

Abstract
Inborn errors of metabolism (IEM) account for only 5% of all pediatric cardiomyopathy and 15% of those with known causes, but they are of particular interest to clinicians because many have disease-specific treatments. More than 40 different IEM involving cardiomyopathy exist, including fatty acid oxidation defects, organic acidemias, amino acidopathies, glycogen storage diseases, and congenital disorders of glycosylation as well as peroxisomal, mitochondrial, and lysosomal storage disorders. Most IEM present in infancy or early childhood with signs and symptoms of multi-organ system dysfunction. Except for mitochondrial disorders, each IEM is generally associated with one functional type of cardiomyopathy by echocardiography. Disease pathophysiology may include infiltration of cardiac myocytes with stored substrate, impaired energy production, and/or production of toxic intermediary metabolites. Although the diagnosis of an IEM often is evident from certain key clinical, laboratory, and biopsy findings, underdiagnosis is likely because of the lack of a systematic clinical approach to diagnosis and inadequate diagnostic testing. Dietary modification, avoidance of fasting, and anticipatory management during times of stress are the mainstays of treatment for most “small molecule” diseases, whereas treatment options for mitochondrial diseases remain limited and primarily involve vitamin supplements. Several lysosomal storage disorders are now treatable by enzyme replacement therapy and/or bone marrow transplantation. Newborn screening using tandem mass-spectrometry offers the potential for presymptomatic diagnosis and early treatment for a growing number of IEM, which will likely change their prevalence and natural history of cardiomyopathy.
Keywords: Inborn error of metabolism, pediatric, cardiomyopathy, treatment
Introduction
Pediatric cardiomyopathy has a number of different causes, including both genetic and non-genetic, which poses a formidable diagnostic challenge to clinicians.(1-3) Identification of the underlying causes of cardiomyopathy may lead to improved outcomes with disease-specific treatments. This review will focus on the specific group of genetic disorders, the inborn errors of metabolism (IEM), that cause cardiomyopathy. Most IEM are caused by defects in enzymes involved in intermediary metabolism (i.e., the breakdown of proteins, lipids, and carbohydrates) or energy production (e.g., oxidative phosphorylation). Many of the IEM are currently treatable using approaches that target the underlying pathophysiology of the disorder, and not surprisingly, in many cases the cardiomyopathy may be reversed. Topics covered in this article include the epidemiology, genetics, causes, pathophysiology, clinical presentations, diagnosis, and treatment of IEM.
Epidemiology and Genetics of Inborn Errors of Metabolism
Inborn errors of metabolism (IEM) occur in approximately 1 in 4000 newborns and comprise more than 1000 distinct disorders in the Online Mendelian Inheritance of Man database (www.omim.org). IEM generally present during childhood, most often in the immediate neonatal period, but also through the first two of life when growth is rapid and the body’s energy demands are high. Approximately 5% of IEM are associated with cardiomyopathy. In some IEM, cardiomyopathy dominates the clinical picture and is a major cause of death, whereas in others it may be an incidental finding discovered during a multisystem evaluation. Rarely is the heart the only affected organ, as described in cardiac phosphorylase kinase deficiency and certain mitochondrial disorders.
Most IEM are inherited in an autosomal recessive manner in which affected individual possesses two defective copies of a gene, one inherited from each parent. The genetic defect renders the enzyme completely inactive or to have only small amounts of activity. Once a couple has an affected child, their recurrence risk is 25% with each future pregnancy. In addition to being able to optimally treat the affected child (proband), knowing a specific diagnosis is important for accurate genetic counseling, family planning, prenatal diagnosis, and the screening of younger and often presymptomatic siblings.
Only a few IEM that cause cardiomyopathy are X-linked. These disorders are associated with a preponderance of affected males, and often there is a history of affected uncles and grandfathers on the maternal side of the family that appears to skip the female generations. Some follow a classic X-linked recessive pattern such that affected females have never been seen (e.g., Barth syndrome) or have been reported only under exceptional circumstances (e.g., Mucopolysaccharidosis II (MPS II), also known as Hunter syndrome). The few described cases of females with MPS II have been due to skewed X-inactivation or a structurally abnormal X chromosome. In contrast, a high proportion of females are affected in Fabry and Danon diseases. Although their symptoms are typically milder and present later as one would expect with random X-inactivation, females can have symptoms as severe as in males. Finally, mitochondrial disorders may be caused by mutations in nuclear genes and have autosomal recessive inheritance or be caused by mutations in mitochondrial DNA that are passed on only through the mother (matriarchal inheritance). Only the egg contributes mitochondria to offspring, and within each egg there may be a population of mitochondria containing normal DNA and another containing a mutation. The mitochondria containing the DNA mutation may be distributed among tissues in different proportions (heteroplasmy) in the child, leading to highly variable disease manifestations and overall severity.
IEM Causes of Cardiomyopathy
The Pediatric Cardiomyopathy Registry (PCMR) has improved our understanding of the various causes and frequencies of cardiomyopathy in children. (2, 4-7) The retrospective arm of the PCMR collected data on 916 children diagnosed with cardiomyopathy from 1990-1995 in the United States and Canada. Two-thirds of these children had an unknown cause for their cardiomyopathy and were classified as having idiopathic disease (Figure 1). The remaining one-third of cases was diagnosed with myocarditis (29.1%) or a variety of genetic causes, including familial isolated cardiomyopathy (24.2%), neuromuscular disease (22.2%), IEM (15.4%), or genetic syndrome (8.8%). Thus, approximately 5% of all cases of cardiomyopathy and 15% with known causes were due to an IEM.
Figure 1.
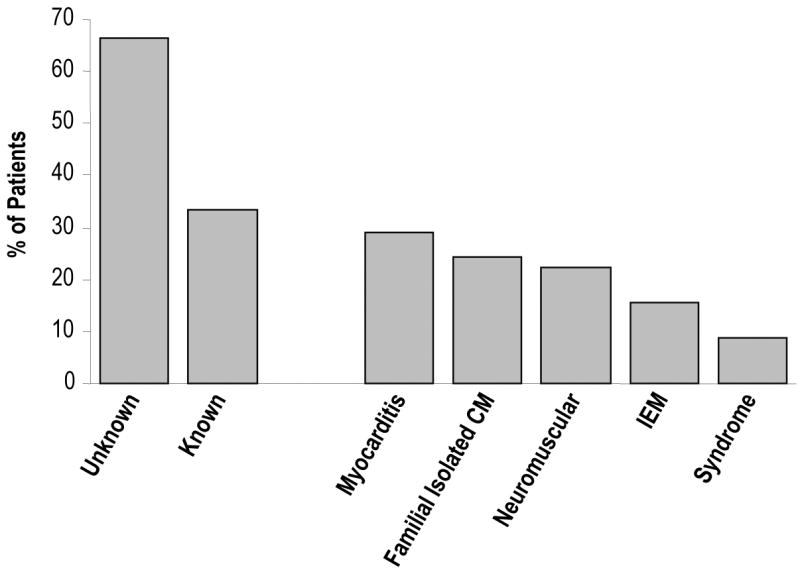
Frequencies of Causes of Cardiomyopathy in the Pediatric Cardiomyopathy Registry, 1990-1995, N=916. CM, Cardiomyopathy; IEM, Inborn Error of Metabolism.
Frequencies of Known Causes of Cardiomyopathy
Since cardiomyopathy is usually diagnosed by echocardiography, the categories of known causes seen in the retrospective arm of the PCMR are presented by functional type of cardiomyopathy (Figure 2). IEM accounted for 26.8% of known causes of hypertrophic cardiomyopathy, 6.8% of cases of dilated cardiomyopathy, 0% of restrictive cardiomyopathy, and 26.9% of mixed/other type of cardiomyopathy. Among children with hypertrophic cardiomyopathy, all four broad genetic categories were equally prevalent at approximately 25% each, and there were no cases of myocarditis. In contrast, myocarditis accounted for more than 50% of the known cause of dilated cardiomyopathy, and among the mixed/other type group, was equally prevalent with inborn errors of metabolism, trailing only familial isolated causes. These data demonstrate that the most common adaptive response of the heart in IEM is hypertrophy, with or without dilatation.
Figure 2.


Distribution of Causes by Type of Cardiomyopathy in the Pediatric Cardiomyopathy Registry, 1990-1995, N=916. CM, Cardiomyopathy; IEM, Inborn Error of Metabolism. Hypertrophic Cardiomyopathy; DCM, Dilated Cardiomyopathy; HCM, RCM, Restrictive Cardiomyopathy; MPS/OS, Mucopolysaccharidoses/Oligosaccharidoses; OxPhos, Oxidative Phosphorylation Defects; FAOD/Carnitine. Fatty Acid Oxidation/Carnitine Transport Defects; AA/OA, Amino Acidopathies/Organic Acidemias. A. All Causes. B. Inborn Errors of Metabolism.
Inborn Error of Metabolism Disease Groups
The IEM that cause cardiomyopathy can be grouped into several categories according to the accumulated substrate or the affected organelle (Table 1). With the exception of mitochondrial disorders, each IEM is generally associated with a specific functional type of cardiomyopathy by echocardiography, which helps to limit the differential diagnosis. Drawing on data from the retrospective arm of the PCMR, nearly half the cases of hypertrophic cardiomyopathy caused by IEM were due to glycogen storage diseases, most commonly Pompe disease, a lysosomal storage disorder caused by a deficiency of the α-glucosidase. The classical infantile form of Pompe disease usually presents in the first few months of life with hypotonia, muscle weakness, an enlarged tongue, and congestive heart failure. The ECG characteristically reveals a short PR interval with tall QRS waves. The cardiac silhouette is massively enlarged and the heart is extremely thickened (left ventricular mass Z-score > 6). Left ventricular outflow tract obstruction may be present. Untreated, children typically die by 12 months of age. Approximately 10% of infants with Pompe disease have mild cardiomyopathy and severe skeletal myopathy. These children with “non-classical infantile Pompe disease” may survive for several years.
Table 1.
Inborn Errors of Metabolism That Cause Cardiomyopathy.
| Disorders of Amino Acid and Organic Acid Metabolism |
|
| Disorders of Fatty Acid Metabolism |
| Carnitine Transport Defects |
|
| Fatty Acid Oxidation Defects |
|
| Disorders of Glycogen Metabolism |
|
| Disorder of Glycoprotein Metabolism |
|
| Lysosomal Storage Disorders |
| Disorders of Mucopolysaccharide (Glycosaminoglycan) Metabolism |
|
| Disorder of Glycogen Metabolism |
|
| Disorders of Glycosphingolipid Metabolism |
| Disorders of Combined Ganglioside, Mucopolysaccharide, and Oligosaccharide Metabolism |
|
| Mitochondrial Disorders |
|
| Peroxisomal Disorder |
|
Reported in adults only
Fatty acid oxidation defects, most commonly VLCAD and LCHAD deficiencies, and oxidative phosphorylation defects each accounted for approximately 25% of IEM in the retrospective arm of the PCMR. Within the dilated cardiomyopathy group, oxidative phosphorylation defects and systemic carnitine deficiency were the most common causes, accounting for 40% of cases each. The mitochondrial syndromes MERFF (myoclonic epilepsy with ragged red fibers), MELAS (mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes), and Kearns-Sayre syndrome affect the synthesis of multiple mitochondrial proteins and respiratory chain complexes, but single gene defects in respiratory chain proteins also may cause cardiomyopathy.
Within the mixed/other group, oxidative phosphorylation defects accounted for 70% of cases, several of whom were boys with Barth syndrome. This condition is thought to be due to a defect in the remodeling of cardiolipin, an unusual lipid with four linoleic-acyl chains that is localized almost exclusively to the inner membrane of mitochondria, where it binds to the respiratory chain complexes and is important for oxidative phosphorylation. This condition is likely under-diagnosed. Classic features include endocardial fibroelastosis, neutropenia with frequent infections, skeletal myopathy, growth retardation, abnormal-appearing mitochondria in biopsies, and 3-methylglutaconic aciduria. However, some boys may only have dilated cardiomyopathy without other systemic findings. Although the cardiomyopathy may be very severe during infancy, it often responds well to standard cardiac medications. In the past, many boys died of infection or cardiomyopathy by age 3, but today, many have entered their teens.
Pathophysiology
The IEM that cause cardiomyopathy may be classified by their non-metabolizable, accumulated substrate or by the affected organelle (Table 1). The named substrate disorders include fatty acid oxidation defects, carnitine transport disorders, organic acidemias, amino acidurias, and glycogen storage diseases. The named organelle disorders include peroxisomal, mitochondrial, and lysosomal storage disorders.
These disorders are thought to cause cardiomyopathy by three main pathophysiological mechanisms that are not mutually exclusive. The first is by bulk storage and infiltration of substrate, which can have a mechanical effect on cardiomyocyte functioning by disrupting the alignment of myofibrils required for efficient contraction. These disorders involve large macromolecules, i.e., triglycerides (fatty acid oxidation defects and carnitine transport disorders), glycogen (hydrolysis), and lysosomal substrates (mucopolysaccharides, oligosaccharides, glycolipids, and glycogen). Eventually, triglycerides and glycogen may occupy a large amount of the cytoplasm, while the lysosomes become greatly increased in size and number.
The second pathophysiologic mechanism is impaired energy production, which results in not enough ATP to meet the needs of the cell. The main sources of energy in the heart are fatty acids and secondarily glycogen. Both substrates are progressively broken down to release hydrogen molecules, whose energy is extracted by the respiratory chain complexes in the mitochondria and converted into ATP through oxidative phosphorylation. Patients who have defects in oxidative phosphorylation are unable to efficiently make ATP from any energy source. Muscle has high energy needs because each cell is packed with myofibrils that are specialized for contraction. Hydrolysis of ATP by the myosin heads provides the force necessary to slide the myosin-containing thick filaments past the actin-containing thin filaments during sarcomere contraction. The adaptive response of the heart to inefficient contraction is hypertrophy, which is why so many IEM are associated with hypertrophic cardiomyopathy.
The third pathophysiologic mechanism is the production of toxic metabolites by organic acidemias, amino acidurias, Refsum disease, and disorders of oxidative phophorylation. The accumulated metabolites exert their deleterious effect by lowering the cellular pH (acids), inhibiting intermediary metabolism (acyl-CoA), or oxidizing mitochondrial components such as DNA, lipids, and proteins structures (free radicals).
Clinical Presentation and Approach to Diagnosis
With the exception of mitochondrial disorders, most IEM are associated with one functional type of cardiomyopathy by echocardiography, which can help narrow down the differential diagnosis (Figure 2). Most IEM present during infancy or early childhood. Diseases associated with the storage of glycogen, fat, or lysosomal substrates generally cause hypertrophic cardiomyopathy. Dilated cardiomyopathy is often caused by diseases associated with excess acidic metabolites as in the organic acidemias, amino acidopathies, and systemic carnitine deficiency. In the latter, the lack of carnitine leads to the accumulation of circulating acyl-CoA species and a decrease in free CoA, which is required for intermediary metabolism.
Cardiomyopathy may be the presenting or dominant clinical feature, but careful searching often reveals other signs of a multisystemic disease as well as abnormal metabolites in blood and urine. Clinical features may include a distinctive physical appearance (e.g., coarse facial features, cloudy corneas, slow growth, and hepatosplenomegaly), neurologic findings (acute or chronic encephalopathy, developmental delay, and seizures), myopathy (hypotonia and weakness), skeletal findings (dysostosis multiplex), or liver dysfunction. In IEM that impair energy production or produce toxic metabolites, congestive heart failure often occurs in the setting of an acute metabolic decompensation triggered by energy stressors, e.g., illness, surgery, infection, fasting, or physical exertion. These inciting events may confound the initial presentation and suggest an alternative diagnosis for the cardiomyopathy, e.g., viral myocarditis. As further described below, it is important to include laboratory testing of blood and urine as part of the initial evaluation. The detection of certain basic laboratory abnormalities, e.g., metabolic acidosis with an increased anion gap, hypoglycemia, hyperammonemia, ketosis or lack thereof, or elevated liver function tests will often point the clinician in the right direction to uncovering the underlying diagnosis.
Previously, we identified several key entry points to clinical diagnostic algorithms based on clinical features and laboratory findings.(1) The algorithms most relevant to IEM include encephalopathy (Figure 3), hypoglycemia (Figure 4), metabolic acidosis (Figure 5), and neuromuscular symptoms (Figure 6). Acute encephalopathy is typically accompanied by metabolic abnormalities in the blood, e.g., metabolic acidosis, hyperammonemia, and hypoglycemia, which are characteristic of small molecule diseases, whereas chronic encephalopathy is often associated with mitochondrial or lysosomal storage disorders. In the former, an event such as an intercurrent illness will often create a state of energy imbalance that leads to ineffectual production of energy and an excess of toxic metabolites.
Figure 3.
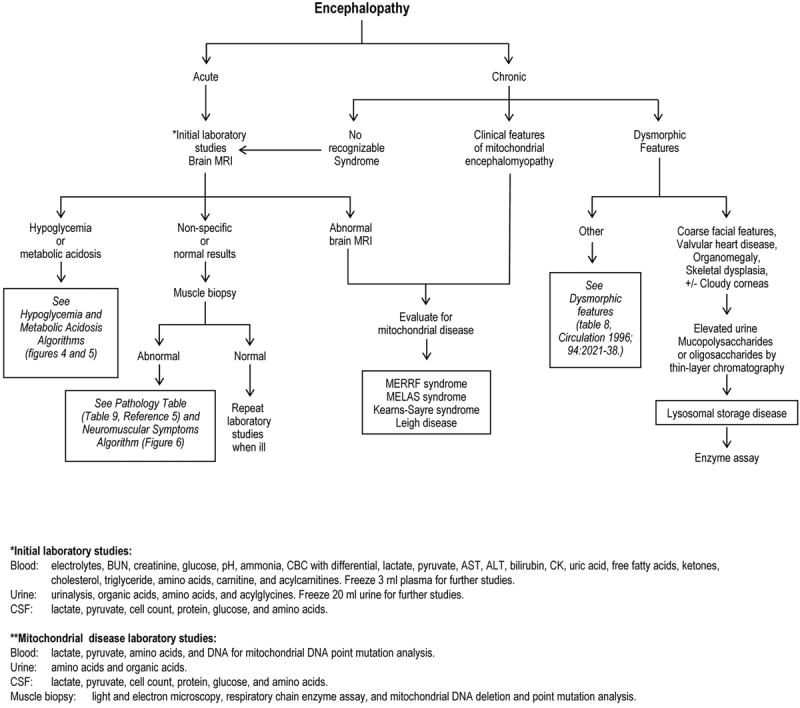
Diagnostic Algorithm: Encephalopathy
Figure 4.
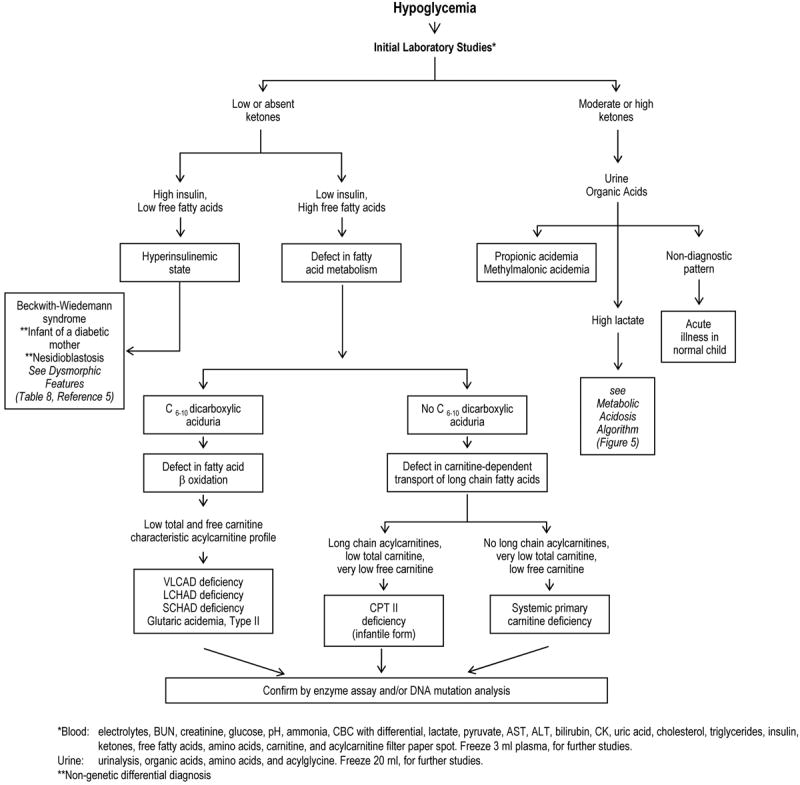
Diagnostic Algorithm: Hypoglycemia
Figure 5.
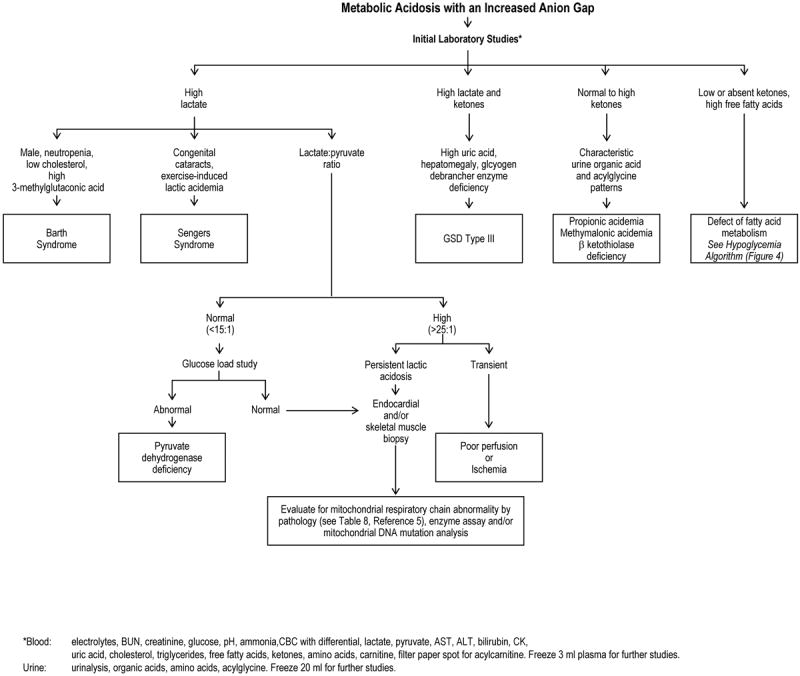
Diagnostic Algorithm: Metabolic Acidosis with an Increased Anion Gap
Figure 6.
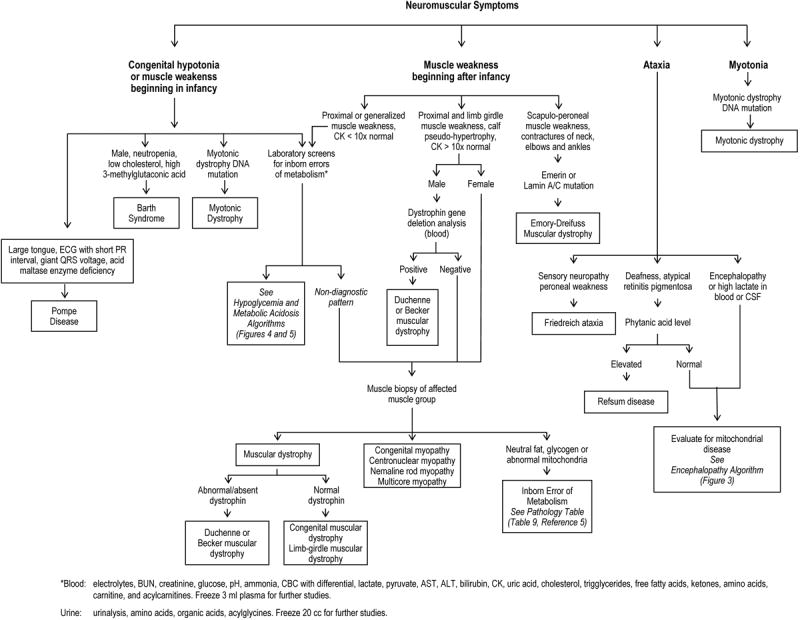
Diagnostic Algorithm: Neuromuscular Symptoms
Hypoglycemia is often the result of ineffective fat metabolism, either from the inability to break down triglycerides, fatty acids, or ketones. Thus, the presence or absence of ketones is a useful starting point for evaluation. Patients with inappropriately low ketones in the setting of low blood sugar (hypoketotic hypoglycemia) have an inability to break down triglycerides into fatty acids and glycerol or to break down fatty acids into ketones through fatty acid oxidation. The latter are associated with low insulin and high free fatty acid levels in blood, dicarboxylic aciduria (from ω-oxidation of fatty acids in peroxisomes), and low carnitine levels in blood. A specific diagnosis is often suggested by the acylcarnitine profile in blood. The absence of dicarboxylic acids points towards the inability of fatty acids to cross the plasma membrane (systemic carnitine deficiency) or into mitochondria (Carnitine-palmitoyl transferase II deficiency). Extremely low carnitine levels (< 10 μmol/L) are seen in systemic carnitine deficiency.
The major diagnostic branch points for the evaluation of hypoglycemia involve the measurement of ketones, insulin, free fatty acids, urine organic acids (including dicarboxylic acids), and carnitine (total and free). These assist in the determination of whether the hypoglycemia is associated with a defect in mobilizing triglycerides (overgrowth syndromes and infant of a diabetic mother), the inability to convert fatty acids into ketones (fatty acid oxidation defects or carnitine-transport defects), organic acidemias, or GSD III. Specific diagnoses are often suggested by specific species identified on the blood acylcarnitine profile.
The major branch points for metabolic acidosis with an increased anion gap (> 15 mEq/L) focus on determining the identity of the offending anion, which can be lactic acid, ketoacids, more complex organic acids (propionic acid), or fatty acids. The different acids are distinguished by direct measurement in blood by routine clinical testing (lactic, ketoacids, free fatty acids) or by tandem-mass spectrometry as acylcarnitine derivatives. When the lactate level is increased, the lactate:pyruvate ratio (L:P) can help to distinguish pyruvate dehydrogenase deficiency (L:P < 15) and oxidative phosphorylation defects (L:P > 25).
The neuromuscular symptoms that point towards an IEM are weakness/hypotonia beginning in infancy and ataxia (Figure 6). Pompe disease is a major cause of “floppy baby syndrome” and the hypotonia can be so severe that it can mimic spinal muscular atrophy. The diagnosis of Barth syndrome is suggested in a male by the presence of neutropenia, a low cholesterol level, and 3-methylglutaconic aciduria. Other metabolic diagnoses presenting with weakness/hypotonia may be guided by laboratory testing and/or skeletal muscle biopsy. Ataxia is a common feature in mitochondrial disorders and Refsum disease. The former is often associated with chronic encephalopathy, cardiac conduction defects, and arrhythmias, whereas Refsum disease is associated with deafness, atypical retinitis pigmentosa, and elevated plasma phytanic acid levels.
Diagnosis
The ideal time to diagnose IEM is in the first few days of life, prior to symptom onset, using comprehensive newborn screening followed by confirmatory enzyme assay.(8, 9) Tandem mass spectroscopy has largely supplanted the Guthrie assays for newborn screening as it can diagnose in a single test more than 40 different IEM on the basis of their characteristic acylcarnitine and amino acid profiles. However, a negative newborn screen should not dissuade the clinician from suspecting an IEM and repeating metabolic testing at a later time since certain abnormal metabolites may not be elevated in the first two days of life (e.g., fatty acid oxidation defects), and not all state laboratories screen for all detectable disorders. Furthermore, several classes of IEM are unable to be diagnosed by current tandem spectrometry assays (e.g., glycogen storage diseases, oxidative phosphorylation defects, and lysosomal storage disorders), although this will likely change in the near future with respect to certain lysosomal storage disorders.(10)
Diagnosis of an IEM in a child presenting with cardiomyopathy requires thoughtful investigation with respect to evaluation of the diagnostic metabolites, demonstration of deficient enzyme activity in the appropriate cell type or bodily fluid, and occasionally, DNA mutation analysis. For the IEM associated with excessive acid production, the best time for testing is when the child’s metabolism is stressed and the underlying metabolic defects are drawn out. Such times include congestive heart failure, infection, illness, post-surgery, fasting, and during a metabolic decompensation (hypoglycemia, metabolic acidosis, acute encephalopathy). At these times, it is good clinical practice to obtain extra serum and urine samples to hold for future metabolic testing rather than waiting until the child is stabilized, as some of the diagnostic metabolites may be transient or present in only small amounts.
While the detection of metabolites can be suggestive (e.g., increased urinary glycosaminoglycans in the mucopolysaccharidoses and an elevated C14:1 acylcarnitine species by tandem mass spectrometry in VLCAD deficiency), the gold standard for diagnosis remains the enzyme assay. Many disorders can easily be confirmed by testing the level of enzymatic activity in white blood cells, serum, or plasma, while others are more easily done in cultured fibroblasts from a skin biopsy. However, the latter requires several weeks for diagnosis, which has led to a push towards developing new assays in white blood cells. For example, the enzyme assay for Pompe disease can now be reliably performed using a dried blood spot on a newborn screening card, which shortens the turn-around time for diagnosis to just a couple of days.(10, 11) Dried blood spots have the added convenience of requiring only a small volume of blood, enzyme stability at room temperature, and easy mailing to the laboratory in an envelope.
Despite the inroads made by newborn screening, IEM are under diagnosed in children due to a lack of awareness that these disorders can cause cardiomyopathy and due to the lack of testing to pursue an underlying cause. We recently showed that children with a known cause of cardiomyopathy are much more likely to have undergone testing of blood, urine, and biopsy specimens as part of their diagnostic evaluation (Table 2). For dilated cardiomyopathy, metabolic urine testing was particularly useful as was endocardial biopsy for diagnosing myocarditis. In hypertrophic cardiomyopathy, metabolic blood and urine testing and skeletal muscle biopsy were significantly associated with establishing a known cause of cardiomyopathy. A positive trend for endocardial biopsy also was seen, but too few patients underwent this procedure. For patients suspected of having a mitochondrial disorder, skeletal muscle biopsy is the gold standard for diagnosis as it can provide supportive pathologic findings (e.g., ragged red fibers by Gomori trichrome staining and abnormal-appearing mitochondria by electron microscopy) and confirm the deficiency of one or more respiratory chain complexes.
Table 2.
Rates of Known Causes of Cardiomyopathy According to Testing Status in the Retrospective Arm of the Pediatric Cardiomyopathy Registry, 1990-1995, Excluding Patients with Familial Isolated Cardiomyopathy, Neuromuscular Disorders, and Malformation Syndromes.
| Type of Testing | No. | Testing (%) | % Known Causes | Odds Ratio | P | |
|---|---|---|---|---|---|---|
| Testing Done | No Testing Done | |||||
| HCM (n= 230)a | ||||||
| Metabolic urine testing | 225 | 22.2 | 28.0 | 8.6 | 4.15 | .001 |
| Metabolic blood testing | 221 | 26.2 | 34.5 | 5.5 | 9.01 | <.001 |
| Biopsy | 227 | 8.8 | 45.0 | 10.1 | 7.25 | <.001 |
| Endomyocardial | 230 | 4.4 | 30.0 | 12.3 | 3.06 | .13 |
| Skeletal | 227 | 4.4 | 60.0 | 11.1 | 12.06 | <.001 |
| Viral serologic testing or culture | 221 | 10.4 | 47.8 | 8.6 | 9.76 | <.001 |
| Chromosome analysis | 224 | 4.9 | 45.5 | 11.3 | 6.56 | .01 |
| Any testing | 219b | 36.5 | 27.5 | 4.3 | 8.41 | <.001 |
| DCM (n=422)c | ||||||
| Metabolic urine testing | 406 | 37.7 | 28.1 | 19.0 | 1.67 | .04 |
| Metabolic blood testing | 407 | 51.6 | 24.8 | 18.8 | 1.42 | .15 |
| Biopsy | 419 | 47.0 | 35.5 | 9.9 | 5.01 | <.001 |
| Endomyocardial | 421 | 44.7 | 37.2 | 9.4 | 5.69 | <.001 |
| Skeletal | 419 | 4.5 | 15.8 | 22.3 | 0.66 | .70 |
| Viral serologic testing or cilture | 408 | 55.6 | 28.6 | 12.7 | 2.76 | <.001 |
| Chromosome analysis | 414 | 2.4 | 10.0 | 22.0 | 0.39 | .70 |
| Any testing | 410d | 78.3 | 26.2 | 6.7 | 4.90 | <.001 |
| Other or Mixed Cardiomyopathy (n=67)e | ||||||
| Metabolic urine testing | 65 | 46.2 | 20.0 | 17.1 | 1.21 | 1.00 |
| Metabolic blood testing | 65 | 53.9 | 25.7 | 10.0 | 3.12 | .12 |
| Biopsy | 67 | 44.8 | 26.7 | 13.5 | 2.33 | .22 |
| Endomyocardial | 67 | 44.8 | 26.7 | 13.5 | 2.33 | .22 |
| Skeletal | 67 | 4.5 | 66.7 | 17.2 | 9.64 | .10 |
| Viral serologic testing or culture | 67 | 44.8 | 16.7 | 21.6 | 0.73 | .76 |
| Chromosome analysis | 67 | 7.5 | 20.0 | 19.4 | 1.04 | 1.00 |
| Any testing | 67f | 68.7 | 21.7 | 14.3 | 1.67 | .74 |
The RCM group (n=22) was too small for any meaningful comparisons and was not included in this table.
See Methods, Reference 5 for definitions of testing components. Testing component information was positive or complete for 219 patients. One patient did not have any testing information available.
A total of 80 (36.5%) of 219 patients underwent ≥ 1 form of testing.
See Methods, Reference 5 for definitions of testing components. Testing component information was positive or complete for 410 patients. Two patients did not have any testing information available.
A total of 321 (78.3%) of 410 patients underwent ≥ 1 form of testing.
See Methods, Reference 5 for definitions of testing components. Testing component information was positive or complete for 67 cases. One patient did not have any testing information available.
A total of 46 (68.7%) of 67 patients underwent ≥ 1 form of testing.
Treatment
Hypoglycemia and metabolic acidosis often correct quickly if an appropriate energy source, usually glucose in the form of 10% dextrose, is provided along with intravenous fluids to flush out the accumulated metabolites. If metabolic acidosis is present, treatment with carnitine will help to replenish low free reserves, bind acidic compounds, liberate CoA from acyl-CoA species, and help to restore intermediary metabolism.
For chronic treatment, there are also several guiding principles. The first is reducing the intake of non-metabolizable substrates from the diet (e.g., branch chain amino acids in propionic academia and long-chain fatty acids in VLCAD, LCHAD, and multiple acyl-CoA dehydrogenase deficiencies). Similarly, avoidance of fasting, which causes the body to attempt to break down its own endogenous stores of energy in the form of fat, glycogen, and protein, some of which may not be metabolizable, is also important. During infancy, overnight fasting beyond 6 hours should be avoided with a middle of the night feeding or continuous tube feedings. After age 1, a cornstarch-supplemented drink at bedtime provides a slow release form of glucose throughout the night. In some disorders, an alternative, metabolizable energy source can be provided in the diet (e.g., medium chain triglycerides for long-chain fatty acid oxidation defects). Certain dietary supplements can decrease secondary toxicity (e.g., antioxidants to neutralize free radicals in oxidative phosphorylation defects, and carnitine to liberate CoA from acylCoA compounds). In some cases, residual enzyme activity can be enhanced by the use of pharmacologic doses of vitamin cofactors (e.g. biotin for propionic acidemia and riboflavin for multiple acyl-CoA dehydrogenase deficiency).
Oxidative phosphorylation defects have been treated empirically with combinations of antioxidants (e.g., water soluble [Vitamin C] and lipid-soluble [CoEnzyme Q10]) to help prevent free radical damage. There are anecdotal reports of improvement following the use of vitamins, but none have been subjected to formal clinical studies. Given the low risk, they are often prescribed empirically upon diagnosis and then monitored for any potential clinical effect.
Carnitine supplements are very effective for treating systemic carnitine deficiency, in which carnitine is unable to be reabsorbed in the kidney. Treatment of dilated cardiomyopathy with high doses of carnitine has produced dramatic improvement within a few days. Enzyme replacement therapy has emerged as a treatment paradigm for several lysosomal storage disorders that are associated with cardiomyopathy, including Gaucher disease, Fabry disease, Pompe disease, and Mucopolysaccharidoses I (Hurler, Hurler-Scheie, and Scheie syndromes), II (Hunter syndrome), and VI (Maroteaux-Lamey), and Pompe disease. In clinical trials, reduction of myocardial hypertrophy has been demonstrated for both Pompe disease and Mucopolysaccharidosis I following enzyme replacement therapy. (12, 13) Beneficial effects of hematopoietic stem cell transplantation also have been seen in Mucopolysaccharidosis I and VI.
Future Directions
Improving the outcomes of children with IEM will require timely diagnosis and the use of disease-specific therapies. Newborn screening will continue to expand as new assays are developed that use tandem mass spectrometry as the readout technology. A tandem mass spectrometry assay for Pompe disease is currently undergoing pilot testing, and assays for other lysosomal storage disorders are under development. The use of dried blood spots for metabolite screening (amino acid and acylcarnitine profiles) and enzyme assays (Pompe disease) will facilitate the early screening of at-risk populations with cardiomyopathy. The approaches to replacing the missing enzymes will be disease-specific and achieved through the use of recombinant enzymes, stem cells, organ transplantation, and gene therapy. The PCMR in the United States, and others like it in Australia (3), are helping to define the epidemiology and natural history of cardiomyopathy in children. This information will help to educate clinicians and measure the impact of newborn screening and disease-specific therapies on outcomes, including survival, quality of life, and cardiomyopathy.
Acknowledgments
I wish to acknowledge my colleagues at the Pediatric Cardiomyopathy Registry for their clear vision, supportive attitude, and stimulating discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schwartz ML, Cox GF, Lin AE, et al. Clinical approach to genetic cardiomyopathy in children. Circulation. 1996;94:2021–38. doi: 10.1161/01.cir.94.8.2021. [DOI] [PubMed] [Google Scholar]
- 2.Lipshultz SE, Sleeper LA, Towbin JA, et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med. 2003;348:1647–55. doi: 10.1056/NEJMoa021715. [DOI] [PubMed] [Google Scholar]
- 3.Nugent AW, Daubeney PE, Chondros P, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. 2003;348:1639–46. doi: 10.1056/NEJMoa021737. [DOI] [PubMed] [Google Scholar]
- 4.Grenier MA, Osganian SK, Cox GF, et al. Design and implementation of the North American Pediatric Cardiomyopathy Registry. Am Heart J. 2000;139:S86–95. doi: 10.1067/mhj.2000.103933. [DOI] [PubMed] [Google Scholar]
- 5.Cox GF, Sleeper LA, Lowe AM, et al. Factors associated with establishing a causal diagnosis for children with cardiomyopathy. Pediatrics. 2006;118:1519–31. doi: 10.1542/peds.2006-0163. [DOI] [PubMed] [Google Scholar]
- 6.Colan SD, Lipshultz SE, Lowe AM, et al. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children. Findings from the Pediatric Cardiomyopathy Registry. Circulation. 2007 doi: 10.1161/CIRCULATIONAHA.106.621185. [DOI] [PubMed] [Google Scholar]
- 7.Towbin JA, Lowe AM, Colan SD, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–76. doi: 10.1001/jama.296.15.1867. [DOI] [PubMed] [Google Scholar]
- 8.Seashore MR. Tandem spectrometry in newborn screening. Curr Opin Pediatr. 1998;10:609–14. doi: 10.1097/00008480-199810060-00013. [DOI] [PubMed] [Google Scholar]
- 9.Naylor EW, Chace DH. Automated tandem mass spectrometry for mass newborn screening for disorders in fatty acid, organic acid, and amino acid metabolism. J Child Neurol. 1999;14(Suppl 1):S4–8. doi: 10.1177/0883073899014001021. [DOI] [PubMed] [Google Scholar]
- 10.Gelb MH, Turecek F, Scott CR, Chamoles NA. Direct multiplex assay of enzymes in dried blood spots by tandem mass spectrometry for the newborn screening of lysosomal storage disorders. J Inherit Metab Dis. 2006;29:397–404. doi: 10.1007/s10545-006-0265-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jack RM, Gordon C, Scott CR, Kishnani PS, Bali D. The use of acarbose inhibition in the measurement of acid alpha-glucosidase activity in blood lymphocytes for the diagnosis of Pompe disease. Genet Med. 2006;8:307–12. doi: 10.1097/01.gim.0000217785.19262.9e. [DOI] [PubMed] [Google Scholar]
- 12.Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- 13.Wraith JE, Beck M, Lane R, et al. Enzyme replacement therapy in patients who have Mucopolysaccharidosis I and are younger than 5 years: Results of a multinational study of recombinant human α-L-iduronidase (Laronidase) Pediatrics. 2007;120:e36–47. doi: 10.1542/peds.2006-2156. [DOI] [PubMed] [Google Scholar]