

Abstract
Diketopiperazines (DKPs) are a well-known class of heterocycles that have recently emerged as a promising biologically active scaffold. Solid-phase organic synthesis has become an important tool in the combinatorial exploration of these privileged structures, expediting the synthesis and, therefore, the discovery of active compounds. To date, certain DKPs have shown potent activities against a range of diseases and biological phenomena, including bacterial infections, various cancers, asthma, infertility, premature labor, and HIV. Recent applications of solid-phase DKP synthesis, with a particular focus on cyclative cleavage and microwave-assisted reactions, are highlighted herein.
Keywords: Diketopiperazine (DKP), microwave (MW)-assisted organic reaction, cyclative cleavage, privileged structure, solid-phase organic synthesis (SPOS), small molecule therapeutic
Introduction
Significant research progress in genomics and proteomics is uncovering an increasing number of biological targets with implications toward disease states [1, 2]. Small molecule activators and inhibitors that modulate the activities of these targets could permit the characterization of their functions and facilitate the development of new therapeutic strategies. The screening of unbiased libraries of compounds is often necessary for the identification of active lead structures. Therefore, an urgent need exists for improved synthetic methods capable of rapidly and reliably generating large collections of small molecules in high purity. Combinatorial solid-phase synthesis incorporating rapid microwave (MW) heating methods represents one approach toward addressing this need [3].
Solid-phase organic synthesis (SPOS) offers a number of advantages over traditional synthetic methods, including the ability to (1) synthesize molecules using new reactions that may be difficult or impossible to perform in solution, and (2) isolate molecules post-synthesis without tedious and often time-consuming purification steps [2, 4]. However, this methodology often suffers from slow reaction rates due to the reduced diffusion of reactants through the polymeric supports [5]. Heating solid-phase reactions, in particular with MW irradiation, can greatly accelerate their reaction rates, as well as decrease side reactions and increase product yields [6]. MW heating is being applied with increasing frequency to SPOS due to the often-dramatic reaction rate enhancements observed (i.e., 10–100-fold), the wider availability of commercial MW reactors, and the overall convenience of this heating method [3, 6]. The strategic combination of these two technologies (SPOS and MW irradiation) constitutes a powerful approach toward combinatorial chemistry and, ultimately, drug discovery (see below) [3].
Despite the advantages outlined above, solid-phase combinatorial synthesis has not yet fully delivered on its promise to produce numerous therapeutic agents [7]. One strategy to improve the effectiveness of combinatorial chemistry and high-throughput screening is to identify and utilize “privileged structures” – small molecule scaffolds that bind to a variety of receptor classes with high affinities [8]. Cyclic dipeptides, or 2,5-diketopiperazines (DKPs), have proven to be one such scaffold [9]. DKPs have diverse medicinal properties, ranging from antibiotics to synthetic vaccines to cancer chemotherapies [10, 11]. Once believed to be only protein artifacts or degradation products, DKPs are now regarded as important metabolic intermediates [12]. Furthermore, these compounds are stable to proteolysis and have been shown to mimic significantly larger peptides by orienting their side chain substituents in a spatially defined manner. The combinatorial synthesis of DKP libraries, therefore, represents an attractive strategy for the identification of novel lead structures and is the broad focus of this review.
Selected Biologically Active DKP Natural Products
To emphasize the interesting and varied biological activities of DKPs, we discuss here two notable DKP natural products that were identified over the past decade and further optimized into useful lead structures. In 1996, the natural product spirotryprostatin B (Fig. 1) was isolated from the fungus, Aspergillus fumigatus [13], and Cui et al. published the first synthesis of this cytotoxic agent that same year [14]. This DKP inhibits the mammalian cell cycle at the G2/M phase and has an IC50 value of 14 μM, making it a promising anticancer lead structure. Three years later, the Danishefsky laboratory published the total synthesis of spirotryprostatin A [15] in addition to DKP analogues (1 and 2) that possessed 104-fold increased activities relative to the natural product (Fig. 1) [16].
Fig. (1).

Chemical structures of DKP-containing natural products spirotryprostatin A and B and analogues 1 and 2 produced by the Danishefsky laboratory.
DKPs 3, isolated from Streptomyces sp., have been shown to inhibit plasminogen activator inhibitor-1 (PAI-1), a potential therapeutic target in cancer and cardiovascular diseases (Fig. 2) [17]. PAI-1 is the key regulatory protein of tissue plasminogen activator, which is responsible for thrombus formation and clearance. Elevated levels of PAI-1 are linked to thromboembolic disease and are implicated in cancer. Researchers at Xenova Ltd. have used these natural products (3) as lead structures in a combinatorial effort to identify more potent inhibitors of PAI-1. These studies generated DKP XR5118 with an IC50 of 3.5 μM against PAI-1. Further optimization and structure streamlining yielded second generation DKP XR11211, displaying an IC50 of 0.2 μM [18] These DKPs have since been used as probes to explore the biochemical mechanism of PAI-1 inhibition [19]. This increased mechanistic understanding will allow for the further optimization of active ligands.
Fig. (2).

Dehydro-DKP natural products 3 and selected DKP plasminogen activator inhibitor-1 antagonists.
Table 1 provides a sampling of additional DKP natural products that are active against a range of important biological targets. These DKPs display both complex structures and an astonishing array of different biological activities. Notably, as combinatorial chemistry requires relatively straightforward chemical reactions and a limited number of steps for the rapid generation of potential lead compounds, the majority of the synthetic DKPs generated in large-scale libraries display significantly simpler structures than those shown in Table 1 (see below). Nevertheless, these natural products and their rich activities provide ample inspiration for the design of new DKP lead structures.
Table 1. Natural Product DKPs and Their Biological Activities (DKP Core Structures are Highlighted in Bold).
| Natural Product | Structure | Isolated from | Biological Activites | Ref. |
|---|---|---|---|---|
| Bicyclomycin |
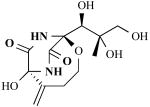
|
Streptomyces sp. (Gram-positive bacteria) | Antibiotic against Gram-negative and Gram-positive bacteria | [20, 21] |
| Brevicompanine |
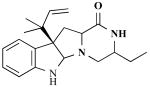
|
Penicillium brevicompactum (fungus) | Plant growth promoter | [22] |
| Chaetocochins |
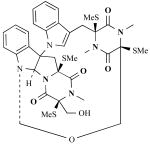
|
Chaetomium cochliodes (fungus) | Cytotoxins (target unknown) | [23] |
| Leptosins |
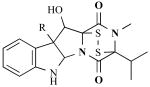
|
Leptoshaeria (marine fungus) | Antitumor agents that inhibit DNA topoisomerases | [24] |
| MPC1001 |
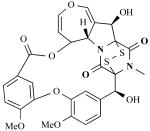
|
Cladorrhinum sp. (fungus) | Antitumor antibiotic | [25] |
| Phenylahistin |
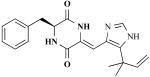
|
Aspergillus ustus (fungus) | Mammalian cell cycle inhibitor | [26] |
| Rhodotorulic acid |
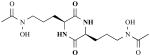
|
Rhodotorula sp. (yeast) | Siderophore in E. coli | [27] |
| Verrucologens |
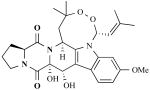
|
Penicillium verruculosum (fungus) | Neurotoxins that block Ca2+-activated K+ channels | [28] |
Previous Reviews of DKPs
There have been several reviews of DKP synthesis in the recent literature, and we briefly summarize them here to put this current review in context. As shown in Table 1, DKPs are ubiquitous in nature and affect a wide variety of biological targets. A number of these effects are outlined by Prasad in a review of cyclo(L-His-L-Pro) activity in humans [9]. The molecular basis for the activity of bicyclomycin has also recently been reviewed by Kohn and Widger [29]. A review by Horton discusses the structural benefits of the DKP scaffold, including its rigid three-dimensional structure, proteolytic stability, synthetic accessibility, and varied biological activity [8]. His review also highlights the linkers and strategies utilized in the solid-phase synthesis of DKPs and other cyclic peptides. Another review by Lambert et al. published shortly thereafter also discusses the synthetic techniques used for the generation of cyclic peptides, albeit DKPs constitute only a small subset of the material covered in the review [30].
A very thorough and informative review on synthetic approaches to all three DKP isomers (2,3-, 2,5-, and 2,6-) was published by Dismore and Beshore in 2002 [31]. The authors state that DKPs can circumvent the limitations of linear peptides and confer more drug-like properties to molecules of interest. It is evident from this review that the presence of biologically active DKPs in nature continues to drive synthetic efforts towards these potential drug candidates. Further, advances in SPOS methodology have paved the way toward the more rapid combinatorial discovery of active DKPs.
In 2003, Fischer wrote an excellent review that focused on DKPs in peptide and combinatorial chemistry and many of the technical advances that allow for rapid DKP synthesis [32]. Only one MW-assisted DKP synthesis had been reported at the time of publication, and this important example was not covered in Fischer's review.
Finally, a review entitled “α,β-dehydroamino acids” was published in 2006 by Bonauer and includes references to the solid- and solution-phase syntheses of naturally occurring dehydro-2,5-DKPs (Fig. 3) [33-35]. These methods were applied to the synthesis of DKP natural product analogues that are capable of inhibiting the mammalian cell cycle and constitute promising lead structures for anti-cancer therapies [11, 35].
Fig. (3).

Selected naturally occurring dehydro-DKPs.
The DKP field currently lacks a review focusing on the pairing of solid-phase synthesis and MW-assisted reactions with an emphasis on medicinal applications. This represents an important and growing area of DKP research and is the topic of the current review. In this review, we seek to provide a brief history of DKP synthesis and recent solid-phase and MW-assisted routes to their construction. Several solid-phase DKP synthetic strategies will be discussed, as well as recent applications of this chemistry toward the discovery of biologically active DKPs. As relatively few reports of MW-assisted DKP synthesis have been published to date, we also include solution-phase examples to provide a comprehensive analysis of the area. Our review concludes with a critique of four solid-phase DKP syntheses that potentially could have benefited from the application of MW heating methods.
1. Brief History of DKP Synthesis
Although scientists are only beginning to characterize the diverse biological functions of DKPs, researchers have been synthesizing these relatively simple compounds for over a century. Curtius and Goebel published the first cyclic dipeptide synthesis in 1888; the structure was later determined to be cyclo(Leu-Leu) [36]. Fischer and Forneau later generated cyclo(Gly-Gly) by treating the dipeptide methyl ester with excess ammonia over multiple days [37]. Although straightforward, these conditions caused a high degree of racemization. Nitecki et al. improved on this synthesis by boiling the formate salts of dipeptide methyl esters in neutral solvents for several hours [38]. Even though optically pure DKPs can be synthesized using this method, it is not amenable to combinatorial efforts focused on sizable library construction.
As introduced above, solid-phase synthetic methods offer significant benefits over solution-phase chemistry, the most notable being the potential for high product purities following cleavage from the solid-support. Gisin and Merrifield first reported DKPs as by-products of solid-phase peptide synthesis in 1972 [39]. They found that 70% of the dipeptide D-Pro-D-Val was cleaved from an ester-linked polystyrene support through an intramolecular aminolysis reaction during treatment with a carboxylic acid. The authors stated that this particular sequence cyclized readily due to “the ease with which acylamino acids can form cis peptide bonds,” and conditions were identified to optimize this reaction. This initial report launched the rich field of solid-phase DKP synthesis, which continues to progress today.
2. Recent Solid-Phase Synthetic Strategies to DKPs and Their Medicinal Applications
The most amenable route to DKP combinatorial synthesis is the cyclization and concomitant cleavage of dipeptide derivatives from solid support. This cyclative cleavage strategy is preferred over a solution-phase cyclization reaction after cleavage, as only DKP product is released from the linker through self-cleavage and thus should be isolated in high purity.
2.1. Central Tertiary Amides Facilitate DKP Cyclization
As highlighted by Gisin and Merrifield, the rate of cyclative cleavage of dipeptides from polymeric supports is highly dependent on their ability to achieve a cis-amide bond [39]. This conformation is favored when Gly, Pro, or N-alkyl groups are present. For this reason, the majority of solid-phase DKP syntheses integrate a central tertiary amide bond. Facile cyclization is generally achieved through three methods: (A) attaching the central amide to the solid support, (B) directly alkylating the central amide nitrogen, or (C) incorporating the tertiary amide via a multiple component reaction (MCR; Fig. 4). Each of these synthetic methods, along with its recent medicinal applications (published within the last decade), is discussed in turn below.
Fig. (4).

Tertiary amide bond integration strategies used in solid-phase DKP syntheses.
2.1.1. Backbone Amide Linkage Strategy to DKPs
Attachment of the central or backbone amide to the solid support (similar to 4 in Fig. 4) allows for the temporary exploitation of a tertiary amide bond (5), yet ultimately releases an N-unsubstituted cyclic dipeptide. This route, however, requires an often-difficult secondary acylation step. While attempting to construct backbone amide-bound polypeptides, Albericio and coworkers observed nearly quantitative DKP formation during deprotection of the dimer, regardless of the side chains present [40]. The authors have since taken advantage of this chemistry for the synthesis of DKPs shown in Fig. 5 [41]. Reductive amination of support-bound aldehyde 10 leads to secondary amide 11, which is then acylated to give the resin-bound, protected methyl ester 12. As stated above, deprotection of the N-Fmoc group with piperidine facilitates cyclization to resin-bound DKP 13. Cleavage with TFA then releases the product DKP 14 in excellent yields and purities (both >95%).
Fig. (5).

Backbone amide linkage DKP synthetic strategy.
This same research group recently reported the synthesis and biological evaluation of a library of guanidine- and amidine-containing DKPs as tryptase inhibitors using the synthetic methods in Fig. 5 [42]. Such inhibitors are valuable, because elevated levels of human β-tryptase are responsible for most allergic inflammatory diseases including asthma. The enzyme consists of four identical subunits with negatively charged active sites pointing toward one central core. This layout justifies the use of divalent/dibasic DKP inhibitors that could simultaneously interact with two vicinal active sites, potentially increasing affinity and selectivity.
The synthesis of these dibasic ligands began with the formation of Lys-, Arg-, and/or Asp-based DKPs according to Fig. 5. The route continued with further elaboration of the side chains through differential deprotection and traditional amide coupling steps to form three sub-libraries totaling 188 compounds. Sub-libraries of cyclo(L-Lys-L-Lys) and cyclo(L-Lys-L-Asp) showed tryptase inhibitory activities ranging from 50% to greater than 80% at 10 μM. Two of the tryptase inhibitors (15 and 16) are shown in Fig. 6 with their corresponding IC50 values. Although these DKP ligands are not sufficiently active to progress to preclinical testing, they do represent important lead compounds that warrant further development.
Fig. (6).

Selected DKP tryptase inhibitors and their corresponding activities.
2.1.2. On-Resin N-Alkylation of Central Amide Route to DKPs
A second strategy for increasing DKP cyclization/cleavage rates from a solid support is to directly alkylate the central amide 6 (as shown in Fig. 4). These N-alkylation reactions can be accomplished under a variety of conditions. Gordon and Steele have used the reductive alkylation of amino acids to increase their DKP yields from solid support [43], while others have utilized amide protecting groups, or have integrated an N-alkylated amino acid for this purpose [31]. In addition, Gong et al. have incorporated iminodiacetic acid into growing peptide chains to allow for the facile and efficient formation of DKPs on solid-phase [44].
Through the synthesis and screening of a 31,372-member combinatorial library, researchers at Pharmacopeia recently found DKPs to be a promising lead structure for follicle stimulating hormone (FSH) receptor agonists [45]. FSH is a 38 kDa heterodimeric protein crucial to human reproduction. Current fertility treatments involve the purification and subsequent subcutaneous or intra-muscular injection of this protein. The high cost and low patient compliance associated with these methods have increased the demand for orally available, small molecule therapeutic development.
Starting with their initial DKP lead, the Pharmacopeia researchers synthesized a focused library of over 300 biaryl DKP derivatives in parallel using two N-alkyl incorporation strategies outlined in Fig. 7 [46]. Route A began by coupling bromoacetic acid to Wang-linker derivatized beads to generate 17, followed by nucleophilic displacement with a primary amine to yield 18. In contrast, route B involved alkylation of 19 with a range of primary alcohols under Mitsunobu conditions followed by deprotection of the 2-nitro-benzenesulfonyl group to give amine 20. These two syntheses then converged with the condensation of iodophenylalanine, and subsequent Suzuki reactions with various arylboronic acids. The incorporated formylphenyl groups were reductively aminated and reacted further with various electrophiles (i.e., acid chlorides, chloroformates, or isocyanates; not shown in Fig. 7). The DKP products were then released from the solid support by cyclative cleavage, purified, and tested for FSH activity in vitro.
Fig. (7).

N-alkyl incorporation methods used by Guo et al. in solid-phase DKP synthesis.
Through analysis of these smaller libraries and further structural optimization, the Pharmacopeia researchers identified several potent DKP FSH receptor agonists. An initial hit (21) and the best agonist from each sublibrary (22–24) are shown in Fig. 8 with their corresponding EC50 values. This research underscores the utility of solid-phase synthesis for lead optimization in drug discovery.
Fig. (8).

Selected DKP follicle stimulating hormone receptor agonists identified at Pharmacopeia and their corresponding activities.
2.1.3. Ugi/De-Boc/Cyclize Route to DKPs
A synthetic route that introduces a tertiary amide bond, while circumventing the often problematic secondary acylation step required by many other syntheses, was reported by Affymax scientists in 1997 (see structure 9, Fig. 4) [47]. This solid-phase strategy utilized the well-studied Ugi four-component reaction (4CR) to simultaneously introduce three additional points of diversity onto resin-bound amine 25 (Fig. 9). This MCR route allowed for a cyclative cleavage when N-Boc-protected amino acid components were used (as in 26), releasing only the pure cyclic products (27). Scientists have used this versatile chemistry, termed the Ugi/De-Boc/Cyclize (UDC) method, to develop a range of lead compounds, including matrix metalloproteinase inhibitors [48], colleganse-1 inhibitors [49], and most recently, oxytocin antagonists [50].
Fig. (9).

The Ugi/De-Boc/Cyclize (UDC) method for solid-phase DKP synthesis.
Oxytocin (OT) is a cyclic nonamer peptide hormone responsible for smooth muscle contraction and preterm birth – the leading cause of infant morbidity and mortality [51]. Small molecule OT inhibitors have been reported; however, these compounds possess neither adequate in vivo potency nor appropriate pharmacokinetics to be progressed toward clinical trials. DKPs, synthesized at GlaxoSmithKline using the UDC method introduced above, were screened in an OT receptor binding assay and exhibited a range of activities as seen in Fig. 10. DKPs containing aromatic aldehyde and R-amino acid components showed the greatest potency. One drawback to this synthetic route, however, is the mixture of stereoisomers generated at the aldehyde position (28). Therefore, determining the activity of each diastereomer requires either separation after synthesis or an alternative stereoselective synthesis.
Through one such solution-phase, stereospecific DKP synthesis [52], the all R DKP stereoisomers (i.e., 30 and 31) were found to be the most potent OT antagonists. Additional solution-phase structure-activity studies of these compounds were pursued, and the authors identified a difluoro-benzyl DKP derivative with excellent pharmacokinetics and an IC50 value of 1.25 nM against OT [53, 54]. Preliminary animal model studies have recently shown that these DKP derivatives can indeed inhibit OT-induced and spontaneous contractions in late term pregnant rats, and clinical trials are on going [55]. This work provides further evidence of the value of DKPs as a new class of potent therapeutics.
Ono Pharmaceutical Co. reported another intriguing example of DKP synthesis in 2006 that further demonstrated the value of the DKP scaffold and MCR routes for their synthesis [56]. These researchers found that certain DKPs were potent inhibitors of HIV infection and replication. This work originated from a small molecule screen for chemokine antagonists, a class of inhibitors that could have implications as anti-inflammatory drugs, anti-allergic drugs, immunosuppressants, and/or anti-HIV drugs. The Ono scientists identified the novel spiro-DKP scaffold 32 to be structurally similar to the type I β-turn structure required for chemokine receptor binding, and therefor, a promising lead (33, Fig. 11). Thereafter, they developed a solid-phase UDC route that gave DKPs (36) in high purities (81–100%) following cyclative cleavage from the solid support. A library of 576 spiro-DKPs was synthesized via an Ugi 4CR with various amines, N-Boc-protected amino acids, and symmetric ketones on isonitrile-derivatized support 34, as depicted in Fig. 12.
Fig. (11).

Structures of spiro-DKP 32 and β-turn 33.
Fig. (12).

Solid-phase UDC method to spiro-DKPs.
The spiro-DKP library members were tested for inhibition of calcium mobilization in several human cell lines over expressing the chemokine receptor. Two active compounds were selected from the initial library, resynthesized in optically pure forms, and retested (e.g., DKP 37, Fig. 13). Because there were no significant differences in the activities of the enantiomers, subsequent spiro-DKPs were synthesized as racemates. These initial hits were optimized through the iterative evaluation of individual sites on the scaffold (R1, R2, R3; Fig. 13). An R1 library (38) of 80 derivatives was designed to explore the optimal substituent at the piperinal site through subsequent reductive amination and acylation steps. A second library (39), in which only the amine components were altered, revealed that n-butyl groups were significantly more active than the initial n-propyl groups. Eighty commercially available N-Boc-protected amino acids were then used to construct the R3 library (40). Finally, all three sites were examined simultaneously in a final 14-member library. These potent chemokine receptor antagonists were then tested for in vitro anti-HIV activities. Three of these compounds (41–43, shown in Fig. 13) were able to potently inhibit replication of various multidrug-resistant HIV-1 strains. These data support the hypothesis that potent chemokine receptor antagonists can effectively inhibit HIV proliferation, and further accentuate the versatility of the DKP as a privileged structure. A patent was filed in 2006 for the co-administration of such DKP compounds with the anti-HIV drug tipranavir in order to increase the efficacy of treatment [57].
Fig. (13).

Selected DKP HIV inhibitors identified by Ono Pharmaceutical Co. and their corresponding activities.
Kennedy et al. published another variation of solid-phase Ugi 4CRs in 2002 (Fig. 14) [58]. These researchers at Array BioPharma used the resin-bound “universal” isonitrile 44 as one of the components to generate over 4000 dipeptide derivatives (45). Treatment with base formed an N-acyloxazolidone intermediate 46, cleaving the Ugi product from the solid support. Sodium methoxide then cleaved this group to give the methyl ester analogue 47, and DKPs 48 were formed via cyclization under acidic conditions. Purification post-cleavage was accomplished using silica-based scavengers. Libraries of both DKPs and 1,4-benzodiaezepine-2,5-diones were constructed with an average mass recovery of 83% or 95%, respectively; however, building block structures and product purities were not reported. To the best of our knowledge, neither medicinal test results nor applications for these compounds have been published.
Fig. (14).

Synthesis used by Kennedy et al. to generate DKPs from a resin-bound universal isonitrile.
2.1. Silyl Linker Generates Cyclic Dipeptides Without Using a Tertiary Amide
In 2001, Wang et al. published the first solid-phase synthesis of Tryprostatin B, a novel DKP natural product that prevents the interaction of microtubule-associated proteins with the carboxy-terminal domain of tubulin, and is close in structure to spirotryprostatin B (Fig. 1) [59]. This solid-phase synthesis was accomplished using novel polymer-supported 2-(trialkylsilyl)ethanol linker 49 (Fig. 15). Cyclization of dipeptides from this linker under mild conditions (30% AcOH/MeOH at RT) gave DKPs 52 in 76–88% yield with greater than 99% purity. In contrast, traditional solid-phase supports (e.g., Wang-liner derivatized polystyrene beads) gave DKPs in good purities (80–95%) but with significantly lower yields (8–23%). This new silyl linker therefore represents an advance for solid-phase DKP synthesis, and could be utilized in the future to generate libraries of DKPs in good purities and yields. In addition, it can be accessed on gram scale in just three synthetic steps.
Fig. (15).

A silyl linker-based synthesis used at Hoffmann-La Roche to generate N-unsubstituted DKPs.
3. MW-Assisted Syntheses of DKPs
We now wish to provide a short review on the use of MW-assisted reactions in DKP synthesis. The majority of this work has been performed on structurally simple cyclic dipeptides. This area, similar to most in the MW field, is relatively young, emerging only six years ago. To the best of our knowledge, less than one dozen articles have been published on the use of MW irradiation in DKP synthesis. For this reason, we will review all reported examples of MW-assisted DKP synthesis here, including solvent-free, solution-, solid-, and fluorous-phase methods.
3.1. MW-Heated, Silica-Adsorbed DKP Reactions
In 2001, Pandey et al. reported the MW-assisted, stereo-specific synthesis of DKP natural product analogues on silica (Fig. 16). Compounds of this structure have cGMP-phosphodiesterase inhibitory activities and could have value in the treatment of impotence. Because N-Boc-protecting groups are unstable at elevated temperatures (>90°C), the researchers reasoned that MW-irradiation could deprotect the amine and facilitate spontaneous intramolecular aminolysis of 53 [60]. These silica-adsorbed reactions were complete after 5 min at 500 W in excellent yields (>85%). In contrast, the room temperature (RT) reactions using TFA gave lower yields of DKP products (60–75%). The authors used this fast, efficient, and “green” route to synthesize a set of five enantiomerically pure DKPs 54 with greater than 95% ee (Fig. 16).
Fig. (16).

MW heating method to DKPs from silica-adsorbed starting materials.
3.2. Solid-State, MW-Assisted DKP Synthesis
Due to the importance of the DKP scaffold, Lopez-Cobenas et al. sought to develop an improved, environmentally benign method for DKP synthesis. In 2005, they published both a communication and a full paper on their solvent-free, MW-assisted route to DKPs (Fig. 17) [61, 62]. Similar to the silica-based route of Pandey et al., this synthesis involved the one-pot deprotection and cyclization of N-Boc-protected dipeptide esters and improved reaction time, yield, and stereocenter integrity relative to the RT control reactions. As epimerization is the most frequent problem encountered in DKP synthesis, this route is potentially very beneficial. In this method, the reaction vessel (containing neat starting material 55) was buried in alumina powder to facilitate heating, and short MW pulses of 600 W were applied to the system (Fig. 17). The sequence of these MW pulses appeared to be critical; six 2-min pulses gave 66% yield, while a 3-min pulse followed by two 2-min pulses afforded 98% product. Conventional heating conditions were also tested and reactions took 2–5 h at 200°C (64–99%), giving a mixture of diastereomers in all cases (e.g., 1:1 mixture of diastereomers of cyclo(L-Ile-L-Ile)). In contrast, the short MW protocol gave improved stereochemical retention with only 10% epimerization of cyclo(L-Ile-L-Ile). Overall, this MW-assisted method gave the researchers fast and efficient access to a range of DKPs (i.e., 56 (Fig. 17).
Fig. (17).

Solvent-free dipeptide methyl ester cyclization strategy to DKPs.
3.3. Solution-Phase, MW-Assisted DKP Routes
In 2006, Tullberg et al. reported the solution-phase cyclization of 11 dipeptide methyl esters in various solvents and performed a systematic comparison of the yields obtained with conventional vs MW heating [63]. Their synthetic method differed from those previously discussed (Figs. 16,17), because it required the acid-mediated deprotection of the N-Boc protecting group prior to MW-assisted cyclization. Test reactions were run in 13 different solvents that were either refluxed for 12 h or irradiated with MWs for 10 min at temperatures 40°C above their boiling points. MW irradiation in water gave the best yields of all of the initial screening conditions (67%; compared to 38% in refluxing toluene). All 11 dipeptide methyl esters were then cyclized to DKPs under conventional and MW heating in four selected solvents (water, t-butanol, toluene/2-butanol, and toluene) and yields were reported for each condition (Tables 2 and 3). Based on these systematic studies, MW-assisted DKP cyclization in water appeared to be independent of amino acid sequence and gave good to excellent yields. The DKP products tended to be insoluble in water and were filtered to give DKPs with greater than 98% purity and no observable epimerization. The most striking data in this report was that for cyclo(L-Phe-L-Nle), which was produced in less than 10% yield by conventional heating in any solvent and less than 15% in the MW in non-aqueous solvents, yet was formed in 97% yield under MW irradiation in water.
Table 2. Structures of DKPs Synthesized in Solution by Tullberg et al. [63].
 | ||
|---|---|---|
| DKP | R1 | R2 |
| a | benzyl | (3-indolyl)-CH2 |
| b | (3-indolyl)-CH2 | CH2OH |
| c | (3-indolyl)-CH2 | CH2OBn |
| d | benzyl | butyl |
| e | benzyl | isobutyl |
| f | (3-indolyl)-CH2 | CH2CONH2 |
| g | L-isopropyl | butyl |
| h | D-isopropyl | butyl |
| i | H | butyl |
| j | isobutyl | 4-OH-benzyl |
| k | benzyl | __ |
Table 3. Comparison of Isolated Yields Obtained Through Conventional or MW-Assisted Heating in Various Solvents [63].
| DKP | Yield (%) Δa | Yield (%) MWb | ||||||
|---|---|---|---|---|---|---|---|---|
| H2O | Toluene/2-BuOH | Toluene | t-BuOH | H2O | Toluene/2-BuOH | Toluene | t-BuOH | |
| a | 41 | 8 | 20 | 62 | 73 | 14 | 18 | 10 |
| b | <5 | 9 | 5 | 41 | 81 | <5 | 10 | 4 |
| c | 57 | 6 | 5 | 10 | 70 | 6 | <5 | 12 |
| d | 5 | 5 | 10 | 10 | 97 | 12 | 12 | 15 |
| e | 7 | 32 | 24 | 37 | 68 | 17 | 14 | 24 |
| f | <5 | <5 | 7 | 10 | 71 | 21 | 7 | 6 |
| g | 7 | 5 | <5 | 8 | 63 | 15 | 8 | 8 |
| h | 35 | 7 | 22 | 41 | 84 | 12 | <5 | 6 |
| i | 33 | 51 | 29 | 88 | 70 | 10 | 14 | 10 |
| j | 28 | 23 | 12 | 37 | 83 | 5 | 12 | <5 |
| k | 89 | 88 | 83 | 93 | 93 | 73 | 70 | 76 |
Reactions were heated to reflux for 12 h.
Reactions were heated to 40°C above solvent boiling point for 10 min.
This same research group also published a MW-assisted DKP synthesis en route to the Schollkopf chiral auxiliaries. In this work, the Boc group of an N-protected dipeptide methyl ester was cleaved in 150°C water, and the intramolecular cyclization was reported to occur in only 15 min [64]. Reaction scale-up in 12 g batches was possible if the reaction mixture was heated to 200°C for 15 min in a sealed vessel. This scale-up method delivered cyclo(Gly-D-Val) in 73% yield. Overall, this MW-assisted, water-based methodology of Tullberg et al. represents an additional green and efficient route to DKPs [65].
Musiol and coworkers have recently reported the results of solution-phase, MW-assisted glycine condensations to form the simplest DKP, cyclo(Gly-Gly), containing neither side chains nor stereocenters [66]. Glycine was dissolved in ethylene glycol and irradiated in an open beaker for 3 min at 300 W. The resulting residue was poured into water and filtered, and the filtrate was then triturated with acetone. The resulting white precipitate was filtered and washed with acetone to afford the pure DKP. These reactions were run in an undergraduate chemistry laboratory, and the average student yield was reported to be 97%. These excellent results underscore the simplicity of these reactions and the utility of MW irradiation in their synthesis.
Cho et al. reported the optimization of solution-phase, MW-assisted Ugi 4CRs to generate DKPs. Their method made use of commercially available dipeptides (57) as bi-functional components and ionic liquids to increase MW heating (Fig. 18) [67]. In general, the coupling of these two technologies (MW heating and MCRs) represents a powerful method toward the rapid one-pot synthesis of diverse small molecules. RT conditions gave Ugi products in moderate to good yields (21–87%) in 33–52 h, depending on the isonitrile component involved. The slowest reacting isonitrile was then used to optimize MW reaction conditions, which doubled the yield of DKPs 58 (compared to conventional methods) in just 15 min (Fig. 18). Due to the poor solubility of the reagents in the ionic liquid, trifluoroethanol was added to the reaction mixture. Even though a 1:1 mixture of these solvents gave a 33% increase in DKP yield (60% vs 45%), trifluoroethanol alone produced similar results, making the ionic liquid unnecessary. As stated by the authors, MW-assisted MCRs are an “excellent tool for the library synthesis of interesting pharmacophores”.
Fig. (18).

MW-assisted, solution-phase Ugi four-component reaction to DKPs.
3.4. Solid-Phase, MW-Assisted Routes to DKPs
Our laboratory recently reported synthetic methods for constructing “macroarrays” of DKP products 61 [68]. This efficient DKP library synthesis involved MW-assisted reactions and a water-accelerated MCR on a planar cellulose support derivatized with a photocleavable linker (59, Fig. 19). The support was constructed and the initial building blocks were loaded using MW irradiation. The Ugi reaction was performed according to our previously reported method [69] with a few alterations: Armstrong's “universal” isonitrile was used as the isonitrile component [70], N-Fmoc-protected amino acids served as the carboxylic acid components, and reagents were spotted three times onto the array instead of once in order to achieve full conversion [68]. Interestingly, we found that MW irradiation did not improve the Ugi reaction conversion compared to RT conditions on our planar support system; instead, the use of water as a solvent at RT gave excellent conversions to the DKP precursors (60) (Fig. 19). Water has been recently reported to accelerate several solution-phase MCRs at RT, and in this work, we observed that these effects were also achievable on support-bound substrates [71].
Fig. (19).

Ugi four-component reactions involving Armstrong's “universal” isonitrile yield DKPs on planar support.
We found it was necessary to transform the “universal” isonitrile fragment into a methyl ester in order to achieve DKP cyclization on planar supports, and this methanolysis reaction worked best under MW irradiation (10–30 min at 80°C; Fig. 19). Piperidine-mediated RT cyclization, followed by photo-cleavage provided the desired DKP products in good purities (70% of the library was greater than 80% pure). This research demonstrated the compatibility of the small molecule macroarray platform with DKP synthesis. Furthermore, it showcased the use of both MW- and water-assisted reactions to generate arrays of diverse, biologically relevant, and spatially addressed compounds. Methods recently developed in our laboratory have allowed various biological screens to be performed directly on the macroarrays after compound cleavage [72]. Studies of the biological relevance of these DKP products (61) are on going.
Recently, Tullberg et al. reported the first “traditional” solid-phase synthesis of DKPs using MW-assisted conditions [73]. In this study, the researchers systematically compared the yields of DKPs synthesized on four different solid-supports (Fig. 20) and in four different solvents (water, t-butanol, toluene/2-butanol, and toluene) to determine the effect of the support on DKP formation. Dipeptides were constructed on the four ester-linked polymeric supports using standard coupling procedures, and cyclative cleavage similar to that shown in Fig. 15 afforded the final DKP products. The product yields for DKPs made by thermal heating at 80°C for 35 h were compared to those obtained after 30 min of MW irradiation at 120°C. Also, the DKPs formed in high yields (73–93%) on the PEG-grafted supports (ArgoGel MB OH and TentaGel S AC) in non-aqueous solvents independent of heating method. Indeed, conventional heating methods gave comparably high yields of all DKPs independent of resin and solvent, with the exception of water. Under MW irradiation, the yields of DKPs in water were low or moderate (45–73%) if less polar supports were utilized. In contrast, the polar PEGA-Ser beads gave high yields of DKPs (79–87%) in water under MW irradiation. Collectively, the results of this study show that MW heating methods can proffer a rapid and efficient route to DKPs relative to conventional heating methods.
Fig. (20).

Solid-supports used by Tullberg et al. to test the effects of the polymer matrix on DKP cyclization.
Scientists in the materials field have also utilized MW-assisted DKP formation reactions. Gupta et al. have generated hetero and homo spiro ladder oligomers with controlled three-dimensional structures (Fig. 21) for future biomimetic applications [74]. These nanoscale ladders were built in two steps from cyclic monomers by (1) coupling the bis-amino acid monomers 62 and 63 on solid-phase, and (2) rigidifying the oligomer through simultaneous DKP formation to yield 64 or 65. The researchers sought conditions for rapid and pure DKP formation without side products. Because DKP stereocenters are sensitive to basic conditions, the researchers were unable to use 20% piperidine at elevated temperatures (RT reactions give minor to undetectable amounts of product). Instead, the researchers developed an acid-catalyzed, MW-assisted route to these complex structures. They were able to achieve fast DKP formation with no epimerization at 130°C in the MW for 30 min in an acetic acid/triethylamime solution. In view of the challenges of DKP synthesis, the work of Gupta et al. represents an excellent example of the utility of MW-assisted reactions for DKP formation.
Fig. (21).

Spiro-DKP oligomers synthesized with MW heating by Gupta et al.
3.5. Fluorous-Phase, MW-Heated DKP Synthesis
Zhang et al. recently reported another unique approach to DKPs in which they coupled MW-assisted, fluorous MCRs with solid-phase extractions [75]. Fluorous, bicyclic proline derivatives 66 were generated via a MW-assisted, one-pot, three-component [3+2] cycloaddition of azomethine ylides in good yields and purities following fluorous solid-phase extraction (F-SPE, 130°C for 20 min; 75–90% yields, >90% purities; Fig. 22). These starting materials then were converted to one of three structure classes, including DKPs 69. Efforts to directly acylate 66 with α-amino acids and α-amino chlorides were unsuccessful, and thus the authors used chloroacetyl chloride to generate 67 (RT, 30 min) followed by amination (MW, 120°C, 10 min) to set up the cyclization. MW irradiation of 68 at 180°C for 15 min affected the cyclization and cleavage of the fluorous tag to give the DKP 69 in 45% yield. Although this yield was not optimal, the product was isolated as a single stereoisomer, with all stereocenters being set in the cycloaddition step, and contained four points of diversity. The combination of MW assistance and fluorous-tagged reagents greatly accelerated reaction times and simplified product purification, respectively. The authors also claim that their “solution-phase” strategy required less development time than corresponding solid-phase routes.
Fig. (22).

Fluorous-phase synthesis of DKPs using MW irradiation.
4. Routes to DKPs that could Potentially Benefit from MWs
As stated previously, solid-phase reactions often suffer from slow reaction kinetics, and MW heating has been shown to greatly increase these reaction rates. Indeed, MW reactors have been touted to become the “Bunsen burners of the 21st century” because of the rapid heating and increased yields and purities obtained by their use [76]. Here, we analyze a selected set of solid-phase DKP syntheses that potentially could benefit from the use of MW-heating, in an attempt to further support this view. This critique is only meant to illustrate that there are many more future opportunities for the incorporation of MW-assisted reactions into synthetic routes toward DKPs, and does not reflect our opinion of the science behind these reports. We review one key reaction at the outset in this section, esterification, and then examine four articles as case studies in chronological order.
4.1. MW-Assisted Solid-Phase Esterification Protocols
As most solid-phase DKP syntheses rely on a cyclative cleavage, the majority of these routes begin with an ester linkage to the solid support. However, it is surprising how few MW-assisted ester-loading conditions have been reported in the literature, and to our knowledge, these reactions are yet to be applied in DKP synthesis. We will discuss three reported strategies here, all of which were performed by the Kappe laboratory and are shown in Table 4. The first method consists of the acetoacetylation of a hydroxyl-based polystyrene support with various β-ketoesters (entry 1) [77]. This reaction proceeds through a highly reactive α-oxoketene intermediate, which is trapped by the resin-bound hydroxyl group. Even though this esterification is an equilibrium, the reaction can be pushed to completion by simply boiling off the resulting alcohol. Strohmeier and Kappe tested a number of β-ketoesters and found (by FTIR of the resulting resin) that greater than 99% loading was achieved after only 1–10 min of MW irradiation at 170°C (Table 4, entry 1).
Table 4. MW-Assisted Ester Loading Conditions.
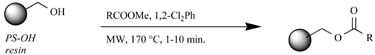


A second ester loading method reported by Stadler and Kappe is based on traditional carbodiimide couplings (Table 4, entry 2). The authors compared results obtained using an activated urea and a symmetric anhydride [78]. Conventional RT reaction conditions gave complete conversion after 48–72 h and seemed “poised” to benefit from MW assistance. However, they found that the one-pot, activated urea coupling procedure was not suited for rate-enhancements by MW heating because the rearranged, inactive urea was the major product formed under MW irradiation. In contrast, the symmetric anhydride protocol gives quantitative loadings onto Wang-linker derived support within 20 min at 150°C or 10 min at 200°C (Table 4, entry 2).
The third and most recent MW-assisted ester-loading method was also reported by Stadler and Kappe in 2001 (Table 4, entry 3) [79]. This protocol involves the coupling of support-bound alkyl halides with cesium carboxylate salts, and typically requires 16–48 h of heating at 50–80°C. The researchers tested the loading of 33 aromatic, heterocyclic, and alkyl carboxylates onto chloro-Wang derived beads under conventional and MW-assisted conditions. They found that their MW conditions gave at least 85% loading after only 15 min of heating for all of the acids tested. On the other hand, even with larger excess of reagents, the addition of KI as a catalyst, and a 48-h reaction time, none of the thermal yields matched the MW-assisted yields.
To the best of our knowledge, no reaction conditions have been published for the MW-assisted ester loading of α-amino acids. Our laboratory, however, has been able to generate ester linkages onto Wang linker-derivatized and hydroxymethyl polystyrene supports in the MW after just 20 min at 60°C using DIC, HOBt, DMAP and DIPEA in DMF. The Fmoc-AA-OH loadings obtained using MW heating are comparable to overnight, RT 1-(2-Mesitylenesulfonyl)-3-nitro-1H-1,2,4-triazole (MSNT) coupling procedures (73 vs 79%, respectively; unpublished results). These MW-assisted ester loading reaction conditions, along with the methods of Kappe and co-workers (Table 4), could be utilized in many of the DKP syntheses reviewed herein to accelerate the initial support-loading step.
4.2. Case Study #1
In 2000, Lin et al. published a straightforward synthesis of tri-substituted DKPs that used Fukuyama's sulfonamide to generate secondary amines on solid support (Fig. 23) [80]. This was accomplished through protection of amine 70 with 2-nitrobenzene-sulfonamide, which activated the amine nitrogen of 71 for a Mitsunobu reaction with primary alcohols to generate 72 (yields >90%). Although this loading sequence was general and high yielding, the Mitsunobu reaction was run at RT for 15 h. Examples of similar solid-phase Mitsunobu reactions have been reported to occur quite cleanly in only 10 min under MW irradiation [81]. Such MW-assisted conditions could have been advantageous in this synthesis.
Fig. (23).

Solid-phase reaction scheme used by Lin et al. to synthesize tri-substituted DKPs.
Another step in this synthesis that may have benefited from MW heating is the bromoacetic acid coupling to secondary amine 73, which was performed at RT for 15 h (Fig. 23). Our laboratory recently published MW-assisted acylation procedures for support-bound secondary amines in the context of N-substituted glycine oligomers, or peptoids [82]. These reactions are typically complete after only 30 sec of MW irradiation. The subsequent amination step to give 74 was also performed at RT and could also benefit from MW assistance. We have also optimized MW-assisted amination conditions for both sterically demanding and electron withdrawing amines (90 sec of MW heating at 95°C [82]). The final steps of this synthesis included TFA-mediated linker cleavage and the heating of linear dipeptides 75 for 2 h to facilitate cyclization to 76 in solution. This method gave a mixture of both cyclized and uncyclized materials. Again here, we speculate that, due to the ester linkage and the flexible tertiary amide bond, DKP formation could occur quickly during the amination step and yield highly pure products under MW irradiation.
4.3. Case Study #2
Golebiowski et al. have published another DKP synthesis that could be accelerated by MW assistance (Fig. 24) [83, 84]. In 2002, they reported the solid-phase synthesis of β-turn mimics using Ugi 4CRs and several post-condensation reactions. The researchers were interested in β-turn mimics because this structural motif is present in many biologically active peptides and mimics could be used to disrupt, or potentially map, protein-protein interactions. Even though the Ugi 4CR gave a 1:1 mixture of diastereomers, this synthetic route allowed for the control of the remaining three stereo-centers. The Ugi 4CR occurred at RT (building blocks: α-N-Boc-diaminopropionic ester resin 77, α-bromoacid, aldehyde, and isocyanide) in 4 h to yield 78. We reason that this reaction potentially could be accomplished in a MW reactor in less than 5 min according to procedures by Hoel and Nielsen [85].
Fig. (24).

Solid-phase route to DKP β-turn mimics.
The first ring closure in this synthesis occurred with the displacement of a secondary bromide by a primary amine to give 79 (Fig. 24). The procedure called for an 18 h RT reaction, but similar reactions are complete in the MW in 90 sec, as described above in Case study #1 [82]. The N-acylation with an α-amino acid could also have been achieved under MW irradiation in 1.5–20 min [86]. The final DKP products 80 were made via cyclative cleavage from the solid support in 2 M AcOH over 18 h at 60°C in 50–90% purity. Once again, we hypothesize that MW heating could have accelerated this step as reported by Tullberg et al. (MW, 30 min, 120°C) [73].
4.4. Case Study #3
In 2003, researchers from Ontogen reported the inhibitory activities of several DKPs against hormone-sensitive lipase (HSL) [87]. HSL inhibition has the potential to reduce the levels of fatty acids in the blood, and serves as a target against obesity or diabetes. The Ontogen researchers performed a high-throughput screen of over 100,000 compounds to identify initial lead inhibitors. These primary screens revealed that certain DKPs had submicromolar activities against HSL.
The researchers generated a library of functionalized DKPs using a synthesis that could have involved numerous MW-assisted reactions. This synthetic route started with pyrrolidines 82 made via a 1,3-dipolar cycloaddition of acrylates and resin-bound azomethine ylides (Fig. 25). The Ontogen researchers performed this reaction in two steps, each at RT for 15 h. However, they could have adapted procedures from Zhang and Chen [88], conducting a one-pot, three-component cycloaddition under MW irradiation in just 15 min. As stated previously, their bromoacetic acid coupling with DIC could have been performed under MW heating in 30 sec as opposed to their 16-h RT procedure, which gave incomplete coupling and greatly reduced their overall yields (12–40%). Likewise, their amination to 83 could have taken 90 sec using MW heating instead of 3 h at RT. Lastly, the researchers performed their cyclization to products 84 at 110°C for three days – again, MW heating may have reduced the reaction time to less than 30 min [73]. Notably, the overall reaction time for the published synthesis was 121 h. We speculate that with the application of MW heating, this potentially could have been shortened to 32 min.
Fig. (25).

Solid-phase synthetic route to hormone-sensitive lipase inhibiting DKPs.
4.5. Case Study #4
Another DKP route that may be amenable to MW heating was published last year by Kuster et al. (Fig. 26) [89]. These researchers synthesized a 56-member library of spiro-2,5-DKPs on solid-phase, and the key step of their synthesis was a high pressure Diels-Alder reaction. The overall yield of the reported synthesis was excellent, giving as much as 68% after eight steps. However, the route was hampered by long reaction times, totaling over 88 h. The first step in the route was the coupling of nitroacetic acid to Wang-derivatized support 85 with DIC and HOBt at RT for 18 h. As discussed above, by applying the methods of Kappe and coworkers, the time required for this esterification reaction could have been reduced to less than 20 min using MW heating [77-79].
Fig. (26).

Solid-phase spiro-DKP synthesis reported by Kuster et al. in 2006.
In the second step, heating the support-bound nitro acetate and Schiff base with acetic anhydride for 24 h generated nitroalkene 87 (Fig. 26); however, solid-phase MW-assisted conditions developed by Strohmeier and Kappe may have been used to accomplish the same reaction in 30–60 min [77]. The key Diels-Alder cycloaddition was then conducted at elevated pressure and RT for over 16 h to give intermediate 88. However, MW heating in sealed tubes has been used to facilitate similar Diels-Alder reactions on solid-phase in less than 40 min [90]. Following a RT reduction of 88, the published protocol calls for a 4-h PyBOP-mediated amino acid coupling at RT. Again this amidation potentially could have been accomplished in 20 min in a MW reactor with the same reagents, or in 1.5 min using HATU [86]. The reduction of the nitroalkane 88 to amine 89 is reported to take 22 h and, to the best of our knowledge, no solid-phase MW-assisted conditions have been published for this transformation. This reduction, and many other reactions, remains to be optimized on solid support under MW irradiation; thus, further research in this field is of vital importance. Finally, the cyclative cleavage step to afford product 92 was performed in 5% acetic acid after 4 h at reflux. Again, as discussed herein, similar reactions have been reported to occur in 30 min under MW irradiation and could be exploited in this synthesis [73].
Conclusions and Outlook
DKPs have emerged as a promising class of heterocycles for the development of small molecule tools and therapeutic agents. Recent advances in solid-phase combinatorial DKP synthesis, including secondary acylation and N-alkylation strategies and MCRs, have provided the tools needed for the rapid synthesis of DKPs. The application of MW-assisted reactions is only further expediting DKP synthesis. Improved access to this important class of molecules should impact the development of potential therapeutics for a range of biological targets. Examples highlighted herein illustrate that DKPs are viable scaffolds for the treatment of a wide range of diseases and biological phenomena, including asthma (tryptase inhibitors), infertility (FSH receptor agonists), premature labor (oxytocin receptor inhibitors), and HIV (chemokine receptor antagonists). We anticipate that the strategic combination of SPOS and MW heating methods will continue to impact the pace and outcomes of future DKP research.
Acknowledgments
We thank the NIH (AI063326-01), Burroughs Welcome Foundation, Johnson & Johnson, DuPont, and Greater Milwaukee Foundation Shaw Scientist Program for support of DKP research in our laboratory. H.E.B. is an Alfred P. Sloan Foundation Fellow, a Camille Dreyfus Teacher-Scholar, and a Research Corporation Cottrell Scholar. J.C.O. is currently supported by a Novartis Graduate Fellowship in Organic Chemistry. Sarah Fowler, Benjamin Gorske, Megan Jacobson, Qi Lin, and Christine McInnis are acknowledged for their critical review of this manuscript.
Abbreviations
- Ac2O
Acetic anhydride
- AcCl
Acetyl chloride
- AcCN
Acetonitrile
- AcOH
Acetic acid
- Arg
Arginine
- Asp
Aspartate
- Boc
Tert-butoxycarbonyl
- BrAcOH
Bromo acetic acid
- CCR5
Chemokine receptor 5
- cGMP
Guanosine 3′,5′-cyclic monophosphate
- DBU
1,8-Diazabicyclo[5.4.0]undec-7-ene
- DIAD
Diisopropyl azodicarboxylate
- DIC
N,N′-diisopropylcarbodiimide
- DIPEA
N,N-diisopropylethylamine
- DKP
Diketopiperazine
- DMAP
4-(Dimethylamino)pyridine
- DMF
N,N-dimethylformamide
- DMSO
Dimethyl sulfoxide
- EC50
Compound concentration that gives half of its maximal effect
- ee
Enantiomeric excess
- Fmoc
(N-(9-fluorenyl) methoxycarbonyl)
- FSH
Follicle stimulating hormone
- F-SPE
Fluorous solid-phase extraction
- Gly
Glycine
- h
Hours
- HATU
O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HIV
Human immunodeficiency virus
- HOBt
1-Hydroxybenzotriazole
- HPLC
High performance liquid chromatography
- HSL
Hormone-sensitive lipase
- IC50
Compound concentration that gives half of its maximal inhibition
- iPrOH
Isopropanol
- KOtBu
Potassium tert-butoxide
- Leu
Leucine
- Lys
Lysine
- MCR
Multiple component reaction
- MePh
Toluene
- MeOH
Methanol
- min
Minutes
- MSNT
1-(2-Mesitylenesulfonyl)-3-nitro-1H-1,2,4-triazole
- MW
Microwave
- NaOMe
Sodium methoxide
- Nle
Norleucine
- NMM
N-methyl morpholine
- NMP
1-Methyl-2-pyrrolidinone
- NO2AcOH
Nitro-acetic acid
- OT
Oxytocin
- PEG
Polyethylene glycol
- Phe
Phenylalanine
- Pro
Proline
- PyBOP
(Benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate
- RT
Room temperature
- SPOS
Solid-phase organic synthesis
- TEA
Triethylamine
- TFA
Trifluoroacetic acid
- TFAA
Trifluoroacetic anhydride
- THF
Tetrahydrofuran
- UDC
Ugi/De-Boc/Cyclize
- Val
Valine
- W
Watts
References
- 1.Kramer R, Cohen D. Nat Rev Drug Discov. 2004;3:965. doi: 10.1038/nrd1552. [DOI] [PubMed] [Google Scholar]
- 2.Burbaum J, Tobal GM. Curr Opin Chem Biol. 2002;6:427. doi: 10.1016/s1367-5931(02)00337-x. [DOI] [PubMed] [Google Scholar]
- 3.Blackwell HE. Org Biomol Chem. 2003;1:1251. doi: 10.1039/b301432k. [DOI] [PubMed] [Google Scholar]
- 4.Ley SV, Baxendale IR. Nat Rev Drug Discov. 2002;1:573. doi: 10.1038/nrd871. [DOI] [PubMed] [Google Scholar]
- 5.Groth T, Grotli M, Meldal M. J Comb Chem. 2001;3:461. doi: 10.1021/cc000106q. [DOI] [PubMed] [Google Scholar]
- 6.Kappe CO. Angew Chem Int Edit. 2004;43:6250. doi: 10.1002/anie.200400655. [DOI] [PubMed] [Google Scholar]
- 7.Bailey DS, Brown D. Drug Discov Today. 2001;6:57. doi: 10.1016/s1359-6446(00)01596-8. [DOI] [PubMed] [Google Scholar]
- 8.Horton DA, Bourne GT, Smythe ML. Mol Divers. 2000;5:289. doi: 10.1023/a:1021365402751. [DOI] [PubMed] [Google Scholar]
- 9.Prasad C. Peptides. 1995;16:151. doi: 10.1016/0196-9781(94)00017-z. [DOI] [PubMed] [Google Scholar]
- 10.Fdhila F, Vazquez V, Sanchez JL, Riguera R. J Nat Prod. 2003;66:1299. doi: 10.1021/np030233e. [DOI] [PubMed] [Google Scholar]
- 11.Nicholson B, Lloyd GK, Miller BR, Palladino MA, Kiso Y, Hayashi Y, Neuteboom STC. Anti-Cancer Drug. 2006;17:25. doi: 10.1097/01.cad.0000182745.01612.8a. [DOI] [PubMed] [Google Scholar]
- 12.McCleland K, Milne PJ, Lucieto FR, Frost C, Brauns SC, Van De Venter M, Du Plessis J, Dyason K. J Pharm Pharmacol. 2004;56:1143. doi: 10.1211/0022357044139. [DOI] [PubMed] [Google Scholar]
- 13.Cui CB, Kakeya H, Osada H. J Antibiot. 1996;49:832. doi: 10.7164/antibiotics.49.832. [DOI] [PubMed] [Google Scholar]
- 14.Cui CB, Kakeya H, Osada H. Tetrahedron. 1996;52:12651. [Google Scholar]
- 15.Edmondson S, Danishefsky SJ. Angew Chem Int Edit. 1998;37:1138. doi: 10.1002/(SICI)1521-3773(19980504)37:8<1138::AID-ANIE1138>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 16.Edmondson S, Danishefsky SJ, Sepp-Lorenzino L, Rosen N. J Am Chem Soc. 1999;121:2147. [Google Scholar]
- 17.Bryans J, Charlton P, Chicarelli-Robinson I, Collins M, Faint R, Latham C, Shaw I, Trew S. J Antibiot. 1996;49:1014. doi: 10.7164/antibiotics.49.1014. [DOI] [PubMed] [Google Scholar]
- 18.Folkes A, Roe MB, Sohal S, Golec J, Faint R, Brooks T, Charlton P. Bioorg Med Chem Lett. 2001;11:2589. doi: 10.1016/s0960-894x(01)00508-x. [DOI] [PubMed] [Google Scholar]
- 19.Einholm AP, Pedersen KE, Wind T, Kulig P, Overgaard MT, Jensen JK, Bodker JS, Christensen A, Charlton P, Andreasen PA. Biochem J. 2003;373:723. doi: 10.1042/BJ20021880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyoshi T, Miyairi N, Aoki H, Kohsaka M, Sakai H, Imanaka H. J Antibiot. 1972;25:569. doi: 10.7164/antibiotics.25.569. [DOI] [PubMed] [Google Scholar]
- 21.Nishida M, Mine Y, Matsubara T, Goto S, Kuwahara S. J Antibiot. 1972;25:582. [PubMed] [Google Scholar]
- 22.Kimura Y, Sawada A, Kuramata M, Kusano M, Fujioka S, Kawano T, Shimada A. J Nat Prod. 2005;68:237. doi: 10.1021/np040178p. [DOI] [PubMed] [Google Scholar]
- 23.Li GY, Li BG, Yang T, Yan JF, Liu GY, Zhang GL. J Nat Prod. 2006;69:1374. doi: 10.1021/np0602970. [DOI] [PubMed] [Google Scholar]
- 24.Yanagihara M, Sasaki-Takahasi N, Sugahara T, Yamamoto S, Shinomi M, Yamashita L, Hayashida M, Yamanoha B, Numata A, Yamori T, Andoh T. Cancer Sci. 2005;96:816. doi: 10.1111/j.1349-7006.2005.00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsumagari N, Nakai R, Onodera H, Hasegawa A, Rahayu E, Ando K, Yamashita Y. J Antibiot. 2004;57:532. doi: 10.7164/antibiotics.57.532. [DOI] [PubMed] [Google Scholar]
- 26.Kanoh K, Kohno S, Asari T, Harada T, Katada J, Muramatsu M, Kewashima H, Sekiya H, Uno I. Bioorg Med Chem Lett. 1997;7:2847. [Google Scholar]
- 27.Hantke K. Mol Gen Genet. 1983;191:301. doi: 10.1007/BF00334830. [DOI] [PubMed] [Google Scholar]
- 28.Cole R, Kirksey J, Moore J, Blankenship B, Diener U, Davis N. Appl Microbiol. 1972;24:248. doi: 10.1128/am.24.2.248-250.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kohn H, Widger W. Curr Drug Targets. 2005;5:273. doi: 10.2174/1568005054880136. [DOI] [PubMed] [Google Scholar]
- 30.Lambert JN, Mitchell JP, Roberts KD. J Chem Soc Perk T 1. 2001:471. [Google Scholar]
- 31.Dinsmore CJ, Beshore DC. Tetrahedron. 2002;58:3297. [Google Scholar]
- 32.Fischer PM. J Pept Sci. 2003;9:9. doi: 10.1002/psc.446. [DOI] [PubMed] [Google Scholar]
- 33.Bonauer C, Walenzyk T, Konig B. Synthesis. 2006:1. [Google Scholar]
- 34.Li WR, Yang JH. J Comb Chem. 2002;4:106. doi: 10.1021/cc010055c. [DOI] [PubMed] [Google Scholar]
- 35.Couladouros EA, Magos AD. Mol Divers. 2005;9:111. doi: 10.1007/s11030-005-1295-9. [DOI] [PubMed] [Google Scholar]
- 36.Curtius T, Goebel F. J Prakt Chem. 1888;37:150. [Google Scholar]
- 37.Fischer E. Ber. 1906;39:530. [Google Scholar]
- 38.Nitecki DE, Halpern B, Westley JW. J Org Chem. 1968;33:864. doi: 10.1021/jo01266a091. [DOI] [PubMed] [Google Scholar]
- 39.Gisin BF, Merrifield RB. J Am Chem Soc. 1972;94:3102. doi: 10.1021/ja00764a036. [DOI] [PubMed] [Google Scholar]
- 40.Jensen KJ, Alsina J, Songster MF, Vagner J, Albericio F, Barany G. J Am Chem Soc. 1998;120:5441. [Google Scholar]
- 41.del Fresno M, Alsina J, Royo M, Barany G, Albericio F. Tetrahedron Lett. 1998;39:2639. [Google Scholar]
- 42.del Fresno M, Fernandez-Forner DB, Miralpeix M, Segarra V, Ryder H, Royo M, Albericio F. Bioorg Med Chem Lett. 2005;15:1659. doi: 10.1016/j.bmcl.2005.01.048. [DOI] [PubMed] [Google Scholar]
- 43.Gordon DW, Steele J. Bioorg Med Chem Lett. 1995;5:47. [Google Scholar]
- 44.Gong X, Yang XX, Wang DX. Chinese Chem Lett. 2006;17:469. [Google Scholar]
- 45.Guo T, Adang AEP, Dolle RE, Dong G, Fitzpatrick D, Geng P, Ho KK, Kultgen SG, Liu R, McDonald E, McGuinness BF, Saionz KW, Valenzano KJ, van Straten NCR, Xie D, Webb ML. Bioorg Med Chem Lett. 2004;14:1713. doi: 10.1016/j.bmcl.2004.01.042. [DOI] [PubMed] [Google Scholar]
- 46.Guo T, Adang AEP, Dong G, Fitzpatrick D, Geng P, Ho KK, Jibilian CH, Kultgen SG, Liu R, McDonald E, Saionz KW, Valenzano KJ, van Straten NCR, Xie D, Webb ML. Bioorg Med Chem Lett. 2004;14:1717. doi: 10.1016/j.bmcl.2004.01.043. [DOI] [PubMed] [Google Scholar]
- 47.Szardenings AK, Burkoth TS, Lu HH, Tien DW, Campbell DA. Tetrahedron. 1997;53:6573. [Google Scholar]
- 48.Szardenings AK, Harris D, Lam S, Shi LH, Tien D, Wang YW, Patel DV, Navre M, Campbell DA. J Med Chem. 1998;41:2194. doi: 10.1021/jm980133j. [DOI] [PubMed] [Google Scholar]
- 49.Szardenings AK, Antonenko V, Campbell DA, DeFrancisco N, Ida S, Shi LH, Sharkov N, Tien D, Wang YW, Navre M. J Med Chem. 1999;42:1348. doi: 10.1021/jm980475p. [DOI] [PubMed] [Google Scholar]
- 50.Wyatt PG, Allen MJ, Borthwick AD, Davies DE, Exall AM, Hatley RJD, Irving WR, Livermore DG, Miller ND, Nerozzi F, Sollis SL, Szardenings AK. Bioorg Med Chem Lett. 2005;15:2579. doi: 10.1016/j.bmcl.2005.03.045. [DOI] [PubMed] [Google Scholar]
- 51.Mitchell BF, Schmid B. J Soc Gynecol Invest. 2001;8:122. [PubMed] [Google Scholar]
- 52.Sollis SL. J Org Chem. 2005;70:4735. doi: 10.1021/jo0501137. [DOI] [PubMed] [Google Scholar]
- 53.Borthwick AD, Davies DE, Exall AM, Livermore DG, Sollis SL, Nerozzi F, Allen MJ, Perren M, Shabbir SS, Woollard PM, Wyatt PG. J Med Chem. 2005;48:6956. doi: 10.1021/jm050557v. [DOI] [PubMed] [Google Scholar]
- 54.Borthwick AD, Davies DE, Exall AM, Hatley RJD, Hughes JA, Irving WR, Livermore DG, Sollis SL, Nerozzi F, Valko KL, Allen MJ, Perren M, Shabbir SS, Woollard PM, Price MA. J Med Chem. 2006;49:4159. doi: 10.1021/jm060073e. [DOI] [PubMed] [Google Scholar]
- 55.McCafferty GP, Pullen MA, Wu C, Edwards RM, Allen MJ, Woollard PM, Borthwick AD, Liddle J, Hickey DMB, Brooks DP, Westfall TD. Am J Physiol Regul Integr Comp Physiol. 2007 doi: 10.1152/ajpregu.00057.2007. [DOI] [PubMed] [Google Scholar]
- 56.Habashita H, Kokubo M, Hamono SI, Hamanaka N, Toda M, Shibayama S, Tada H, Sagawa K, Fukushima D, Maeda K, Mitsuya H. J Med Chem. 2006;49:4140. doi: 10.1021/jm060051s. [DOI] [PubMed] [Google Scholar]
- 57.Kraft MF, Mayers DL. Method for treating HIV infection through co-administration of tipranavir and GW873140. USA: 2005. [Google Scholar]
- 58.Kennedy AL, Fryer AM, Josey JA. Org Lett. 2002;4:1167. doi: 10.1021/ol0256015. [DOI] [PubMed] [Google Scholar]
- 59.Wang B, Chen L, Kim K. Tetrahedron Lett. 2001;42:1463. [Google Scholar]
- 60.Pandey SK, Awasthi KK, Saxena AK. Tetrahedron. 2001;57:4437. [Google Scholar]
- 61.Lopez-Cobenas A, Cledera P, Sanchez JD, Perez-Contreras R, Lopez-Alvarado P, Ramos MT, Avendano C, Menendez JC. Synlett. 2005;7 [Google Scholar]
- 62.Lopez-Cobenas A, Cledera P, Sanchez JD, Lopez-Alvarado P, Ramos MT, Avendano C, Menendez JC. Synthesis. 2005;19:3412. [Google Scholar]
- 63.Tullberg M, Grotli M, Luthman K. Tetrahedron. 2006;62:7484. [Google Scholar]
- 64.Carlsson AC, Jam F, Tullberg M, Pilotti A, Ioannidis P, Luthman K, Grotli M. Tetrahedron Lett. 2007;47:5199. [Google Scholar]
- 65.Dallinger D, Kappe CO. Chem Rev. 2007 doi: 10.1021/cr0509410. [DOI] [PubMed] [Google Scholar]
- 66.Musiol R, Tyman-Szram B, Polanski J. J Chem Educ. 2006;83:632. [Google Scholar]
- 67.Cho S, Keum G, Kang SB, Han SY, Kim Y. Mol Divers. 2003;6:283. doi: 10.1023/b:modi.0000006812.16141.b5. [DOI] [PubMed] [Google Scholar]
- 68.Lin Q, Blackwell HE. Chem Commun. 2006:2884. doi: 10.1039/b604329a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin Q, O'Neill JC, Blackwell HE. Org Lett. 2005;7:4455. doi: 10.1021/ol051684o. [DOI] [PubMed] [Google Scholar]
- 70.Keating TA, Armstrong RW. J Am Chem Soc. 1995;117:7842. [Google Scholar]
- 71.Pirrung MC, Das Sarma K. J Am Chem Soc. 2004;126:444. doi: 10.1021/ja038583a. [DOI] [PubMed] [Google Scholar]
- 72.Bowman MD, O'Neill JC, Stringer JR. Chem Biol. 2007;14:351. doi: 10.1016/j.chembiol.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 73.Tullberg M, Luthman K, Grotli M. J Comb Chem. 2006;8:915. doi: 10.1021/cc0600876. [DOI] [PubMed] [Google Scholar]
- 74.Gupta S, Macala M, Schafmeister CE. J Org Chem. 2006;71:8691. doi: 10.1021/jo0609125. [DOI] [PubMed] [Google Scholar]
- 75.Zhang W, Lu Y, Chen CHT, Curran DP, Geib S. Eur J Org Chem. 2006:2055. doi: 10.1002/ejoc.200600077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bose A, Banik B, Lavlinskaia N, Jayaraman M, Manhas M. Chemtech. 1997;27:18. [Google Scholar]
- 77.Strohmeier GA, Kappe CO. J Comb Chem. 2002;4:154. doi: 10.1021/cc010043r. [DOI] [PubMed] [Google Scholar]
- 78.Stadler A, Kappe CO. Tetrahedron. 2001;57:3915. [Google Scholar]
- 79.Stadler A, Kappe CO. Eur J Org Chem. 2001:919. [Google Scholar]
- 80.Lin X, Dorr H, Nuss JM. Tetrahedron Lett. 2000;41:3309. [Google Scholar]
- 81.Lampariello LR, Piras D, Rodriques M, Taddei M. J Org Chem. 2003;68:7893. doi: 10.1021/jo034785d. [DOI] [PubMed] [Google Scholar]
- 82.Gorske BC, Jewell SA, Guerard EJ, Blackwell HE. Org Lett. 2005;7:1521. doi: 10.1021/ol0502984. [DOI] [PubMed] [Google Scholar]
- 83.Golebiowski A, Jozwik J, Klopfenstein SR, Colson AO, Grieb AL, Russell AF, Rastogi VL, Diven CF, Portlock DE, Chen JJ. J Comb Chem. 2002;4:584. doi: 10.1021/cc020029u. [DOI] [PubMed] [Google Scholar]
- 84.Golebiowski A, Klopfenstein SR, Shao X, Chen JJ, Colson AO, Grieb AL, Russell AF. Org Lett. 2000;2:2615. doi: 10.1021/ol006145s. [DOI] [PubMed] [Google Scholar]
- 85.Hoel AML, Nielsen J. Tetrahedron Lett. 1999;40:3941. [Google Scholar]
- 86.Erdelyi M, Gogoll A. Synthesis. 2002;11:1592. [Google Scholar]
- 87.Slee DH, Bhat AS, Nguyen TN, Kish M, Lundeen K, Newman MJ, McConnell SJ. J Med Chem. 2003;46:1120. doi: 10.1021/jm020460y. [DOI] [PubMed] [Google Scholar]
- 88.Zhang W, Chen CHT. Tetrahedron Lett. 2005;46:1807. [Google Scholar]
- 89.Kuster GJT, van Berkom LWA, Kalmoua M, van Loevezijn A, Sliedregt LAJM, van Steen BJ, Kruse CG, Rutjes FPJT, Scheeren HW. J Comb Chem. 2006;8:85. doi: 10.1021/cc050072s. [DOI] [PubMed] [Google Scholar]
- 90.Kaval N, Van der Eycken J, Caroen J, Dehaen W, Strohmeier GA, Kappe CO, Van der Eycken E. J Comb Chem. 2003;5:560. [Google Scholar]