfer model [4, 5, 45]. Why, then do older lpr/lpr gld/gld mice develop anti-chromatin Abs even though they have high numbers of Treg cells? In other transgenic models we have suggested that high numbers of Th cells can lead to autoantibody production or autoimmunity even in the presence of Treg cells [48, 49]. Furthermore, in the in vivo transfer model that we used to test Th and Treg function, the recipient mice received a single dose of Treg cells at the same time they received Th cells. We have shown that if the injection of Treg cells is delayed by even one day after administration of Th cells, the Treg cells lose their effectiveness at blocking autoAb production [5]. Thus, in the aging lpr/lpr gld/gld mice, Treg cells may be simply overwhelmed by the continual expansion of Th cells. Finally, another possibility is that Treg cells lose their effectiveness as the Fas/FasL-deficient mice age or T effectors become resistant to regulation, thereby allowing Th cells to promote the induction of anti-chromatin Abs. Strong support of the idea that T cells can become resistant to Treg cell suppression comes from a recent study which showed that during experimental autoimmune encephalomyelitis, a population of Treg cells from the CNS could suppress naïve responder T cells but not responder T cells from the CNS [50].
Likely, other factors in the aging lpr/lpr gld/gld mice also play a role in the development of autoAbs. For instance, older Fas/FasL-deficient mice lose normal compartmentalization of the B and T cells in the spleen, which may alter how and when Th, Treg, and B cells interact [14]. Another difference in the autoimmune and non-autoimmune environment may be that the dendritic cells secrete inflammatory cytokines that block Treg cell suppression. Cytokines such as IL-6 and IL-12, known to be secreted by dendritic cells, inhibit Treg cell suppression [51, 52]. As dendritic cells in at least one murine lupus model overexpress IL-6 compared to control C57BL/6 mice [53], the status of these cells in lupus models merits further attention.
Taken together, the data presented here suggests that the interplay between Th and Treg cells is crucial to preventing autoAb production in young Fas/FasL-deficient mice. However, as the mice age and accumulate ever-increasing numbers of Th cells, some Th cells acquire resistance to Treg cells. Our data suggest that IFN-γ-producing cells may be crucial in the process of developing anti-chromatin Abs even in the face of a numerous Treg cell population.
Acknowledgments
We thank Simone Wilms and Michele Metzgar for excellent technical assistance, J. Faust, D. Hussey, and D. Ambrose at the Wistar Institute Flow Cytometry Facility, and James Hayden at the Wistar Institute Microscopy Facility.
Footnotes
This work was supported by grants (to J.E. and A.J.C) from the National Institutes of Health and Commonwealth Universal Research Enhancement Program, Pennsylvania Department of Health.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mandik-Nayak L, Bui A, Noorchashm H, Eaton A, Erikson J. Regulation of anti-double-stranded DNA B cells in nonautoimmune mice: localization to the T-B interface of the splenic follicle. Journal of Experimental Medicine. 1997;186:1257–1267. doi: 10.1084/jem.186.8.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mandik-Nayak L, Seo S-j, Sokol C, Potts KM, Bui A, Erikson J. MRL-lpr/lpr mice exhibit a defect in maintaining developmental arrest and follicular exclusion of anti-double-stranded DNA B cells. Journal of Experimental Medicine. 1999;189:1799–1814. doi: 10.1084/jem.189.11.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fields ML, Sokol CL, Eaton-Bassiri A, Seo S-j, Madaio MP, Erikson J. Fas/Fas Ligand deficiency results in altered localization of anti-double-stranded DNA B cells and dendritic cells. Journal of Immunology. 2001;167:2370–2378. doi: 10.4049/jimmunol.167.4.2370. [DOI] [PubMed] [Google Scholar]
- 4.Seo S-j, Fields ML, Buckler JL, Reed AJ, Mandik-Nayak L, Nish SA, Noelle RJ, Turka LA, Finkelman FD, Caton AJ, Erikson J. The impact of T helper and T regulatory cells on the regulation of anti-double-stranded DNA B cells. Immunity. 2002;16:535–546. doi: 10.1016/s1074-7613(02)00298-4. [DOI] [PubMed] [Google Scholar]
- 5.Fields ML, Hondowicz BD, Metzgar MH, Nish SA, Wharton GN, Picca CC, Caton AJ, Erikson J. CD4+CD25+ Regulatory T Cells Inhibit the Maturation but Not the Initiation of an Autoantibody Response. J Immunol. 2005;175:4255–4264. doi: 10.4049/jimmunol.175.7.4255. [DOI] [PubMed] [Google Scholar]
- 6.Crispin JC, Martinez A, Alcocer-Varela J. Quantification of regulatory T cells in patients with systemic lupus erythematosus. J Autoimmun. 2003;21:273–276. doi: 10.1016/s0896-8411(03)00121-5. [DOI] [PubMed] [Google Scholar]
- 7.Liu MF, Wang CR, Fung LL, Wu CR. Decreased CD4+CD25+ T cells in peripheral blood of patients with systemic lupus erythematosus. Scand J Immunol. 2004;59:198–202. doi: 10.1111/j.0300-9475.2004.01370.x. [DOI] [PubMed] [Google Scholar]
- 8.Wu HY, Staines NA. A deficiency of CD4+CD25+ T cells permits the development of spontaneous lupus-like disease in mice, and can be reversed by induction of mucosal tolerance to histone peptide autoantigen. Lupus. 2004;13:192–200. doi: 10.1191/0961203303lu1002oa. [DOI] [PubMed] [Google Scholar]
- 9.Scalapino KJ, Tang Q, Bluestone JA, Bonyhadi ML, Daikh DI. Suppression of disease in New Zealand Black/New Zealand White lupus-prone mice by adoptive transfer of ex vivo expanded regulatory T cells. J Immunol. 2006;177:1451–1459. doi: 10.4049/jimmunol.177.3.1451. [DOI] [PubMed] [Google Scholar]
- 10.Cuda CM, Wan S, Sobel ES, Croker BP, Morel L. Murine lupus susceptibility locus Sle1a controls regulatory T cell number and function through multiple mechanisms. J Immunol. 2007;179:7439–7447. doi: 10.4049/jimmunol.179.11.7439. [DOI] [PubMed] [Google Scholar]
- 11.Mohamood AS, Trujillo CJ, Zheng D, Jie C, Murillo FM, Schneck JP, Hamad AR. Gld mutation of Fas ligand increases the frequency and up-regulates cell survival genes in CD25+CD4+ TR cells. Int Immunol. 2006;18:1265–1277. doi: 10.1093/intimm/dxl057. [DOI] [PubMed] [Google Scholar]
- 12.Bagavant H, Thompson C, Ohno K, Setiady Y, Tung KS. Differential effect of neonatal thymectomy on systemic and organ-specific autoimmune disease. Int Immunol. 2002;14:1397–1406. doi: 10.1093/intimm/dxf105. [DOI] [PubMed] [Google Scholar]
- 13.Bagavant H, Tung KS. Failure of CD25+ T cells from lupus-prone mice to suppress lupus glomerulonephritis and sialoadenitis. J Immunol. 2005;175:944–950. doi: 10.4049/jimmunol.175.2.944. [DOI] [PubMed] [Google Scholar]
- 14.Fields ML, Hondowicz BD, Wharton GN, Adair BS, Metzgar MH, Alexander ST, Caton AJ, Erikson J. The regulation and activation of lupus-associated B cells. Immunol Rev. 2005;204:165–183. doi: 10.1111/j.0105-2896.2005.00238.x. [DOI] [PubMed] [Google Scholar]
- 15.Hardy RR, Carmack CE, Shinton SA, Kemp JD, Hayakawa K. Resolution and characterization of pro-B and pre-B cell stages in normal mouse bone marrow. J. Exp. Med. 1991;173:1213–1225. doi: 10.1084/jem.173.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roark JH, Kuntz CL, Nguyen K-A, Mandik L, Cattermole M, Erikson J. B cell selection and allelic exclusion of an anti-DNA immunoglobulin transgene in MRL-lpr/lpr mice. J. Immunol. 1995;154:4444–4455. [PubMed] [Google Scholar]
- 17.Chu JL, Ramos P, Rosendorff A, Nikolic-Zugic J, Lacy E, Matsuzawa A, Elkon KB. Massive Upregulation of the Fas ligand in lpr and gld mice: Implications for Fas regulation and the graft-versus-host disease-like wasting syndrome. J. Exp. Med. 1995;181:393–398. doi: 10.1084/jem.181.1.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watanabe D, Suda T, Hashimoto, H, Nagata S. Constitutive activation of the Fas ligand gene in mouse lymphoproliferative disorders. The EMBO J. 1995;14:12–18. doi: 10.1002/j.1460-2075.1995.tb06970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komano H, Ikegami Y, Yokoyama M, Suzuki R, Yonehara S, Yamasaki Y, Shinohara N. Severe impairment of B cell function in lpr/lpr mice expressing transgenic Fas selectively on B cells. International Immunology. 1998;11:1035–1042. doi: 10.1093/intimm/11.7.1035. [DOI] [PubMed] [Google Scholar]
- 20.Zhu B, Beaudette BC, Rifkin IR, Marshak-Rothstein A. Double mutant MRL-lpr/lpr-gld/gld cells fail to trigger lpr-graft-versus-host disease in syngeneic wild-type recipient mice, but can induce wild-type B cells to make autoantibody. Eur J Immunol. 2000;30:1778–1784. doi: 10.1002/1521-4141(200006)30:6<1778::AID-IMMU1778>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki I, Fink PJ. The dual functions of fas ligand in the regulation of peripheral CD8+ and CD4+ T cells. Proc Natl Acad Sci U S A. 2000;97:1707–1712. doi: 10.1073/pnas.97.4.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morse HC, Davidson WF, Yater RA, Murphy ED, Roths JB, Coffman RL. Abnormalities induced by the mutant lpr gene: expression of a unique lymphocyte subset. JI. 1982;129:2612. [PubMed] [Google Scholar]
- 23.Dumont FJ, Habbersett RC, Nichols EA, Treffinger JA, Tung AS. A monoclonal antibody (100C5) to the Lyt-2-T cell population expanding in MRL/Mp-lpr/lpr mice detects a surface antigen normally expressed on Lyt-2+ cells and B cells. Eur J Immunol. 1983;13:455–459. doi: 10.1002/eji.1830130605. [DOI] [PubMed] [Google Scholar]
- 24.Asano T, Tomooka S, Serushago BA, Himeno K, Nomoto K. A new T cell subset expressing B220 and CD4 in lpr mice: defects in the response to mitogens and in the production of IL-2. Clin Exp Immunol. 1988;74:36–40. [PMC free article] [PubMed] [Google Scholar]
- 25.Kariyone A, Takiguchi M, Igarashi S, Kano K. Ontogeny and function of B220+ L3T4+ T-cell subset of MRL/Mp-lpr/lpr mice. Cell Immunol. 1988;115:112–120. doi: 10.1016/0008-8749(88)90166-9. [DOI] [PubMed] [Google Scholar]
- 26.Shirai T, Abe M, Yagita H, Okumura K, C. M. H, III, Davidson WF. The expanded populations of CD4-CD8- T cell receptor a/b+ T cells associated with the lpr and gld mutations are CD2- Journal of Immunology. 1990;144:3756–3761. [PubMed] [Google Scholar]
- 27.Davidson WF, Calkins C, Hugins A, Giese T, Holmes KL. Cytokine secretion by C3H-lpr and -gld T cells. Hypersecretion of IFN-gamma and tumor necrosis factor-alpha by stimulated CD4+ T cells. Journal of Immunology. 1991;146:4138–4148. [PubMed] [Google Scholar]
- 28.Giese T, Davidson WF. Evidence for early onset, polyclonal activation of T cell subsets in mice homozygous for lpr. Journal of Immunology. 1992;149:3097–3106. [PubMed] [Google Scholar]
- 29.Chan O, Shlomchik MJ. A new role for B cells in systemic autoimmunity: B cells promote spontaneous T cell activation in MRL-lpr/lpr mice. J. Immunol. 1998;160:51–59. [PubMed] [Google Scholar]
- 30.Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. Journal of Experimental Medicine. 1999;189:1639–1648. doi: 10.1084/jem.189.10.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ansel KM, McHeyzer-Williams LJ, Ngo VN, McHeyzer-Williams MG, Cyster JG. In Vivo-activated CD4 T cells upregulate their CXC Chemokine receptor 5 and reprogram their response to lymphoid chemokines. Journal of Experimental Medicine. 1999;190:1123–1134. doi: 10.1084/jem.190.8.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 33.Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- 34.Gett AV, Hodgkin PD. Cell division regulates the T cell cytokine repertoire, revealing a mechanism underlying immune class regulation. Proc Natl Acad Sci U S A. 1998;95:9488–9493. doi: 10.1073/pnas.95.16.9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bird JJ, Brown DR, Mullen AC, Moskowitz NH, Mahowald MA, Sider JR, Gajewski TF, Wang CR, Reiner SL. Helper T cell differentiation is controlled by the cell cycle. Immunity. 1998;9:229–237. doi: 10.1016/s1074-7613(00)80605-6. [DOI] [PubMed] [Google Scholar]
- 36.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 37.King C, Tangye SG, Mackay CR. T Follicular Helper (T(FH)) Cells in Normal and Dysregulated Immune Responses. Annu Rev Immunol. 2008 doi: 10.1146/annurev.immunol.26.021607.090344. [DOI] [PubMed] [Google Scholar]
- 38.Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol. 2005;6:331–337. doi: 10.1038/ni1179. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi T, Kuniyasu Y, Toda M, Sakaguchi N, Itoh M, Iwata M, Shimizu J, Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. International Immunology. 1998;10:1969–1980. doi: 10.1093/intimm/10.12.1969. [DOI] [PubMed] [Google Scholar]
- 40.Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21:589–601. doi: 10.1016/j.immuni.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 41.Shevach EM. Regulatory T cells in autoimmunity. Annual Review of Immunology. 2000;18:423–449. doi: 10.1146/annurev.immunol.18.1.423. [DOI] [PubMed] [Google Scholar]
- 42.Suri-Payer E, Amar AZ, Thornton AM, Shevach EM. CD4+CD25+ T cells inhibit both the induction and effector function of autoreactive T cells and represent a unique lineage of immunoregulatory cells. J Immunol. 1998;160:1212–1218. [PubMed] [Google Scholar]
- 43.Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J Immunol. 2003;170:3939–3943. doi: 10.4049/jimmunol.170.8.3939. [DOI] [PubMed] [Google Scholar]
- 44.Smith KM, Brewer JM, Rush CM, Riley J, Garside P. In vivo generated Th1 cells can migrate to B cell follicles to support B cell responses. J Immunol. 2004;173:1640–1646. doi: 10.4049/jimmunol.173.3.1640. [DOI] [PubMed] [Google Scholar]
- 45.Fields ML, Nish SA, Hondowicz BD, Metzgar MH, Wharton GN, Caton AJ, Erikson J. The influence of effector T cells and Fas ligand on lupus-associated B cells. J Immunol. 2005;175:104–111. doi: 10.4049/jimmunol.175.1.104. [DOI] [PubMed] [Google Scholar]
- 46.Takahashi S, Fossati L, Iwamoto M, Merino R, Motta R, Kobayakawa T, Izui S. Imbalance towards Th1 predominance is associated with acceleration of lupus-like autoimmune syndrome in MRL mice. J Clin Invest. 1996;97:1597–1604. doi: 10.1172/JCI118584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Balomenos D, Rumold R, Theofilopoulos AN. Interferon-gamma is required for lupus-like disease and lymphoaccumulation in MRL-lpr mice. J Clin Invest. 1998;101:364–371. doi: 10.1172/JCI750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guay HM, Larkin J, 3rd, Picca CC, Panarey L, Caton AJ. Spontaneous autoreactive memory B cell formation driven by a high frequency of autoreactive CD4+ T cells. J Immunol. 2007;178:4793–4802. doi: 10.4049/jimmunol.178.8.4793. [DOI] [PubMed] [Google Scholar]
- 49.Rankin AL, Reed AJ, Oh S, Cozzo Picca C, Guay HM, Larkin J, 3rd, Panarey L, Aitken MK, Koeberlein B, Lipsky PE, Tomaszewski JE, Naji A, Caton AJ. CD4+ T Cells Recognizing a Single Self-Peptide Expressed by APCs Induce Spontaneous Autoimmune Arthritis. J Immunol. 2008;180:833–841. doi: 10.4049/jimmunol.180.2.833. [DOI] [PubMed] [Google Scholar]
- 50.Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Backstrom BT, Sobel RA, Wucherpfennig KW, Strom TB, Oukka M, Kuchroo VK. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 52.King IL, Segal BM. Cutting edge: IL-12 induces CD4+CD25- T cell activation in the presence of T regulatory cells. J Immunol. 2005;175:641–645. doi: 10.4049/jimmunol.175.2.641. [DOI] [PubMed] [Google Scholar]
- 53.Wan S, Xia C, Morel L. IL-6 produced by dendritic cells from lupus-prone mice inhibits CD4+CD25+ T cell regulatory functions. J Immunol. 2007;178:271–279. doi: 10.4049/jimmunol.178.1.271. [DOI] [PubMed] [Google Scholar]