

Abstract
Mutation of Bcr-Abl is an important mechanism by which chronic myelogenous leukemia (CML) cells become resistant to Gleevec. The T315I mutation is clinically significant since CML cells harboring this mutation are insensitive to Gleevec and other Bcr-Abl-targeted drugs. Identification of new agents capable of effectively killing CML cells with T315I mutation would have important therapeutic implications in Gleevec-resistant CML. Here, we showed that β-phenylethyl isothiocyanate (PEITC), a natural compound found in vegetables, is effective in killing CML cells expressing T315I BCR-ABL. Treatment of leukemia cell lines harboring wild-type or mutant Bcr-Abl with 10 μm PEITC resulted in an elevated ROS stress and a redox-mediated degradation of the BCR-ABL protein, leading to massive death of the leukemia cells. Antioxidant NAC attenuated the PEITC-induced oxidative stress in CML cells and prevented the degradation of BCR-ABL, caspase-3 activation and cell death. We further showed that the ROS-induced degradation of BCR-ABL was mediated partially by caspase-3 and the proteasome pathway. The ability of PEITC to effectively kill T315I-positive CML cells was further confirmed using primary leukemia cells isolated from CML patients. Our results suggest that PEITC is a promising compound capable of killing Gleevec-resistant CML cells through a ROS-mediated mechanism and warrants further investigations.
Keywords: CML, BCR-ABL, T315I mutation, drug resistance, PEITC, ROS
Introduction
Chronic myelogenous leukemia (CML) is a myeloproliferative disorder characterized by unregulated growth of myeloid leukemia cells in the bone marrow and accumulation of these cells in the blood. CML represents approximately 15−20% of all adult leukemia and the disease development is clearly linked to the constitutively active tyrosine kinase of the chimeric protein BCR-ABL, which is encoded by the Bcr-Abl fusion gene sequence as the result of chromosome 9/22 translocation (Philadelphia chromosome) or other aberrant cytogenetic events.1–3 Conventional therapeutic agents for CML treatment include interferon-alpha and cytotoxic agents, such as hydroxyurea, ara-C and busulfan. Although these drugs are effective to various degrees in treating CML, the toxic side effects of these agents may limit the dosage and duration of the clinical treatment. The development of targeted agents that specifically inhibit the tyrosine kinase activity of BCR-ABL has revolutionized the treatment of CML, and perhaps marks the successful beginning of molecularly targeted therapy.
Imatinib mesylate (Gleevec) represents the first generation of BCR-ABL tyrosine kinase inhibitors that are very effective in the clinical treatment of CML. Due to its high therapeutic activity and relatively low toxicity, Gleevec has replaced other cytotoxic agents and become the front-line agent for CML.4,5 This compound interacts with the ATP-binding pocket of BCR-ABL, inhibits the tyrosine kinase activity and effectively kills CML cells.4,5 However, a set of BCR-ABL mutants, especially the T315I mutation, leads to alteration in the three-dimensional structure of the enzyme active site and exhibits constitutive kinase activity and resistance to Gleevec.6–9 Such Bcr-Abl mutations impose new challenges in treatment of CML and have prompted the development of second generation of tyrosine kinase inhibitors to overcome this drug resistance. Dasatinib (BMS0354825) is one of such second-generation tyrosine kinase inhibitor approved by the FDA for the treatment of CML. Clinical trials demonstrated that dasatinib induced clinical responses in all BCR-ABL genotypes including various mutations with the exception of the T315I mutation.10,11 In vitro studies confirmed that the T315I mutation confers resistance to both imatinib and dasatinib.12 These findings underscore the importance and urgency to develop alternative strategies.13,14 The use of imatinib and dasatinib in the clinical treatment of CML will inevitably lead to a selection of CML cells with T315I mutation and development of drug resistance, for which no effective tyrosine kinase inhibitor is currently available in the clinic. Thus, identification of novel compounds and development of new strategies for the effective treatment of CML with T315I mutation are important and challenging tasks.
Alternative strategies to effectively kill CML cells with T315I mutation would be to trigger cell death process through a mechanism different from the inhibition of tyrosine kinase activity and to abolish the function of this oncoprotein by inducing its rapid degradation. Based on the previous observations that the BCR-ABL oncogenic signal can promote ROS generation and induce cellular redox imbalance,15–18 we postulated that such oxidative stress might serve as a biochemical basis to preferentially trigger reactive oxygen species (ROS)-mediated damage to these cells by further oxidative stress with exogenous ROS-generating agents. Furthermore, since many proteins, including BCR-ABL, contain redox-sensitive cysteine residues that can be oxidized by ROS leading to changes in protein structure and stability, we speculated that induction of severe ROS stress in CML cells might potentially alter the redox status of BCR-ABL and render it vulnerable to degradation. In fact, recent studies suggest that N-acetylcysteine (NAC, an antioxidant) and BSO (an inhibitor of glutathione synthesis) seem to affect BCR-ABL stability.19,20 The present study was designed to test these possibilities using β-phenylethyl isothiocyanates (PEITC, a natural compound found in edible vegetables) as an agent to induce further oxidative stress in CML cells. PEITC has previously been shown to effectively disable the cellular glutathione system by inducing depletion of cellular glutathione and inhibition of glutathione peroxidase activity, leading to massive ROS accumulation in cancer cells.18 We hypothesized that the increase of ROS generation in CML under the stimulation of BCR-ABL oncogenic signal might render these cells highly dependent on glutathione to maintain redox balance, and that depletion of glutathione by PEITC would cause severe ROS accumulation and trigger cell death. A possible induction of BCR-ABL degradation by redox-mediated mechanism would add to the potency of cell killing. Since T315I mutation retains constitutive tyrosine kinase activity, CML cells with this mutation should, like the CML cells with wild-type BCR-ABL, exhibit increased ROS stress and sensitivity to PEITC.
Materials and methods
Chemicals and reagents
β-phenylethyl isothiocyanate, NAC and bovine catalase were purchased from Sigma-Aldrich (St Louis, MO, USA). Gleevec was a product of Novartis Pharmaceuticals (East Hanover NJ, USA). Caspase inhibitors Z-VAD-FMK and Z-DEVD-FMK were obtained from BD Biosciences (San Jose, CA, USA). The proteasome inhibitor MG132 was acquired from EMD biosciences (Calbiochem, San Diego, CA, USA). PEITC was dissolved in dimethylsulfoxide and further diluted with culture medium before use. The final dimethylsulfoxide concentrations in the cell culture medium were less than 0.1% (v/v). Bovine catalase was freshly dissolved in culture media and sterilized by passing through a 0.2-μm sterile syringe filter (Corning, Inc., Corning, NY, USA) before use.
Cell culture and isolation of primary CML cells
All human and murine BCR-ABL positive cells were maintained in RPMI 1640 medium with 10% fetal bovine serum at 37 °C in an atmosphere of 5% CO2 balanced with humidified air. KBM5 cell line expressing the 210 kD BCR-ABL protein and lacking normal c-ABL was derived from a female myeloid CML patient in blast crisis as described previously.21,22 KBM5-T315I line was derived from KBM5 by exposing to increasing concentrations of Gleevec, leading to the selection of survival clone harboring T315I mutation as previously described.23 KBM5-T315I cells were routinely maintained in culture medium containing 1 μm Gleevec. In the study where KBM5-T315I cells were compared with the parental KBM5 cells, Gleevec was washed off and KBM5-T315I cells were cultured in drug-free medium for several days before the experiments. BaF3-BCR-ABL cells and BaF3-BCR-ABL/T315I cells were derived from the murine BaF3 cells by transfection using Bcr-Abl or T315I-mutated Bcr-Abl. 32D-BCR-ABL cells were derived from the murine 32D cells by transfection using Bcr-Abl. The methods to establish these cell lines and their culture conditions were described previously.24–26 Primary CML cells were isolated from fresh peripheral blood samples from CML patients after obtaining informed consent in accordance with a research protocol approved by MD Anderson Cancer Center institutional review board (IRB). Cells were isolated using gradient centrifugation with Fico/Lite lymphoH (d = 1.077, Atlanta Biologicals, Atlanta, GA, USA). After isolation, cells were washed with phosphate-buffered saline and suspended in fresh culture medium. All drug treatments started after the cells were precultured in fresh medium for 24 h.
Cytotoxicity assays
Two different methods were used to evaluate the cytotoxic effect of PEITC and Gleevec under various experimental conditions. Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described previously.27 Briefly, cells were treated with various concentrations of PEITC or Gleevec in 96-well plates for 72 h and MTT reagent was added and incubated during the last 4 h of the drug treatment. After washing off the culture medium, the cell pellets were dissolved in 200 μl dimethylsulfoxide and OD570 nm was quantified using a 96-well plate reader. Drug sensitivity was compared using the cell-survival curves and the IC50 values, defined as the drug concentration that induced 50% loss of cell viability. Cell death was determined by flow cytometric analysis after the cells were double-stained with annexin-V-FITC and propidium iodide (PI), using an assay kit from BD Biosciences (San Diego, CA, USA). After leukemia cells were incubated with various concentrations of compounds as indicated in the figure legends, the samples were stained with annexin-V/PI and analyzed using a BD FACSCalibur flow cytometer equipped with the CellQuest Pro software as described previously.18,28
Measurement of cellular ROS and glutathione
Cellular ROS contents were measured by incubating the control and drug-treated cells with 2.5 μm of the CM-H2DCF-DA (Invitrogen-Molecular Probe, Carlsbad, CA, USA) for 60 min as a chemical probe for ROS. CM-H2DCF-DA is cell-membrane permeable and once inside the cells, it reacts with H2O2 and other ROS species and generates green fluorescence. The intensity of green fluorescence, reflecting cellular ROS contents, was measured by flow cytometric analysis using a BD FACSCalibur equipped with Becton Dickson CellQuest Pro software as described previously.18 Total cellular glutathione contents were measured using a DTNB (5,5’-dithiobis (2-nitrobenzoic acid)) -enzyme-cycling glutathione assay kit (Cayman Chemical, Ann Arbor, MI, USA) and calculated using the standard curves generated in parallel experiments as described previously.18
Immunoblot analysis
Protein lysates were prepared from the control and drug-treated cells, separated by electrophoresis on 8 or 10% SDS-PAGE and transferred to nitrocellulose membranes. The molecules of interest were detected using specific antibodies as described previously.29 Primary rabbit polyclonal antibodies against full-length and cleaved BCR-ABL or caspase-3 were obtained from Cell Signaling Technology (Boston, MA, USA). Mouse monoclonal antibody against poly (ADP-ribose) polymerase (PARP) was purchased from BD Transduction Laboratories (San Diego, CA, USA). β-actin was also probed as a loading control.
Results and discussions
Effective killing of Gleevec-resistant CML cells expressing T315I-mutant BCR-ABL by PEITC
The observations that CML cells carrying the T315I mutation are resistant to Gleevec and to the second-generation tyrosine kinase inhibitors, such as dasatinib, indicate an urgent need for new agents that can effectively kill the drug-resistant CML cells by alternative mechanisms. Based on our previous observations that malignant transformation by Bcr-Abl causes increased ROS generation, and that this redox imbalance renders the leukemia cells vulnerable to further ROS stress by exogenous agents, such as PEITC,18 we reasoned that the aberrantly active tyrosine kinase of T315I-mutant BCR-ABL is likely to cause a constant increase in ROS generation and render these CML cells sensitive to PEITC. To test this possibility, we first compared the cytotoxic effect of Gleevec and PEITC in the KBM5 CML cell line harboring wild-type BCR-ABL and a variant line (KBM5-T315I) that was previously demonstrated to carry the T315I mutation.23 As shown in Figure 1a, KBM5-T315I cells were significantly less sensitive to Gleevec (IC50 = 5.4 ± 0.07 μm) compared to the parental KBM5 cells (IC50 = 0.28 ± 0.05 μM, P>0.0001). In contrast, both cell lines were equally sensitive to PEITC, with the IC50 values of 3.1 ± 0.1 for KBM5 cells and 2.8 ± 0.4 for KBM5-T315I cells (Figure 1b). It should be noted that the plasma PEITC concentrations in the micromolar range are clinically achievable through oral administration.30 At 10 μm, PEITC caused complete inhibition of cell growth in both KBM5 and KBM5-T315I cells, whereas approximately 30% of KBM5-T315I cells remained viable after treatment with 10 μm Gleevec. The potent activity of PEITC in killing both KBM5 and KBM5-T315I cells was further demonstrated using flow cytometric analysis after the cells were double-stained with annexin-V and PI (Figure 1c). Exposure of CML cells to 10 μm PEITC for 24 h resulted in 66 and 63% acute cell death in KBM5 and KBM5-T315I cells, respectively.
Figure 1.
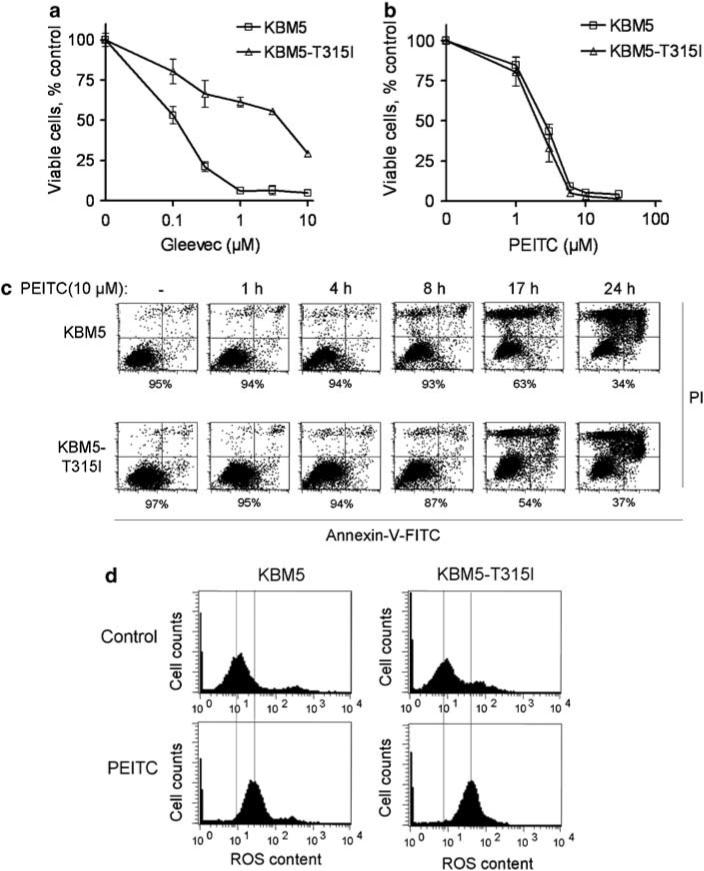
Cytotoxic effect of β-phenylethyl isothiocyanate (PEITC) in chronic myelogenous leukemia (CML) cells expressing either wild-type BCR-ABL (KBM5) or T315I-mutant BCR-ABL (KBM5-T315I). (a) Dose-dependent inhibition of cell proliferation by Gleevec. (b) Dose-dependent inhibition of cell proliferation by PEITC (0, 1, 3, 6, 10 and 30 μm). Cell viability was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. (c) Time-dependent induction of cell death by PEITC in KBM5 and KBM5-T315I cells. Cell death was detected by annexin-V/propidium iodide (PI) assay. The number shown below each panel indicates the percentage of the annexin-V and PI double-negative cells (viable). (d) Induction of ROS increase by PEITC (10 μm, 1.5 h) in KBM5 and KBM5-T315I cells. Cellular ROS contents were measured by flow cytometric analysis after the cells were stained with CM-H2DCF-DA fluorescence dye.
Consistent with the ability of PEITC to abrogate the glutathione antioxidant system,18 we showed that incubation of KBM5 and KBM5-T315I cells with PEITC led to a significant increase in cellular ROS, as evidenced by flow cytometric analysis after the cells were labeled with ROS-sensitive fluorescent probe CM-H2DCF-DA (Figure 1d). Incubation of KBM5 and KBM5-T315I cells with 10 μm of PEITC for 90 min caused an increase of cellular ROS by 129 and 282% respectively.
Induction of rapid cleavage of wild-type and T315I-mutant BCR-ABL by PEITC
Based on the observations that PEITC caused a substantial increase in cellular ROS and that BCR-ABL protein contains multiple redox-sensitive cysteine residues, we tested the possibility that the ROS stress induced by PEITC might alter the redox states of the wild-type and T315I-mutant BCR-ABL proteins and affect their stability. As illustrated in Figure 2a, exposure of KBM5 and KBM5-T315I cells to 10 μm PEITC caused a time-dependent decrease of the 210-kD BCR-ABL protein, which occurred as early as 4 h following drug incubation, with a concurrent appearance of a 52-kD cleavage product in both cell lines (Figure 2a). Interestingly, it took longer time for the T315I-mutant protein to be completely cleared from the KBM5-T315I cells, possibly due to a higher basal level of the mutant BCR-ABL protein in these cells. The increase in T315I-mutant protein may be due to a decrease in its turnover or an increase of its synthesis in KBM5-T315I cells. Since the PEITC-induced degradation of the T315I BCR-ABL protein was slower than that of the wild-type BCR-ABL, it is likely that stability of this oncoprotein is higher when it carries the T315I mutation. The observation that PEITC induced more ROS increase in the mutant cells but caused similar cytotoxicity in the wild-type and mutant KBM5 cells is consistent with the notion that T315I mutation might confer the BCR-ABL protein higher tolerability to ROS-induced degradation. The ability of PEITC to induce degradation of wild-type and mutant BCR-ABL proteins was further confirmed in a pair of murine cells (BaF3) stably transfected with either the wild-type or T315I-mutant Bcr-Abl (Figure 2b). As expected, PEITC was also effective in inducing apoptosis in both murine cell lines, whereas Gleevec caused cell death only in BaF3-BCR-ABL cells but not in BaF3-BCRABL-T315I cells (Figure 2c).
Figure 2.
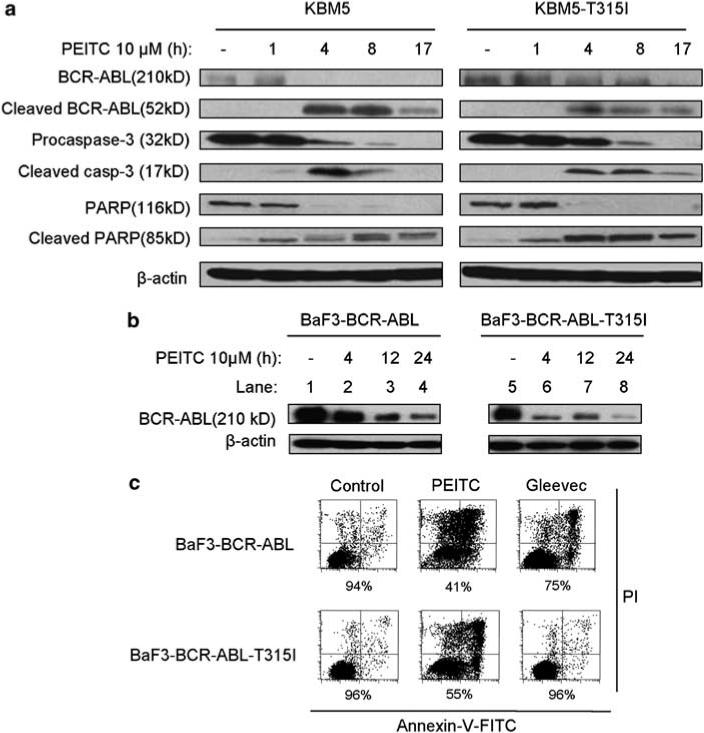
β-phenylethyl isothiocyanate (PEITC)-induced cleavage of wild-type and T315I-mutant BCR-ABL and its correlation with caspase-3 activation and the onset of cell death. (a) Time-dependent cleavage of BCR-ABL, caspase-3 and poly (ADP-ribose) polymerase induced by PEITC in KBM5 and KBM5-T315I cells. Cells were incubated with 10 μm PEITC for the indicated times, and protein cleavage was detected by immunoblotting using the respective specific antibodies. (b) PEITC treatment induces a time-dependent decrease of BCR-ABL levels in BaF3-BCRABL and BaF3-BCR-ABL-T315I cells as detected by immunoblotting. (c) Cell death induced by PEITC or Gleevec in the murine leukemia cells expressing BCR-ABL or T315I-mutant BCR-ABL. Cells were incubated with 10 μm PEITC or 2 μm Gleevec for 24 h, and cell death was measured by the annexin-V/propidium iodide (PI) assay. The number shown below each panel indicates the percentage of the annexin-V and PI double-negative cells (viable).
Since caspase activation and the cleavage of PARP are hallmarks of apoptosis, we measured the cleavage of caspase-3 (activation) and PARP at various time points after cells were treated with PEITC and evaluated the temporal relationship between the protein cleavage, BCR-ABL degradation and apoptotic cell death detected by annexin-V reactivity. As shown in Figure 2a, it appeared that caspase-3 activation and PARP cleavage occurred slightly before the degradation of BCR-ABL protein and that the cleavage of all three of these proteins proceeded well before the cells became annexin-positive (Figure 1c). These data together suggest that the cleavage of BCR-ABL might not be the primary event that triggered apoptosis and that the biochemical changes, such as glutathione depletion and ROS stress induced by PEITC,18 might be the initial factors that caused cell death. However, the rapid degradation of BCR-ABL proteins may significantly enhance the potency of PEITC in killing CML cells.
Effect of antioxidants on PEITC-induced BCR-ABL cleavage and cell death
We then used the antioxidant NAC and the H2O2-scavenging enzyme catalase to evaluate the role of ROS in mediating PEITC-induced BCR-ABL degradation and apoptosis. As shown in Figure 3a, treatment of 10 μm PEITC induced a rapid increase of ROS within 90 min and subsequently led to significant cell death in both cell lines (Figure 3b). Preincubation of cells with the antioxidant NAC (2 mm) almost completely suppressed PEITC-induced ROS increase and prevented cell death in both cell lines (Figure 3b), suggesting that ROS stress might play a critical role in mediating the cytotoxic effect of PEITC. Addition of catalase (500 unit/ml) to the cell culture medium only partially reduced the PEITC-induced ROS increase and did not significantly prevent cytotoxicity. This was likely due to the ineffectiveness of catalase to enter KBM5 cells and scavenge intracellular hydrogen peroxide. In contrast, the same concentrations of catalase fully prevented cell death induced by exogenous H2O2 (100 μm, Figure 3b), suggesting that exogenous catalase is effective in scavenging H2O2 in the medium but not sufficient to prevent the intracellular ROS increase induced by PEITC in these cells under the conditions used. Consistent with this observation, western blot analysis showed that only NAC could prevent the PEITC-induced cleavage of BCR-ABL, whereas 500 U/ml of catalase did not suppress BCR-ABL degradation (Figure 3c).
Figure 3.
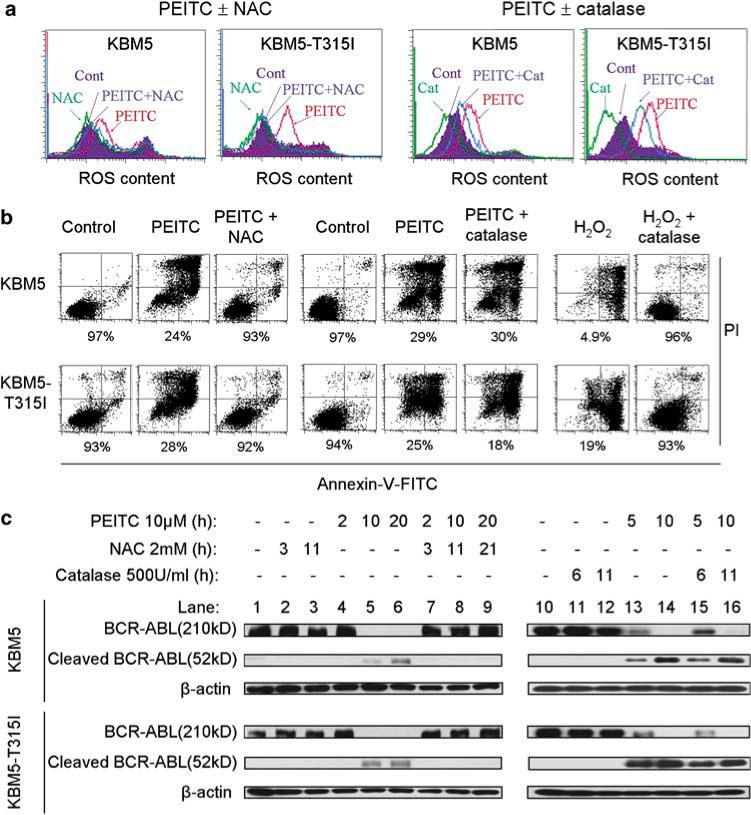
Induction of cell death and BCR-ABL cleavage by β-phenylethyl isothiocyanate (PEITC) through a ROS-mediated mechanism. (a) Changes in ROS content following treatment with 10 μm PEITC for 1.5 h with and without a 1-h pretreatment with 2 mm NAC or 500 units/ml bovine catalase. Intracellular ROS was detected by CM-H2DCF-DA fluorescent dye (control, purple; N-acetylcysteine (NAC) or catalase, green; PEITC, pink and PEITC + NAC or catalase, blue). (b) Cell death induced by 10 μm PEITC for 40 h with or without a 1-h pretreatment with 2 mm NAC or 500 units/ml bovine catalase. For comparison, cells were also treated with 100 μm H2O2 in the presence or absence of 500 units/ml catalase as a positive control for catalase activity. Cell death was determined by the annexin-V/propidium iodide (PI) assay. The number shown below each panel indicates the annexin-V and PI double-negative cells (viable). (c) Time-dependent cleavage of BCR-ABL following PEITC treatment with or without a 1-h pretreatment with 2 mm NAC or 500 units/ml catalase. BCR-ABL protein expression was detected by immunoblotting.
Because NAC is a precursor for glutathione synthesis, preincubation of cells with NAC would increase cellular glutathione (GSH) and counteract the glutathione-depleting effect of PEITC. As shown in Figure 4, incubation of cells with 10 μm PEITC caused about 30−40% depletion of glutathione at 5 h and a complete depletion at 10 h in both KBM5 and KBM5-T315I cells. The severe loss of cellular glutathione was unlikely due to cell death, since significant apoptosis was not detected until 17 h of drug incubation (Figure 1c). A preincubation with 2 mm NAC largely restored cellular glutathione, even at the 10-h time point. This may explain why NAC was very effective in preventing PEITC-induced ROS increase and cell death.
Figure 4.
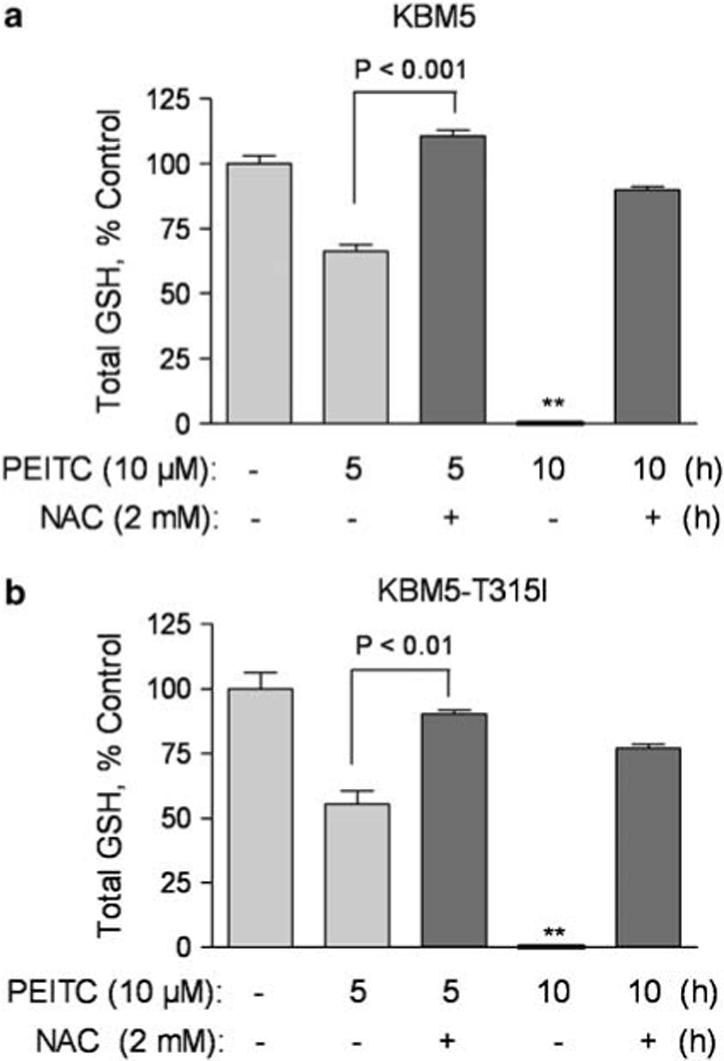
N-acetylcysteine (NAC) prevents depletion of cellular glutathione induced by β-phenylethyl isothiocyanate (PEITC) in chronic myelogenous leukemia (CML) cells. KBM5 and KBM5-T315I cells were incubated with 10 μm PEITC in the presence or absence of 2 mm NAC for 5−10h as indicated. Total cellular glutathione contents were then measured as described in Material and methods.
Caspase-3 mediates PEITC-induced cleavage of BCR-ABL
To test if caspases or/and the proteasome might be involved in mediating PEITC-induced BCR-ABL degradation, we used chemical inhibitors of caspases or proteasome to evaluate the role of these enzymes. As shown in Figure 5a, pretreatment of cells with 20 μm Z-VAD-FMK, a common inhibitor of multiple caspases, effectively prevented the cleavage of the wild-type and T315I-mutant BCR-ABL protein, as evidenced by the preservation of the full-length (210 kD) BCR-ABL and suppression of the generation of the 52-kDa BCR-ABL (Figure 5a, lanes 4−6). Interestingly, the proteasome inhibitor MG132 (5 μm) did not prevent the cleavage of BCR-ABL to a 52-kDa fragment (lanes 7−9), and only delayed the loss of full-length protein at the early time point (lane 8). This held true for both the wild-type and T315I-mutant BCR-ABL. Thus, it appeared that the PEITC-induced cleavage of BCR-ABL into a 52-kD fragment was largely mediated by caspase activity, whereas the proteasome might only play a complementary role to accelerate further degradation. This is in agreement with the previous observations that caspase-3 is a redox-sensitive enzyme31 and that inhibition of proteasome by MG132 may affect the caspase activity in a ROS-dependent manner.32 Furthermore, we demonstrated that Z-VAD-FMK was able to significantly reduce PEITC-induced cell death in both KBM5 and KBM5-T315I cells, whereas MG132 exhibited little effect in preventing apoptosis (Figure 5b), consistent with the important role of caspases in mediating degradation of BCR-ABL.
Figure 5.
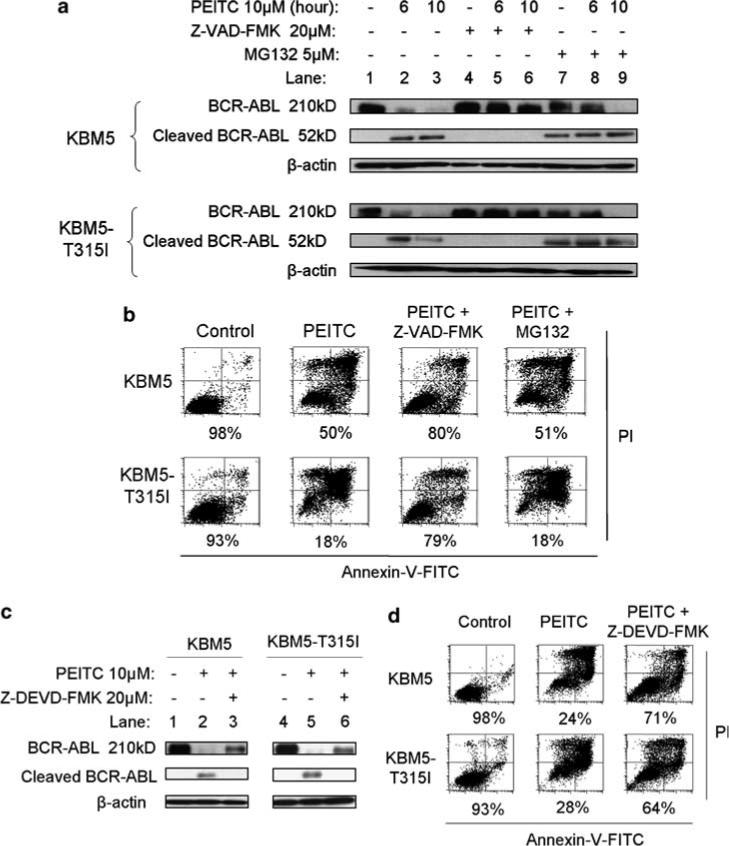
Effects of caspase inhibition or proteasome inhibition on β-phenylethyl isothiocyanate (PEITC)-induced BCR-ABL degradation and cell death in KBM5 and KBM5-T315I cells. (a) Time-dependent cleavage of BCR-ABL protein following PEITC treatment with or without a 1-h pretreatment with 20 μm Z-VAD-FMK or MG132. BCR-ABL expression was detected by immunoblotting. (b) Cell death induced by 10 μm PEITC with or without a 1-h pretreatment with 20 μm Z-VAD-FMK or MG132 for 36 h. Cell viability was measured by annexin-V/propidium iodide (PI) assay. The number shown below each panel indicates the annexin-V/PI double-negative cells (viable). (c) Cleavage of BCR-ABL protein induced by PEITC treatment with or without a 1-h pretreatment with 20 μm Z-DEVD-FMK, measured by immunoblotting. (d) Cell death induced by 10 μm PEITC (40 h) with or without a 1-h pretreatment with 20 μm Z-DEVD-FMK. Cell viability was measured by annexin-V/PI assay. The number shown below each panel indicates the annexin-V/PI double-negative cells (viable).
Because Z-VAD-FMK is a broad spectrum caspase inhibitor, the results in Figure 4a did not allow the identification of the specific caspase responsible for the cleavage of BCR-ABL. We then tested if Z-DEVD-FMK, a specific inhibitor of caspase-3, could prevent BCR-ABL cleavage and suppress PEITC-induced cell death in both KBM5 and KBM5-T315I cells. As shown in Figures 5c–d, inhibition of caspase-3 by Z-DEVD-FMK exhibited similar preventive effects on BCR-ABL cleavage and cell death as the pan-caspase inbitor Z-VAD-FMK, suggesting that caspase-3 may be a key effector protease that mediates PEITC-induced degradation of BCR-ABL and apoptosis. This is in line with the previous observations that the normal c-ABL protein is a substrate of capase-3 and that this protein structural features are preserved in the BCR-ABL chimeric protein.33–36
PEITC is effective against Gleevec-resistant primary CML cells
Based on the observations that PEITC was equally effective against cultured cell lines expressing either the wild-type or mutant BCR-ABL, we further tested the ability of this compound to kill primary CML cells isolated from patients who were clinically resistant to Gleevec treatment. As shown in Figure 6a, the primary CML cells from two Gleevec-resistant patients also exhibit resistance to Gleevec in vitro. The CML cells from a patient with T315I mutation were particularly resistant to Gleevec, with the IC50 value of more than 50 μm. In contrast, the CML cells from both patients were equally sensitive to PEITC in vitro, with the IC50 values of 8.3 and 10.5 μm, respectively (Figure 6b). Consistently, flow cytometry analysis revealed that incubation with 10 μm PEITC for 24 h caused more than 50% cell death in the primary CML cells of both patient samples (Figure 6c). MTT assay of a total of six CML patient samples showed that the IC50 values of PEITC were 12.6 ± 3.3 μm (mean ± s.d.).
Figure 6.
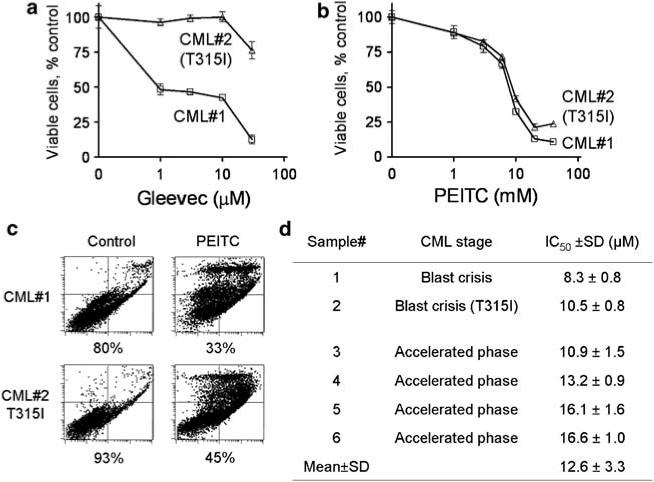
Cytotoxic effect of β-phenylethyl isothiocyanate (PEITC) in primary chronic myelogenous leukemia (CML) cells isolated from the peripheral blood samples of six CML patients clinically refractory to Gleevec. (a–b) representative dose-dependent cytotoxic effect of Gleevec (a) and PEITC (b) in primary CML cells with or without T315I mutation. Cells were incubated with the indicated concentrations of Gleevec or PEITC for 72 h, and cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. (c) Induction of cell death by PEITC (10 μm, 24 h) in primary CML cells with or without T315I mutation. Cell viability was measured by annexin-V/propidium iodide (PI) assay. The number shown below each panel indicates the percentage of viable cells. (d) IC50 values of PEITC in primary CML cells from six patients clinically refractory to Gleevec. The IC50 values were estimated by MTT assay.
Relationship between BCR-ABL expression and cellular sensitivity to Gleevec and PEITC
To further evaluate the relationship between BCR-ABL expression and cellular sensitivity to Gleevec and PEITC, we used an isogenic pair of murine cells, 32D cells (not harboring Bcr-Abl) and the stable Bcr-Abl-transfected cells (32D-BCR-ABL)37 for comparison. As shown in Figure 7a, Gleevec induced a massive cell death in 32D-BCR-ABL cells but not in parental 32D cells. In contrast, PEITC caused significant cell death in both cell lines, indicating different mechanisms of action between Gleevec and PEITC. Analysis of cellular ROS levels revealed that PEITC induced a substantial increase of ROS in both cell lines, consistent with its ROS-mediated mechanism of action (Figure 7b). Interestingly, the basal ROS level in 32D-BCR-ABL cells (10 units) was slightly higher than that in 32D cells (7 units), consistent with the oxidative stress induced by Bcr-Abl expression. Gleevec did not cause a significant change in ROS levels in either cell line (Figure 7b). These data together suggest that Gleevec is highly specific in killing cells expressing BCR-ABL, whereas PEITC can be cytotoxic to cells with or without expression of BCR-ABL, as long as a severe increase of ROS can be induced. This suggests a possibility that PEITC could also be toxic to normal cells without BCR-ABL expression. Interestingly, recent data suggest that compared to cancer cells, normal cells that seem to have a lower basal ROS generation are less dependent on the glutathione antioxidant system, and thus are less sensitive to PETIC.18 Nevertheless, the therapeutic selectivity and potential toxicity of PEITC to normal bone marrow cells should be carefully evaluated in future studies.
Figure 7.
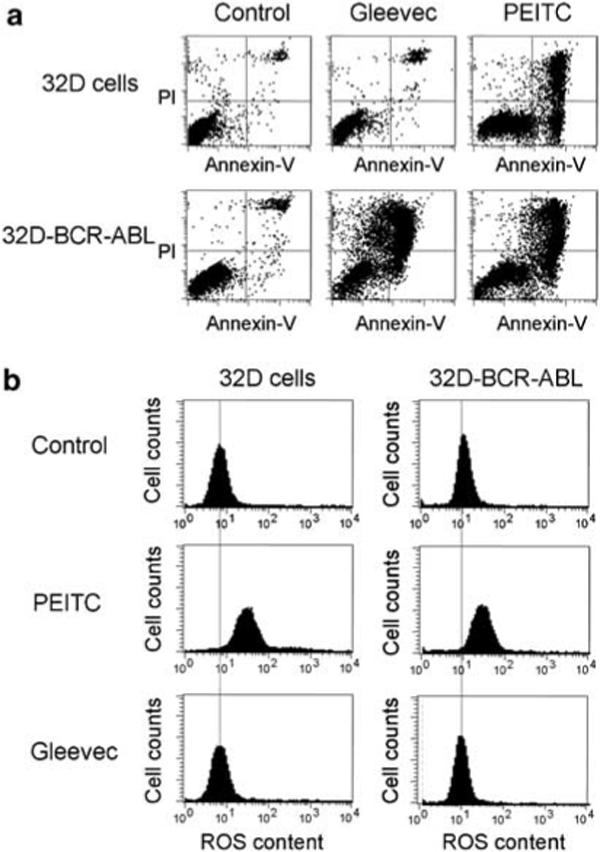
Cytotoxic effects of Gleevec and β-phenylethyl isothiocyanate (PEITC) in cells with or without BCR-ABL expression and relationship with ROS generation. (a) The murine myeloid 32D cells (without BCR-ABL) and the Bcr-Abl stable transfectant line (32D-BCR-ABL cells) were incubated with 10 μm PEITC or 5 μm Gleevec for 24 h, and cell death was measured by the annexin-V/PI double staining followed by flow cytometry analysis. (b) Effect of PEITC (10 μm, 2 h) and Gleevec (5 μm, 2 h) on cellular ROS contents. Cellular ROS were measured by flow cytometric analysis after the cells were stained with CM-H2DCF-DA fluorescence dye.
In summary, our study showed that the increase of ROS stress in CML cells expressing either wild-type or mutant BCR-ABL can be exploited for therapeutic purposes and that the natural compound PEITC is effective in killing CML cells harboring the T315I mutation, which confers resistance to Gleevec and the second-generation tyrosine kinase inhibitors. A key mechanism by which PEITC kills CML cells is through the induction of further ROS stress leading to oxidative damage, which could be prevented by antioxidants, such as NAC. Depletion of cellular glutathione seems to be an important biochemical event that mediated the cytotoxic action of PEITC, and replenishment of GSH by NAC abolished the cytotoxicity. The potent activity of PEITC to cause rapid increase of ROS and massive death of leukemia cells is consistent with the known mechanisms of action of this compound, which induces glutathione depletion by promoting its export from the cells and inhibits glutathione peroxidase activity, leading to a rapid disabling of the glutathione antioxidant system.18 However, these data do not necessarily prove that the depletion of glutathione is the sole mechanism responsible the cytotoxic activity of PEITC. It would be interesting to test if other compounds, such as the glutathione synthesis inhibitor buthionine sulfoximine might have similar effects on cellular ROS and viability in CML cells. It is also interesting to note that PEITC was able to induce a rapid degradation of BCR-ABL protein including the T315I mutant protein. Our data suggest that the severe ROS stress induced by PEITC might alter the redox state of BCR-ABL protein and render it vulnerable to degradation by caspase-3. This notion is supported by the observation that either NAC or a specific inhibitor of caspase-3 could significantly suppress the cleavage of BCR-ABL. It should be pointed out that the degradation of BCR-ABL might not be the primary cause of PEITC-induced cell death, which is likely triggered by direct oxidative damage to mitochondria and other critical cellular molecules. However, the ability of PEITC to induce rapid degradation BCR-ABL (wild type and mutant) may effectively abolish the prosurvival signal of this oncoprotein, thus add to the potency of this compound in killing CML cells. Furthermore, because normal cells have a lower basal ROS output and possess intact redox regulatory mechanisms, they are less vulnerable to ROS stress imposed by PEITC.18 Based on the promising activity of PEITC against Gleevec-resistant CML cells and its unique mechanism of action, we conclude that this compound may be useful to overcome CML resistance to kinase inhibitors and merits further clinical evaluation.
Acknowledgements
This work was supported in part by Grants CA085563, CA100428, CA109041 and CA16672 from the National Institutes of Health. DT is a recipient of a scholarship from Anandamahidol Foundation under the royal patronage of His Majesty the King of Thailand.
References
- 1.Faderl S, Talpaz M, Estrov Z, O'Brien S, Kurzrock R, Kantarjian HM. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341:164–172. doi: 10.1056/NEJM199907153410306. [DOI] [PubMed] [Google Scholar]
- 2.Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–183. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- 3.Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7:441–453. doi: 10.1038/nrc2147. [DOI] [PubMed] [Google Scholar]
- 4.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 5.Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346:645–652. doi: 10.1056/NEJMoa011573. [DOI] [PubMed] [Google Scholar]
- 6.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 7.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 8.Branford S, Rudzki Z, Walsh S, Parkinson I, Grigg A, Szer J, et al. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood. 2003;102:276–283. doi: 10.1182/blood-2002-09-2896. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto M, Kurosu T, Kakihana K, Mizuchi D, Miura O. The two major imatinib resistance mutations E255K and T315I enhance the activity of BCR/ABL fusion kinase. Biochem Biophys Res Commun. 2004;319:1272–1275. doi: 10.1016/j.bbrc.2004.05.113. [DOI] [PubMed] [Google Scholar]
- 10.Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–2541. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 11.Cortes J, Rousselot P, Kim DW, Ritchie E, Hamerschlak N, Coutre S, et al. Dasatinib induces complete hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in blast crisis. Blood. 2007;109:3207–3213. doi: 10.1182/blood-2006-09-046888. [DOI] [PubMed] [Google Scholar]
- 12.Bradeen HA, Eide CA, O'Hare T, Johnson KJ, Willis SG, Lee FY, et al. Comparison of imatinib mesylate, dasatinib (BMS-354825), and nilotinib (AMN107) in an N-ethyl-N-nitrosourea (ENU)-based mutagenesis screen: high efficacy of drug combinations. Blood. 2006;108:2332–2338. doi: 10.1182/blood-2006-02-004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Druker BJ. Circumventing resistance to kinase-inhibitor therapy. N Engl J Med. 2006;354:2594–2596. doi: 10.1056/NEJMe068073. [DOI] [PubMed] [Google Scholar]
- 14.Quintas-Cardama A, Kantarjian H, Cortes J. Flying under the radar: the new wave of BCR-ABL inhibitors. Nat Rev Drug Discov. 2007;6:834–848. doi: 10.1038/nrd2324. [DOI] [PubMed] [Google Scholar]
- 15.Sattler M, Verma S, Shrikhande G, Byrne CH, Pride YB, Winkler T, et al. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J Biol Chem. 2000;275:24273–24278. doi: 10.1074/jbc.M002094200. [DOI] [PubMed] [Google Scholar]
- 16.Kim JH, Chu SC, Gramlich JL, Pride YB, Babendreier E, Chauhan D, et al. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood. 2005;105:1717–1723. doi: 10.1182/blood-2004-03-0849. [DOI] [PubMed] [Google Scholar]
- 17.Koptyra M, Falinski R, Nowicki MO, Stoklosa T, Majsterek I, Nieborowska-Skorska M, et al. T BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 2006;108:319–327. doi: 10.1182/blood-2005-07-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Q, Tsukahara F, Maru Y. N-acetyl-cysteine enhances growth in BCR-ABL-transformed cells. Cancer Sci. 2005;96:240–244. doi: 10.1111/j.1349-7006.2005.00038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konig H, Hartel N, Schultheis B, Schatz M, Lorentz C, Melo JV, et al. Enhanced Bcr-Abl-specific antileukemic activity of arsenic trioxide (Trisenox) through glutathione-depletion in imatinib-resistant cells. Haematologica. 2007;92:838–841. doi: 10.3324/haematol.10955. [DOI] [PubMed] [Google Scholar]
- 21.Beran M, Pisa P, O'Brien S, Kurzrock R, Siciliano M, Cork A, et al. Biological properties and growth in SCID mice of a new myelogenous leukemia cell line (KBM-5) derived from chronic myelogenous leukemia cells in the blastic phase. Cancer Res. 1993;53:3603–3610. [PubMed] [Google Scholar]
- 22.Wetzler M, Talpaz M, Van Etten RA, Hirsh-Ginsberg C, Beran M, Kurzrock R. Subcellular localization of Bcr, Abl, and Bcr-Abl proteins in normal and leukemic cells and correlation of expression with myeloid differentiation. J Clin Invest. 1993;92:1925–1939. doi: 10.1172/JCI116786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ricci C, Scappini B, Divoky V, Gatto S, Onida F, Verstovsek S, et al. Mutation in the ATP-binding pocket of the ABL kinase domain in an STI571-resistant BCR/ABL-positive cell line. Cancer Res. 2002;62:5995–5998. [PubMed] [Google Scholar]
- 24.Deng M, Daley GQ. Expression of interferon consensus sequence binding protein induces potent immunity against BCR/ABL-induced leukemia. Blood. 2001;97:3491–3497. doi: 10.1182/blood.v97.11.3491. [DOI] [PubMed] [Google Scholar]
- 25.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 26.Samanta AK, Lin H, Sun T, Kantarjian H, Arlinghaus RB. Janus kinase 2: a critical target in chronic myelogenous leukemia. Cancer Res. 2006;66:6468–6472. doi: 10.1158/0008-5472.CAN-06-0025. [DOI] [PubMed] [Google Scholar]
- 27.Zhou Y, Achanta G, Pelicano H, Gandhi V, Plunkett W, Huang P. Action of (E)-2′-deoxy-2′-(fluoromethylene)cytidine on DNA metabolism: incorporation, excision, and cellular response. Mol Pharmacol. 2002;61:222–229. doi: 10.1124/mol.61.1.222. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood. 2003;101:4098–4104. doi: 10.1182/blood-2002-08-2512. [DOI] [PubMed] [Google Scholar]
- 29.Xu RH, Pelicano H, Zhang H, Giles FJ, Keating MJ, Huang P. Synergistic effect of targeting mTOR by rapamycin and depleting ATP by inhibition of glycolysis in lymphoma and leukemia cells. Leukemia. 2005;19:2153–2158. doi: 10.1038/sj.leu.2403968. [DOI] [PubMed] [Google Scholar]
- 30.Liebes L, Conaway CC, Hochster H, Mendoza S, Hecht SS, Crowell J, et al. High-performance liquid chromatography-based determination of total isothiocyanate levels in human plasma: application to studies with 2-phenethyl isothiocyanate. Anal Biochem. 2001;291:279–289. doi: 10.1006/abio.2001.5030. [DOI] [PubMed] [Google Scholar]
- 31.Wu SJ, Ng LT, Lin CC. Effects of antioxidants and caspase-3 inhibitor on the phenylethyl isothiocyanate-induced apoptotic signaling pathways in human PLC/PRF/5 cells. Eur J Pharmacol. 2005;518:96–106. doi: 10.1016/j.ejphar.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 32.Bang JH, Han ES, Lim I, Lee CS. Differential response of MG132 cytotoxicity against small cell lung cancer cells to changes in cellular GSH contents. Biochem Pharmacol. 2004;68:659–666. doi: 10.1016/j.bcp.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 33.Di Bacco AM, Cotter TG. p53 expression in K562 cells is associated with caspase-mediated cleavage of c-ABL and BCR-ABL protein kinases. Br J Haematol. 2002;117:588–597. doi: 10.1046/j.1365-2141.2002.03468.x. [DOI] [PubMed] [Google Scholar]
- 34.Barila D, Rufini A, Condo I, Ventura N, Dorey K, Superti-Furga G, et al. Caspase-dependent cleavage of c-Abl contributes to apoptosis. Mol Cell Biol. 2003;23:2790–2799. doi: 10.1128/MCB.23.8.2790-2799.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Machuy N, Rajalingam K, Rudel T. Requirement of caspase-mediated cleavage of c-Abl during stress-induced apoptosis. Cell Death Differ. 2004;11:290–300. doi: 10.1038/sj.cdd.4401336. [DOI] [PubMed] [Google Scholar]
- 36.Podar K, Raab MS, Tonon G, Sattler M, Barila D, Zhang J, et al. Up-regulation of c-Jun inhibits proliferation and induces apoptosis via caspase-triggered c-Abl cleavage in human multiple myeloma. Cancer Res. 2007;67:1680–1688. doi: 10.1158/0008-5472.CAN-06-1863. [DOI] [PubMed] [Google Scholar]
- 37.Ling X, Wang Y, Dietrich MF, Andreeff M, Arlinghaus RB. Vaccination with leukemia cells expressing cell-surface-associated GM-CSF blocks leukemia induction in immunocompetent mice. Oncogene. 2006;25:4483–4490. doi: 10.1038/sj.onc.1209477. [DOI] [PubMed] [Google Scholar]