

Abstract
The antithrombotic activity of heparin has largely been credited with the success found in some cancer treatment by heparin. There are, however, many potent growth factors involved in tumor and blood vessel growth that bind to heparin with high affinity and their regulation by heparin may play a role in heparin’s efficacy. We therefore chose to study the activity of a heparin analog, sucrose octasulfate (SOS), which has been similarly shown to interact with heparin-binding growth factors. Using mouse melanoma and lung carcinoma models, we demonstrate in vivo inhibition of tumor growth by SOS. SOS, however, showed little effect in coagulation assays indicating that this activity was not a primary mechanism of action for this molecule. Studies were then performed to assess the effect of SOS on basic fibroblast growth factor (FGF-2) activity, a growth factor which promotes tumor and blood vessel growth and is produced by B16 melanoma cells. SOS potently inhibited FGF-2 binding to endothelial cells and stripped pre-bound FGF-2 from cells. SOS also regulated FGF-2 stimulated proliferation. Further, SOS facilitated FGF-2 diffusion through Descemet’s membrane, a heparan sulfate-rich basement membrane from the cornea, suggesting a possible role in FGF-2 clearance. Our results suggest that molecules such as SOS have the potential to remove growth factors from tumor microenvironments and the approach offers an attractive area for further study.
Heparin, long used as an anticoagulant in clinical settings, has been shown to have a number of other important biological activities that allow it to influence a wide range of physiologic and pathologic processes (Lindahl, 1999). For example, many growth factors bind to heparin with high affinity and it has been demonstrated that this interaction alters growth factor signaling and biological response (Rapraeger et al., 1991; Yayon et al., 1991; Folkman and Shing, 1992; Fannon and Nugent, 1996; Fannon et al., 2000, 2003). For over 70 years the efficacy of heparin therapy in the inhibition of tumor growth has been studied and debated (Shear, 1935; Lippman, 1957; Elias et al., 1975; Edlis et al., 1976). Clinical trials have shown that heparin therapy can prolong the lives of some cancer patients (Prandoni et al., 1992; von Tempelhoff et al., 2000; Altinbas et al., 2004), including patients with low risk of thrombosis (Klerk et al., 2005), however, a recent clinical trial showed no significant effect (Sideras et al., 2006) when advanced, incurable patients were treated with low molecular weight heparin (LMWH). Factors such as the type of cancer, stage of the disease, dose ranges, and treatment schedules could play a part in the conflicting results. Along with its acknowledged antithrombotic role, heparin activity could be due, in part, to its ability to regulate the activity of heparin-binding growth factors such as fibroblast growth factor-2 (FGF-2), which is crucial to tumor and blood vessel growth (see (Folkman, 2001) for review).
Heparan sulfate proteoglycans, physiological analogs of heparin that consist of a protein core and glycosaminoglycan (GAG) chains, are ubiquitous throughout the body. They reside in most cell membranes and are prominent in extracellular matrix (ECM). To a large extent, mass action can dictate where a growth factor will tend to reside based on its abundance and availability of binding sites. In addition to transport through the circulatory system many growth factors are stored and transported locally by diffusion through the ECM (Vlodavsky et al., 1987). A growth factor such as FGF-2 is labile and has a limited half-life in circulation (Whalen et al., 1989; Edelman et al., 1993). Heparin-binding growth factors can, however, reside for long periods of time in matrices by binding to heparan sulfate moieties. The GAG chains protect them from proteolytic degradation (Coltrini et al., 1993) thus increasing their half-life (Saksela et al., 1988). When matrix is degraded in the case of injury, for example, growth factors can be released and become available to the injured tissue (Bashkin et al., 1989; Buczek-Thomas et al., 2002, 2004). Studies indicate that heparinase-release of ECM-bound heparin-binding growth factors may be particularly relevant in tumor progression (Ilan et al., 2006). Further, guided migration in response to growth factors in development is also regulated by heparan sulfate expression (Fayein et al., 1992; Izvolsky et al., 2003a,b). While heparan sulfate and heparin have the ability to extend the half-life of these factors, however, the bound GAG-protein complex may exhibit different characteristics from the unconjugated molecules. Diffusion, binding, and signaling events may be altered based on the coupling of sites that may no longer be easily accessible for interaction with other molecules (Dowd et al., 1999).
The maximum dose attainable for in vivo heparin administration is limited by anticoagulative properties that can lead to bleeding complications. The reduced complications and the pharmokinetics associated with LMWH have made it the preferred therapy to unfractionated heparin (Mousa, 2006) and small-scale experimental studies have suggested that its effect in cancer therapy is related to growth factor regulation (Castelli et al., 2004). Ideal therapy, when antithrombotic properties are not desired, might consist of a synthetic heparin analog with little to no anticoagulative properties that is still capable of interacting with and regulating the growth factor activity of tumor and blood vessel-promoting factors. We chose to study the potential use of the synthetic disaccharide sucrose octasulfate (SOS). Its insoluble form, aluminum sucralfate, has been used for years as an ulcer treatment (Nagashima and Hirano, 1980; Fisher, 1981; Nakazawa et al., 1981; Folkman et al., 1991) and we have previously shown that SOS binds with high affinity to FGF-2 (Folkman et al., 1991; Forsten et al., 2000b). It contains eight sulfate residues, the primary moieties on heparin and heparan sulfate required for growth factor binding (Fig. 1). In this study we test the efficacy of SOS in murine melanoma and lung carcinoma models to measure its potential in inhibiting tumor growth. We then compare its anticoagulative activity to that of heparin in partial thromboplastin time (PTT), prothrombin time (PT), and thrombin activation studies. Based on these results, we hypothesize that inhibitory regulation by SOS might be indirect and thus explore possible mechanisms related to growth factor regulation that might explain this activity: SOS regulation of FGF-2 binding to endothelial cells, its ability to release pre-bound growth factor from endothelial surfaces, its effect on FGF-2 transport through HSPG-rich basement membrane, and its regulation of growth factor-stimulated cell proliferation. Our results suggest unique and novel diffusion characteristics for SOS-bound growth factors worthy of future studies targeted at investigating its potential as a growth factor regulating therapeutic.
Fig. 1.
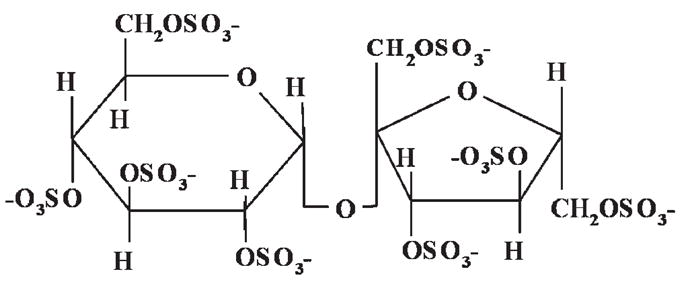
SOS: shown is the chemical structure of SOS.
Materials and Methods
Heparin
Heparin was obtained from two sources, Sigma (St. Louis, MO) with an average molecular weight of 17–19,000 Da, and Hepar (Franklin, OH) with an average molecular weight of 12–15,000 Da. Preliminary studies showed similar activities for both reagents (data not shown).
Tumor studies
C57/BL6 male mice, 9–10 weeks old were used and studies were performed in accordance with institutional, state and federal guidelines. Mice were injected subcutaneously with 1 × 106 B16/F10 cells (ATCC Culture Collection, Manassas, VA) in the hind flank. Treatment was begun on the first day of implantation. SOS (100 mg/kg/day) or saline was delivered once daily through intraperitoneal injection. Tumor volume was measured by vernier calipers and calculated using the formula a2 × b ×0.52 (where a equals the shortest side and b the longest)(Kisker et al., 2001).
For the Lewis lung carcinoma model, Alzet® osmotic pumps containing Sigma heparin, SOS, or saline were implanted in the peritoneal cavity of mice 3 days prior to subcutaneous tumor implantation to ensure a steady state circulating concentration of molecules prior to the introduction of the tumor. 1 ×106 LLC cells were injected subcutaneously in the hind flank. Tumor volume was measured as described above.
Radiolabeled growth factor-cell binding studies
Bovine capillary endothelial (BCE) cells were isolated and maintained for nine passages in DMEM (penicillin 100 U/ml/, streptomycin 100 μg/ml) 10% calf serum (Invitrogen, Carlsbad, CA). Cells were plated at a density of 50,000 cells/well in 24-well plates (Costar, Cambridge, MA) and cultured overnight prior to initiating studies as previously described (Fannon et al., 2003). Briefly, wells were washed once with binding buffer (DMEM, 25 mM HEPES, and 0.05% gelatin) at 4°C and fresh binding buffer added (0.5 ml/well). Cells were incubated at 4°C for 10 min and then 125I-FGF-2 (1 ng/ml) was added +/− SOS (Bukh Meditec, Copenhagen, Denmark). Cells were incubated at 4°C for 1.5 h and then washed three times with ice-cold binding buffer. HSPG-bound 125I-FGF-2 was removed with a high salt wash (2 M NaCl in 20 mM HEPES (pH 7.4)) followed by a wash with phosphate buffered saline (PBS). Cell surface receptor-bound 125I-FGF-2 was subsequently extracted with two washes (one 5 min wash and one rapid wash) at room temperature using salt-acid wash (2 M NaCl in 20 mM sodium acetate (pH 4.0)). The level of non-specific binding was measured in the presence of excess unlabeled FGF-2 and subtracted from all data.
Studies to determine the release of pre-bound 125I-FGF-2 were performed in a similar fashion. After the initial growth factor binding period (1 ng/ml), cells were washed three times to remove unbound growth factor and SOS or heparin (Sodium Heparin-Hepar® Industries, Inc., Franklin, OH) was added in fresh binding buffer. Cells were incubated an additional 1.5 h at 4°C. Unbound radiolabeled growth factor was quantitated by measuring binding buffer from each condition in a gamma counter.
Proliferation assays
BCE or Human umbilical vein endothelial cells (HUVEC) were plated at a density of 15,000/well in 24-well plates and incubated overnight at 37°C. BCE cells were cultured in supplemented DMEM (penicillin (100 U/ml), streptomycin (100 U/ml) (p/s), 10% calf serum (CS) (Invitrogen). Supplemented medium was changed to reduce the serum concentration (DMEM, p/s, 1% CS) +/− FGF-2 (1 ng/ml) with or without heparin or SOS. Cells were incubated at 37°C for 72 h. Quantification was done using a Coulter counter (Model Z1).
Hematology studies
PT (Thromboplastin C Plus), PTT (Actin FSL), and thrombin (BC Thrombin) time assays were performed in the Hematology laboratory at Children’s Hospital-Boston using a BCS System (Dade Behring, West Sacramento, CA). Pooled normal plasma was used for assays (George King Bio-Medical, Overland Park, KA).
Heparin/SOS calcium binding studies
The binding of heparin and SOS to Ca2+ was demonstrated by competitive binding against the fluorescent Ca2+ binding dye Oregon Green® 488 BAPTA-2 (Molecular Probes, Eugene, OR). Standard solutions of SOS and heparin were prepared with 0.007 mM CaCl2 (Sigma–Aldrich, St. Louis, MO) and 0.0024 mM Oregon Green® BAPTA-2, along with a blank solution consisting of 0.007 mM CaCl2 and 0.0024 mM Oregon Green® 488 BAPTA-2. A solution of 0.0024 mM Oregon Green® 488 BAPTA-2 was used for background correction. Four replicates of each standard (200 μl volume) were added to a 96-well plate, and fluorescence was measured on a Per Septive Biosystems (Framingham, MA) Cyto Fluor® Series 4000 Multi-Well Plate Reader (excitation at 485 nm, emission at 535 nm). All readings were corrected for background fluorescence.
Growth factor transport through Descemet’s membrane
Transport of growth factors through Descemet’s membrane was measured essentially as described (Dowd et al., 1999). Briefly, Descemet’s membranes were isolated from adult bovine corneas by dissection and membrane pieces were placed between two glass slides with matching central holes (0.5 cm diameter). The mounted membrane was placed between two diffusion chambers (Crown Glass, Somerville, MA) and held in place with a clamp. Each chamber was filled with 3 ml of PBS containing 1 mg/ml BSA and the membrane allowed to equilibrate overnight with stirring (500 rpm). Prior to initiating growth factor transport, each 125I-growth factor was purified on a PD-10 Sephadex G-25 column to isolate 125I-growth factor from unincorporated label. Transport was initiated by replacing the buffer in each chamber of the diffusion cell with fresh PBS, 1 mg/ml BSA with or without SOS (0.5 mg/ml), and then ~1 μCi of 125I-growth factor (0.83 nM FGF-2; 2.2 nM heparin binding-epidermal growth factor (HB-EGF); 0.02 nM EGF) was added to the source chamber. At various time points, 200 μl samples were removed from the receiver chamber and a fresh 200 μl aliquot of buffer added. Each 200 μl receiver sample was subjected to precipitation with trichloroacetic acid (12.5% final concentration) to isolate 125I-growth factor from free label. 125I-growth factor levels were quantitated by counting TCA pellets in a gamma counter.
Results
SOS inhibits the growth of both Lewis lung carcinoma and B16 melanoma in vivo
We hypothesized that SOS, based on the similarity to heparin, would have an effect on tumor seeding and growth. C57 mice were implanted with B16 melanoma, a murine melanoma strain previously shown to produce both FGF-2 and vascular endothelial growth factor (VEGF) (Torcia et al., 1999; Shan et al., 2004). Mice were treated from day one with SOS or control (saline) via a once daily intraperitoneal injection as described in the Materials and Methods Section. As shown in Figure 2A, SOS had a significant inhibitory activity on tumor growth in this model by day 12 with the average volume of tumors treated with SOS being only 30% of those treated with saline (P <0.05). A similar reduction was found at day 17 (32%).
Fig. 2.

SOS reduces tumor volume. A: C57/BL6 male mice were injected subcutaneously with 1 ×106 B16/F10 cells and treated with SOS ((●) 100 mg/kg/day) or saline (○) daily as described in the Materials and Methods Section. The mean volume of the tumor(n =5) was determined on the day indicated. Note direct overlap of mean values for SOS and salineonday7.B:Osmotic pumps containing saline (○), heparin (□), or SOS (●) releasing 5 μg /h were implanted in the peritoneal cavity of C57/BL6 male mice. After 72 h mice were injected subcutaneously with 1 × 106 Lewis lung carcinoma cells. Mean tumor volume values (n =5) are shown. Error bars are SEM for both A and B.
A second model system was then investigated. Osmotic pumps with SOS, heparin, or saline were implanted in the peritoneal cavities of mice 72 h prior to implantation of Lewis lung carcinoma tumors. These tumors have been shown to produce high levels of VEGF another potent heparin-binding growth factor responsible for blood vessel growth (Folkman, 2001). The pumps provide a continuous release of drug in contrast to a single daily bolus. As shown in Figure 2B, heparin and SOS showed similar dynamics with regard to tumor volume regulation with both significantly inhibiting tumor growth (approximately 50%) compared to control by day 16 ( P <0.05). Interestingly, when the tumors were resected, all of the heparin-treated mice died of bleeding whereas all of the SOS-treated mice survived (data not shown).
SOS does not act as an anticoagulant
Given the similarity of SOS to heparin and the theory that heparin’s impact on cancer is via its anticoagulant properties, we next investigated whether these properties were shared by SOS. We hypothesized that the small size of SOS would preclude it from influencing the classic coagulation factors IIa (thrombin) and Xa which are the basis for much of heparin’s anticoagulative properties since these activities require a specific pentasaccharide sequence within heparin (Thunberg et al., 1982). PTT, PT, and thrombin times were determined for heparin and SOS to address this hypothesis. As shown in Table 1, both heparin and SOS had only minimal effects on PT times exhibiting a less than 3 sec increase from control. In contrast, heparin had a dramatic effect on PTT results, as expected, doubling the reaction time from that in the absence of heparin after an increase in concentration to 2.4 μg/ml. A similar increase was found with SOS but at a concentration 50 times higher (125 μg/ml). The most dramatic differences were seen with the thrombin assay. In the presence of 6 μg/ml heparin, no evident clot formed within the assay time period while the addition of SOS had no effect on clot time with concentrations as high as 750 μg/ml.
TABLE 1.
Effect of heparin and SOS on PT, PTT, and thrombin assays
| Concentration (μg/ml) | PT (sec) | PTT (sec) | Thrombin (sec) |
|---|---|---|---|
| Heparin | |||
| 0 | 13.0 | 27.3 | 19.3 |
| 0.6 | 12.8 | 29.5 | 21.9 |
| 1.2 | 13.1 | 37.3 | 25.4 |
| 1.8 | 13.4 | 47.2 | 30.6 |
| 2.4 | 3.5 | 57.9 | 35.8 |
| 3 | 13.2 | 73.1 | 49.1 |
| 3.6 | 13.4 | 87.6 | 57.5 |
| 4.2 | 13.3 | 102.7 | 95.5 |
| 4.8 | 13.6 | 119.1 | 111.7 |
| 5.4 | 14.9 | 139.6 | no clot |
| 6 | 14.8 | 178.3 | no clot |
| SOS | |||
| 0 | 12.4 | 30.3 | 17.9 |
| 50 | 12.9 | 44.5 | 16.1 |
| 125 | 13.2 | 57.1 | 15.9 |
| 250 | 13.6 | 76.0 | 15.9 |
| 375 | 13.8 | 95.5 | 16.5 |
| 500 | 14.4 | 115.9 | 16.8 |
| 750 | 14.9 | 165.4 | 17.3 |
We suspected that the modest anticoagulant activity we observed with SOS was due to the ability of this charged disaccharide to chelate calcium. In order to test this possibility, chelation curves were generated in a binding competition assay by titrating SOS or heparin using an Oregon green-BAPTA conjugated substrate in the presence of calcium. Both heparin and SOS were able to chelate calcium as evident by the sharp reduction in fluorescent intensity at 530 nm. However, heparin was active in the nanomolar concentration range with an IC50 less than 500 ng/ml whereas SOS showed activity only in the micromolar to millimolar range with an IC50 of ~800 μg/ml, similar to the range in the PTT assay that shows activity (Fig. 3).
Fig. 3.

SOS chelates calcium poorly. Both heparin and SOS chelate calcium but effective concentrations differ significantly. Heparin (□) and SOS (●) were mixed with CaCl2 and Oregon Green® BAPTA-2 and assayed for fluorescence intensity at 530 nm as described in the Materials and Methods Section. Values shown are mean ± SD from four samples and figure is representative of three independent experiments.
SOS inhibits FGF-2 binding and releases pre-bound FGF-2 from endothelial surfaces
Since anticoagulation does not seem to be a strong property of SOS and, hence, unlikely to play a role in the tumor growth inhibition evident (Fig. 2), we next looked at the effect of SOS on the heparin-binding growth factor FGF-2 given its relevance to endothelial and tumor cell proliferation (Folkman, 2001). It has been shown previously by our group (Forsten et al., 1997, 2000a; Fannon et al., 2000), and others (Moscatelli, 1992), that heparin and heparan sulfate can inhibit FGF-2 steady-state binding to cells. Using iodinated FGF-2 and BCE cells, a potent inhibitory effect on 125I-FGF-2 steady-state binding by SOS at 4° C was observed (Fig. 4A). An IC50 of ~2 μg/ml was found and essentially complete inhibition of binding was obtained with concentrations three orders of magnitude higher.
Fig. 4.
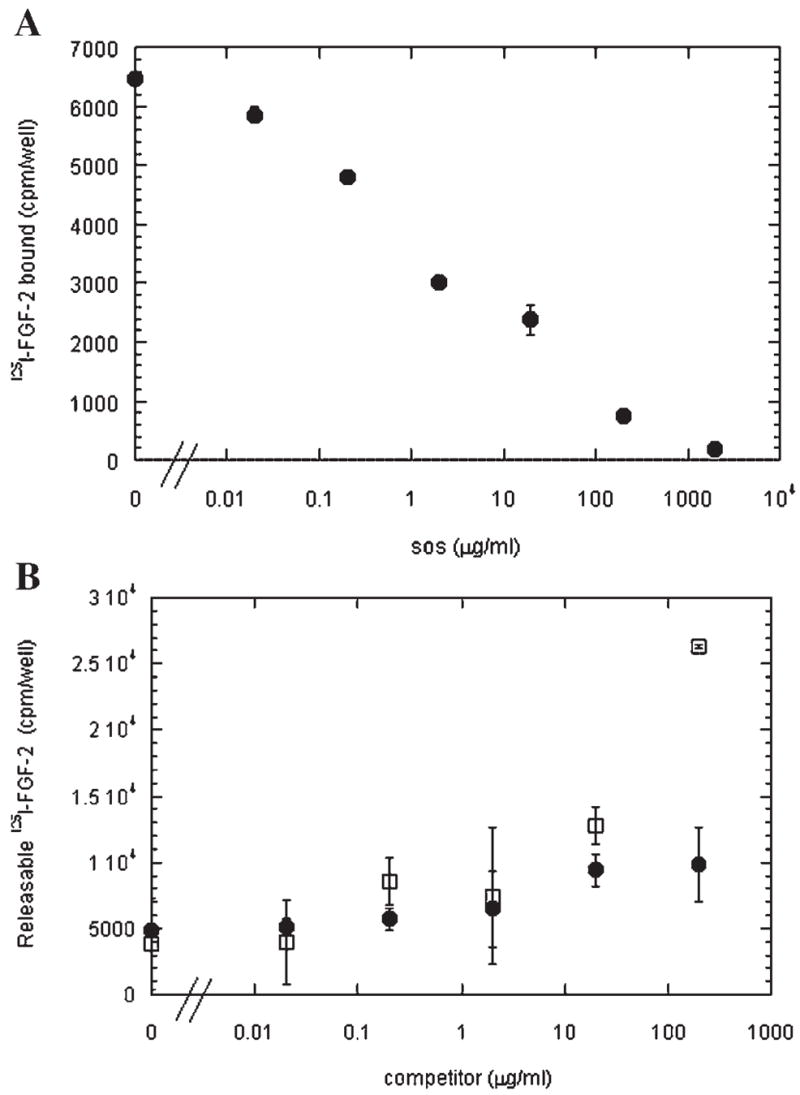
SOS inhibits binding and stimulates release of FGF-2 from BCE cells. A: Total binding of 125I-FGF-2 (1 ng/ml) to BCE cells at 4°C in the presence of SOS was performed as described in the Materials and Methods Section. Values shown are mean ± SD of the sum of HSPG-bound and receptor-bound from triplicate wells. These data are representative of three independent experiments. B: 125I-FGF-2 (1 ng/ml) was bound to BCE cells at 4°C and then switched to growth factor-free binding buffer supplemented with SOS (●) or heparin (□) as described in the Materials and Methods Section.
Heparin has been shown to have the capability to ‘‘strip’’ growth factors from vessel surfaces and extracellular matrices by competing for endogenous growth factor binding sites within the matrix (Whalen et al., 1989). Given the similar effect of SOS to heparin on FGF-2 steady-state binding, a competition experiment was conducted. 125I-FGF-2 was allowed to bind to cells at 4°C until steady state was achieved. Unbound 125I-FGF-2 was then washed away, SOS or heparin was added, and the cells were allowed to incubate for an additional 1.5 h period at 4°C to measure release. As shown in Figure 4B, both heparin and SOS were able to compete pre-bound 125I-FGF-2 from the endothelial cell surfaces and the inhibition was similar at concentrations less than 100 μg/ml. At 200 μg/ml, however, SOS released approximately double the FGF-2 released in the absence of competitor while heparin released close to seven times as much.
SOS has a biphasic effect on growth-factor-mediated BCE cell proliferation
Inhibition of steady-state growth factor binding and release of pre-bound FGF-2 suggest that cell proliferation would be inhibited by SOS based on loss of receptor activation. However, heparin has been shown to inhibit growth factor binding in equilibrium assays (Tessler et al., 1994) while increasing the proliferation rate of endothelial cells at similar concentrations in other studies (Weatherford et al., 1996). These findings are not necessarily at odds. The time frame for proliferation assays is typically on the order of days and performed at physiological temperature while steady-state binding assays are performed at 4°C and represent a ‘‘snapshot’’ of cellular activity. We therefore investigated how heparin and SOS would impact FGF-2-mediated proliferation of BCE cells. As shown in Figure 5, there was some potentiation of proliferation with both heparin and SOS at concentrations below 20 μg /ml followed by a reduction in proliferative response at higher concentrations. At 2,000 μg/ml, SOS-treated wells had cell numbers equivalent to that seen in the absence of FGF-2 indicating a complete inhibition of FGF-2 mediated proliferation. Similar effects were seen with SOS treatment of FGF-2-stimulated HUVEC (data not shown).
Fig. 5.
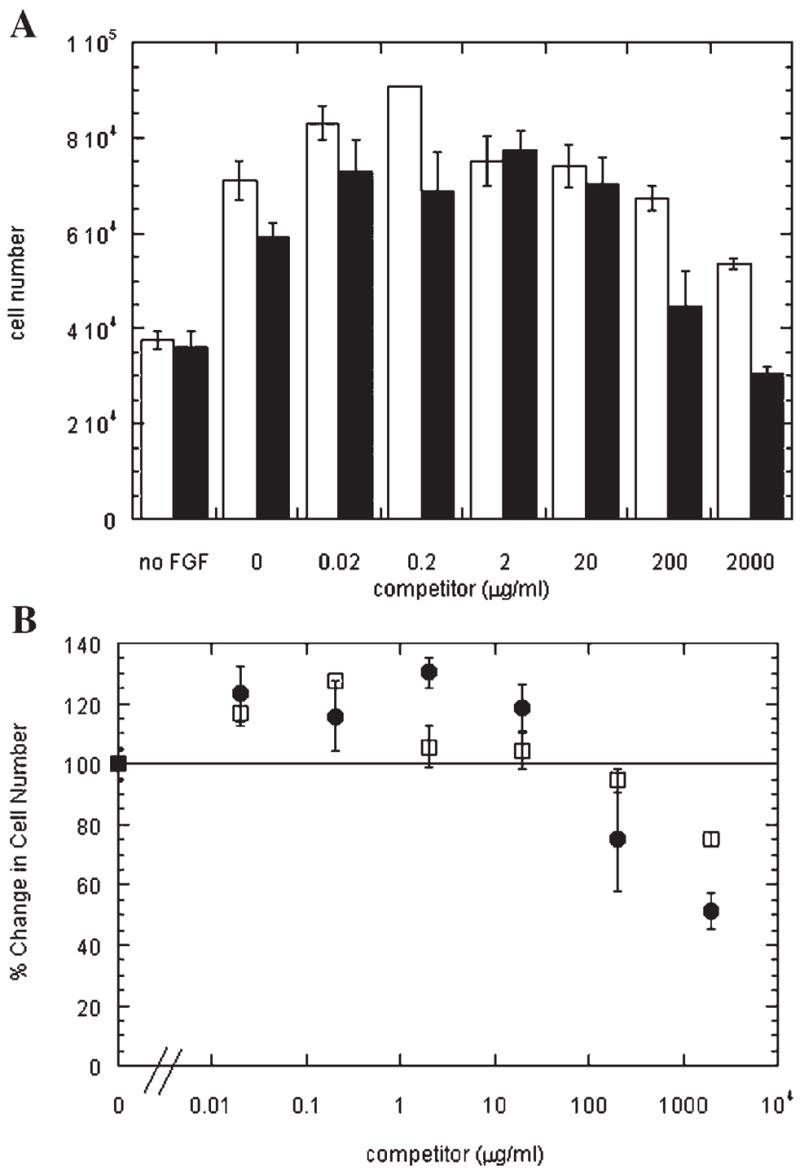
Both SOS and heparin have a biphasic effect on FGF-2 stimulated proliferation. A:BCE cells were seeded and stimulated with FGF-2 (1 ng/ml) in the presence of SOS (●) or heparin (□) at the given concentration as described in the Materials and Methods Section. Cell density was determined 72h later. B: Results from (A) are scaled to the percentage of BCE cells found following 72 hof stimulation with FGF-2. Note that FGF-2 stimulation in the absence of SOS (●) and heparin (□) is 240 ± 9% of control compared to basal growth in (A). Values shown are mean ± SD from triplicate wells and figure is representative of three independent experiments.
SOS increases growth factor transport across descemet’s membrane
Growth factor availability within the tumor environment is also a likely regulator of proliferation. Retention of FGF-2 within the ECM by heparan sulfate proteoglycans has been shown to control FGF-2 diffusion (Rosenbaum et al., 1986; Dowd et al., 1999) and we therefore hypothesized that, based on its small size, SOS could facilitate transport of heparin-binding growth factors across the matrix by competing with the larger matrix-bound HSPG for binding. The deposition of FGF-2 within the ECM might be a critical mechanism used to create diffusion gradients to drive and direct angiogenesis and other physiological and pathological events. Thus, the ability of SOS to modulate the interactions of FGF-2 with ECM could reflect an ability to remove FGF-2 from the tumor microenvironment, which could contribute to decreased tumor growth. Descemet’s membrane, the ECM layer sandwiched between the endothelium and the stroma in the cornea, was isolated from bovine cornea and used to study growth factor transport as it is a macroscopic easily obtainable natural ECM shown to contain HSPG (Dowd et al., 1999). Growth factor diffusion in the presence of heparin was not informative in our experimental model because heparin itself moves too slowly through the matrix likely due to its large hydrodynamic volume (data not shown). SOS, however, is much smaller (Fig. 1) and its impact on 125I-FGF-2 transport was investigated. In the absence of SOS, negligible 125I-FGF-2 crossed the membrane as indicated by the low levels found in the receiver chamber during the 10 h experimental period (Fig. 6A). In contrast, addition of SOS resulted in a dramatic increase in the amount of 125I-FGF-2 found within the receiver chamber. A delay of ~90 min was observed and this was followed by approximately first order transport over the 10 h period indicating that neutralization of binding and simple transport mechanisms were active. A similar enhancement of transport was observed with 125I-HB-EGF like growth factor) (Fig. 6B). No effects of SOS were observed with the non-heparin-binding analog, EGF, indicating that the effect of SOS was not the result of a general alteration of the transport properties of the membranes (Fig. 6C).
Fig. 6.

SOS increases transport of FGF-2 and HB-EGF but not EGF across Descemet’s membrane. 125I-FGF-2 (0.83 nM) (A), 125I-HB-EGF (2.2 nM)(B), or 125I-EGF (0.02 nM)(C) were added to the source side of the transport chamber in the presence (●) or absence (○) of SOS (0.5 mg/ml) and samples were taken from the receiver chamber as a function of time. Values shown represent the total amount of radio labeled growth factor within the receiver side. Figure is representative of three independent experiments.
Discussion
Tumor studies done over the course of many years have resulted in disparate findings as to the ability of heparin to inhibit tumor growth. The conflicting data may be due, in part, to a variety of conditions including tumor type, scheduling, dosage, and stage of disease. Early studies proposed that heparin interrupted mitosis by affecting the viscosity of the cytoplasm (Heilbrun, 1949). Other groups have proposed that fibrin was crucial for tumor cell adhesion, and heparin’s ability to inhibit fibrin formation at primary and secondary sites was responsible for reduction in tumor size and number of metastases when combined with chemotherapy (Elias et al., 1975). We demonstrate in this study that a heparin analog with little ability to inhibit coagulation or inhibit fibrin formation is nonetheless able to inhibit in vivo growth of an FGF-2 secreting tumor (Fig. 2). Based solely on these studies, however, we cannot preclude the possibility that SOS is able to influence thrombosis at other levels in our tumor models. Our in vitro studies support the hypothesis that SOS, through binding of FGF-2, is able to regulate FGF-2 activity. Whether this is the sole mechanism of activity with regard to tumor regulation is still under study but this work forms the foundation for those types of investigations.
The importance of heparin and heparan sulfate in growth factor interactions was recognized by us (Shing et al., 1984; Lobb et al., 1986; Nugent and Edelman, 1992) and others (Rapraeger et al., 1991; Yayon et al., 1991; Wong et al., 1995; Faham et al., 1996; Venkataraman et al., 1996) based on early work showing that some growth factors bind to heparin with high affinity. Since then, the family of heparin-binding growth factors has grown considerably. Although charged groups are important to heparin/heparan sulfate functionality (Rusnati et al., 1994), it has been shown that they are not sufficient for growth factor binding in some instances. For example, FGF-2 binds tightly to heparin and heparan sulfate but has little affinity for other proteoglycans such as chondroitin sulfate and keratan sulfate that have similar charge densities (Walicke, 1988; Caldwell and Svendsen, 1998). In addition, specificity of interaction with other functional groups within the GAG chain has also been demonstrated (Lapierre et al., 1996; Pye et al., 2000; Pankonin et al., 2005).
When studying GAGs it is often difficult to assess experiments based on equimolar concentrations. The heterogeneity of length, number of charged groups, and other modifications on heparin and heparan sulfate chains can complicate what would otherwise be straightforward analysis. Often it is informative to use mass equivalents rather than molar because of the differences in chain lengths in any heparin preparation. The use of units to normalize heparin activity presumes that the activity being measured has equal correlation to what are now known to be a multitude of heparin interactions in vivo. In our studies there is a significant difference between full-length heparin chains and the SOS disaccharide. This difference is certainly a factor in the inability of SOS to efficiently inhibit coagulation. Our PTT studies showed activity approximately 50-fold stronger for heparin than SOS based on mass. A comparison of molar equivalents would increase the difference by an order of magnitude. This underscores the advantage of using a molecule that has decreased anticoagulation properties with homogeneous size and activity, thus enabling much higher and predictable tolerated doses to be administered.
We have previously shown that SOS binds to FGF-2 with high affinity (Folkman et al., 1991) and here we show that it possesses additional characteristics formerly demonstrated for heparin. Two notable exceptions—minimal inhibition of blood coagulation (Table 1) and facilitated transport of growth factors through matrices (Fig. 6)—are shown here. Our proliferation assay results (Fig. 5) suggest that heparin and SOS, although only inhibitory in equilibrium binding studies, can slightly enhance FGF-2 proliferation at low concentration and progressively inhibit at high concentration. Biphasic responses such as this have been described for many molecules (Ilondo et al., 1994; Trinh et al., 2000). Local conditions such as the relative concentrations of the growth factor and SOS should have a significant influence on how molecules like SOS act. For example, studies by Yeh et al. (2002) show that SOS can promote dimerization of FGF-2-FGFR complexes in vitro in a concentration-dependent manner and also demonstrate that SOS, in combination with FGF-2, can increase osteocalcin expression and stimulate closure of coronal and saggital sutures of mouse calvaria explants in vitro. Their data certainly supports our enhanced proliferation result at low SOS concentrations and does not necessarily conflict with our inhibitory data at high SOS concentrations. Their in vitro studies included a 9 day incubation period in culture with a high concentration of FGF-2 where the kinetics of the SOS-growth factor binding and trafficking could lead to a similar regime with regard to FGF-2: SOS levels that we have during our 3 day period. Our studies finding growth inhibition were done at high ratios of SOS to FGF-2 which could lead to reduced receptor binding and altered signaling dynamics due to competition by free SOS. Further work is needed to better quantify and understand how SOS regulates proliferation by FGF-2 and other heparin-binding proteins. Another significant effect of SOS on growth factor activity was found in the diffusion studies across ECM. SOS significantly increased the transport of FGF-2 and HB-EGF through ECM while transport of EGF, a non-heparin-binding growth factor, was not significantly altered (Fig. 6). The addition of heparin did not result in a similar enhancement of heparin-binding growth factor transport likely because of slowed diffusion due to size constraints with heparin or potential interactions between heparin and the ECM (data not shown). SOS, by chaperoning heparin-binding growth factors, could inhibit storage of growth factors in the tumor microenvironment ultimately leading to their clearance from the area and disruption of potential growth factor gradients potentially necessary for tumor metastasis. Although the degradation of matrix and release of growth factors is generally associated with tumor growth and metastasis, the removal of growth factors from the matrix environment by a molecule such as SOS, without degradation or disruption of the matrix structure, might conversely ‘‘drain’’ the local environment of stimulating factors. Alternatively, one could envision use of SOS as a chaperone to enhance the tissue penetration of exogenous heparin-binding growth factors potentially increasing the effective delivery of growth factors to designed targets, thus serving to enhance cell activity. Ultimately the effect will likely be dependent on the desired application, the local cell and ECM environment, and the ability to obtain the desired dose of SOS within the correct location.
In conclusion, therapy that includes high concentrations of heparin is always limited by bleeding complications. LMWH, although less prone to complications, can still have side effects. We have demonstrated that SOS shares some characteristics of heparin including an ability to inhibit tumor growth and growth factor binding to cells, yet does not possess the potent anticoagulation activity that has limited the use of heparin for cancer therapy. From a therapeutic standpoint, SOS also has the advantage of being synthesized rather than being obtained from an animal source, which allows chemical homogeneity of product that is not possible with unfractionated heparin or LMWH. We have shown that SOS has many desirable characteristics indicating that the use of such molecules to manipulate endogenous or exogenous heparin-binding growth factor activity and bioavailability should be investigated further.
Acknowledgments
Contract grant sponsor: NIH;
Contract grant numbers: HL086644, HL56200.
Contract grant sponsor: Research to Prevent Blindness Challenge Grant.
M.A.N. is a consultant for Momenta Pharmaceuticals, Inc.
Literature Cited
- Altinbas M, Coskun HS, Er O, Ozkan M, Eser B, Unal A, Cetin M, Soyuer S. A randomized clinical trial of combination chemotherapy with and without low-molecular-weight heparin in small cell lung cancer. J Thromb Haemost. 2004;2:1266–1271. doi: 10.1111/j.1538-7836.2004.00871.x. [DOI] [PubMed] [Google Scholar]
- Bashkin P, Doctrow S, Klagsbrun M, Svahn CM, Folkman J, Vlodavsky I. Basic fibroblast growth factor binds to subendothelial extracellular matrix and is released by heparitinase and heparin-like molecules. Biochemistry. 1989;28:1737–1743. doi: 10.1021/bi00430a047. [DOI] [PubMed] [Google Scholar]
- Buczek-Thomas JA, Chu CL, Rich CB, Stone PJ, Foster JA, Nugent MA. Heparan sulfate depletion within pulmonary fibroblasts: Implications for elastogenesis and repair. J Cell Physiol. 2002;192:294–303. doi: 10.1002/jcp.10135. [DOI] [PubMed] [Google Scholar]
- Buczek-Thomas JA, Lucey EC, Stone PJ, Chu CL, Rich CB, Carreras I, Goldstein RH, Foster JA, Nugent MA. Elastase mediates the release of growth factors from lung in vivo. Am J Respir Cell Mol Biol. 2004;31:344–350. doi: 10.1165/rcmb.2003-0420OC. [DOI] [PubMed] [Google Scholar]
- Caldwell MA, Svendsen CN. Heparin, but not other proteoglycans potentiates the mitogenic effects of FGF-2 on mesencephalic precursor cells. Exp Neurol. 1998;152:1–10. doi: 10.1006/exnr.1998.6815. [DOI] [PubMed] [Google Scholar]
- Castelli R, Porro F, Tarsia P. The heparins and cancer: Review of clinical trials and biological properties. Vasc Med. 2004;9:205–213. doi: 10.1191/1358863x04vm566ra. [DOI] [PubMed] [Google Scholar]
- Coltrini D, Rusnati M, Zoppetti G, Oreste P, Isacchi A, Caccia P, Bergonzoni L, Presta M. Biochemical bases of the interaction of human basic fibroblast growth factor with glycosaminoglycans. New insights from trypsin digestion studies. Eur J Biochem. 1993;214:51–58. doi: 10.1111/j.1432-1033.1993.tb17895.x. [DOI] [PubMed] [Google Scholar]
- Dowd CJ, Cooney CL, Nugent MA. Heparan sulfate mediates bFGF transport through basement membrane by diffusion with rapid reversible binding. J Biol Chem. 1999;274:5236–5244. doi: 10.1074/jbc.274.8.5236. [DOI] [PubMed] [Google Scholar]
- Edelman ER, Nugent MA, Karnovsky MJ. Perivascular and intravenous administration of basic fibroblast growth factor: Vascular and solid organ deposition. Proc Natl Acad Sci U S A. 1993;90:1513–1517. doi: 10.1073/pnas.90.4.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlis HE, Goudsmit A, Brindley C, Niemetz J. Trial of heparin and cyclophosphamide (NSC-26271) in the treatment of lung cancer. Cancer Treat Rep. 1976;60:575–578. [PubMed] [Google Scholar]
- Elias EG, Shukla SK, Mink IB. Heparin and chemotherapy in the management of inoperable lung carcinoma. Cancer. 1975;36:129–136. doi: 10.1002/1097-0142(197507)36:1<129::aid-cncr2820360109>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Faham S, Hileman RE, Fromm JR, Linhardt RJ, Rees DC. Heparin structure and interactions with basic fibroblast growth factor. Science. 1996;271:1116–1120. doi: 10.1126/science.271.5252.1116. [DOI] [PubMed] [Google Scholar]
- Fannon M, Nugent MA. Basic fibroblast growth factor binds its receptors, is internalized, and stimulates DNA synthesis in Balb/c3T3 cells in the absence of heparan sulfate. J Biol Chem. 1996;271:17949–17956. doi: 10.1074/jbc.271.30.17949. [DOI] [PubMed] [Google Scholar]
- Fannon M, Forsten KE, Nugent MA. Potentiation and inhibition of bFGF binding by heparin: A model for regulation of cellular response. Biochemistry. 2000;39:1434–1445. doi: 10.1021/bi991895z. [DOI] [PubMed] [Google Scholar]
- Fannon M, Forsten-Williams K, Dowd CJ, Freedman DA, Folkman J, Nugent MA. Binding inhibition of angiogenic factors by heparan sulfate proteoglycans in aqueous humor: Potential mechanism for maintenance of an avascular environment. FASEB J. 2003;17:902–904. doi: 10.1096/fj.02-0935fje. [DOI] [PubMed] [Google Scholar]
- Fayein NA, Courtois Y, Jeanny JC. Basic fibroblast growth factor high and low affinity binding sites in developing mouse brain, hippocampus and cerebellum. Biol Cell. 1992;76:1–13. doi: 10.1016/0248-4900(92)90189-8. [DOI] [PubMed] [Google Scholar]
- Fisher RS. Sucralfate: A review of drug tolerance and safety. J Clin Gastroenterol. 1981;3:181–184. [PubMed] [Google Scholar]
- Folkman J. Harrison’s textbook of internal medicine. 15. Columbus, OH: McGraw-Hill; 2001. Angiogenesis; pp. 517–530. [Google Scholar]
- Folkman J, Shing Y. Control of angiogenesis by heparin and other sulfated polysaccharides. Adv Exp Med Biol. 1992;313:355–364. doi: 10.1007/978-1-4899-2444-5_34. [DOI] [PubMed] [Google Scholar]
- Folkman J, Szabo S, Stovroff M, McNeil P, Li W, Shing Y. Duodenal ulcer. Discovery of a new mechanism and development of angiogenic therapy that accelerates healing. Ann Surg. 1991;214:414–425. doi: 10.1097/00000658-199110000-00006. discussion 426–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsten KE, Courant NA, Nugent MA. Endothelial proteoglycans inhibit bFGF binding and mitogenesis. J Cell Physiol. 1997;172:209–220. doi: 10.1002/(SICI)1097-4652(199708)172:2<209::AID-JCP8>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Forsten KE, Fannon M, Nugent MA. Potential mechanisms for the regulation of growth factor binding by heparin. J Theor Biol. 2000a;205:215–230. doi: 10.1006/jtbi.2000.2064. [DOI] [PubMed] [Google Scholar]
- Forsten KE, Wang N, Robinson RM, Nugent MA. A simple assay for evaluating inhibitors of proteoglycan-ligand binding. Ann Biomed Eng. 2000b;28:119–127. doi: 10.1114/1.260. [DOI] [PubMed] [Google Scholar]
- Heilbrun LV. The effect of heparin on cell division. Proc Soc Exp Biol Med. 1949;70:179–182. doi: 10.3181/00379727-70-16866. [DOI] [PubMed] [Google Scholar]
- Ilan N, Elkin M, Vlodavsky I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int J Biochem Cell Biol. 2006;38:2018–2039. doi: 10.1016/j.biocel.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Ilondo MM, Damholt AB, Cunningham BA, Wells JA, De Meyts P, Shymko RM. Receptor dimerization determines the effects of growth hormone in primary rat adipocytes and cultured human IM-9 lymphocytes. Endocrinology. 1994;134:2397–2403. doi: 10.1210/endo.134.6.8194466. [DOI] [PubMed] [Google Scholar]
- Izvolsky KI, Shoykhet D, Yang Y, Yu Q, Nugent MA, Cardoso WV. Heparan sulfate-FGF10 interactions during lung morphogenesis. Dev Biol. 2003a;258:185–200. doi: 10.1016/s0012-1606(03)00114-3. [DOI] [PubMed] [Google Scholar]
- Izvolsky KI, Zhong L, Wei L, Yu Q, Nugent MA, Cardoso WV. Heparan sulfates expressed in the distal lung are required for Fgf10 binding to the epithelium and for airway branching. Am J Physiol Lung Cell Mol Physiol. 2003b;285:L838–846. doi: 10.1152/ajplung.00081.2003. [DOI] [PubMed] [Google Scholar]
- Kisker O, Onizuka S, Banyard J, Komiyama T, Becker CM, Achilles EG, Barnes CM, O’Reilly MS, Folkman J, Pirie-Shepherd SR. Generation of multiple angiogenesis inhibitors by human pancreatic cancer. Cancer Res. 2001;61:7298–7304. [PubMed] [Google Scholar]
- Klerk CP, Smorenburg SM, Otten HM, Lensing AW, Prins MH, Piovella F, Prandoni P, Bos MM, Richel DJ, van Tienhoven G, Buller HR. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J Clin Oncol. 2005;23:2130–2135. doi: 10.1200/JCO.2005.03.134. [DOI] [PubMed] [Google Scholar]
- Lapierre F, Holme K, Lam L, Tressler RJ, Storm N, Wee J, Stack RJ, Castellot J, Tyrrell DJ. Chemical modifications of heparin that diminish its anticoagulant but preserve its heparanase-inhibitory, angiostatic, anti-tumor and anti-metastatic properties. Glycobiology. 1996;6:355–366. doi: 10.1093/glycob/6.3.355. [DOI] [PubMed] [Google Scholar]
- Lindahl U. What else can ‘Heparin’ do? Haemostasis. 1999;29:38–47. doi: 10.1159/000054111. [DOI] [PubMed] [Google Scholar]
- Lippman M. The growth-inhibitory action of heparin on the Ehrlich ascites tumor in mice. Cancer Res. 1957;17:11–14. [PubMed] [Google Scholar]
- Lobb R, Sasse J, Sullivan R, Shing Y, D’Amore P, Jacobs J, Klagsbrun M. Purification and characterization of heparin-binding endothelial cell growth factors. J Biol Chem. 1986;261:1924–1928. [PubMed] [Google Scholar]
- Moscatelli D. Basic fibroblast growth factor (bFGF) dissociates rapidly from heparan sulfates but slowly from receptors. Implications for mechanisms of bFGF release from pericellular matrix. J Biol Chem. 1992;267:25803–25809. [PubMed] [Google Scholar]
- Mousa SA. Role of current and emerging antithrombotics in thrombosis and cancer. Drugs Today (Barc) 2006;42:331–350. doi: 10.1358/dot.2006.42.5.973580. [DOI] [PubMed] [Google Scholar]
- Nagashima R, Hirano T. Selective binding of sucralfate to ulcer lesion. I. Experiments in rats with acetic acid-induced gastric ulcer receiving unlabelled sucralfate. Arzneimittelforschung. 1980;30:80–83. [PubMed] [Google Scholar]
- Nakazawa S, Nagashima R, Samloff IM. Selective binding of sucralfate to gastric ulcer in man. Dig Dis Sci. 1981;26:297–300. doi: 10.1007/BF01308368. [DOI] [PubMed] [Google Scholar]
- Nugent MA, Edelman ER. Kinetics of basic fibroblast growth factor binding to its receptor and heparan sulfate proteoglycan: A mechanism for cooperactivity. Biochemistry. 1992;31:8876–8883. doi: 10.1021/bi00152a026. [DOI] [PubMed] [Google Scholar]
- Pankonin MS, Gallagher JT, Loeb JA. Specific structural features of heparan sulfate proteoglycans potentiate neuregulin-1 signaling. J Biol Chem. 2005;280:383–388. doi: 10.1074/jbc.M402645200. [DOI] [PubMed] [Google Scholar]
- Prandoni P, Lensing AW, Buller HR, Carta M, Cogo A, Vigo M, Casara D, Ruol A, ten Cate JW. Comparison of subcutaneous low-molecular-weight heparin with intravenous standard heparin in proximal deep-vein thrombosis. Lancet. 1992;339:441–445. doi: 10.1016/0140-6736(92)91054-c. [DOI] [PubMed] [Google Scholar]
- Pye DA, Vives RR, Hyde P, Gallagher JT. Regulation of FGF-1 mitogenic activity by heparan sulfate oligosaccharides is dependent on specific structural features: Differential requirements for the modulation of FGF-1 and FGF-2. Glycobiology. 2000;10:1183–1192. doi: 10.1093/glycob/10.11.1183. [DOI] [PubMed] [Google Scholar]
- Rapraeger AC, Krufka A, Olwin BB. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 1991;252:1705–1708. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- Rosenbaum J, Tobelem G, Molho P, Barzu T, Caen JP. Modulation of endothelial cells growth induced by heparin. Cell Biol Int Rep. 1986;10:437–446. doi: 10.1016/0309-1651(86)90039-1. [DOI] [PubMed] [Google Scholar]
- Rusnati M, Coltrini D, Caccia P, Dell’Era P, Zoppetti G, Oreste P, Valsasina B, Presta M. Distinct role of 2-O-, N-, and 6-O-sulfate groups of heparin in the formation of the ternary complex with basic fibroblast growth factor and soluble FGF receptor-1. Biochem Biophys Res Commun. 1994;203:450–458. doi: 10.1006/bbrc.1994.2203. [DOI] [PubMed] [Google Scholar]
- Saksela O, Moscatelli D, Sommer A, Rifkin DB. Endothelial cell-derived heparan sulfate binds basic fibroblast growth factor and protects it from proteolytic degradation. J Cell Biol. 1988;107:743–751. doi: 10.1083/jcb.107.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan S, Robson ND, Cao Y, Qiao T, Li CY, Kontos CD, Garcia-Blanco M, Dewhirst MW. Responses of vascular endothelial cells to angiogenic signaling are important for tumor cell survival. FASEB J. 2004;18:326–328. doi: 10.1096/fj.03-0765fje. [DOI] [PubMed] [Google Scholar]
- Shear MS. Studies on the chemical treatment of tumors: The effect of disturbances in fluid exchange on transplanted mouse tumors. Am J Cancer. 1935;25:66–88. [Google Scholar]
- Shing Y, Folkman J, Sullivan R, Butterfield C, Murray J, Klagsbrun M. Heparin affinity: Purification of a tumor-derived capillary endothelial cell growth factor. Science. 1984;223:1296–1299. doi: 10.1126/science.6199844. [DOI] [PubMed] [Google Scholar]
- Sideras K, Schaefer PL, Okuno SH, Sloan JA, Kutteh L, Fitch TR, Dakhil SR, Levitt R, Alberts SR, Morton RF, Rowland KM, Novotny PJ, Loprinzi CL. Low-molecular-weight heparin in patients with advanced cancer: A phase 3 clinical trial. Mayo Clin Proc. 2006;81:758–767. doi: 10.4065/81.6.758. [DOI] [PubMed] [Google Scholar]
- Tessler S, Rockwell P, Hicklin D, Cohen T, Levi BZ, Witte L, Lemischka IR, Neufeld G. Heparin modulates the interaction of VEGF165 with soluble and cell associated flk-1 receptors. J Biol Chem. 1994;269:12456–12461. [PubMed] [Google Scholar]
- Thunberg L, Backstrom G, Lindahl U. Further characterization of the antithrombin-binding sequence in heparin. Carbohydr Res. 1982;100:393–410. doi: 10.1016/s0008-6215(00)81050-2. [DOI] [PubMed] [Google Scholar]
- Torcia M, Lucibello M, De Chiara G, Labardi D, Nencioni L, Bonini P, Garaci E, Cozzolino F. Interferon-alpha-induced inhibition of B16 melanoma cell proliferation: Interference with the bFGF autocrine growth circuit. Biochem Biophys Res Commun. 1999;262:838–844. doi: 10.1006/bbrc.1999.1292. [DOI] [PubMed] [Google Scholar]
- Trinh L, Noronha SB, Fannon M, Shiloach J. Recovery of mouse endostatin produced by Pichia pastoris using expanded bed adsorption. Bioseparation. 2000;9:223–230. doi: 10.1023/a:1008133914854. [DOI] [PubMed] [Google Scholar]
- Venkataraman G, Sasisekharan V, Herr AB, Ornitz DM, Waksman G, Cooney CL, Langer R, Sasisekharan R. Preferential self-association of basic fibroblast growth factor is stabilized by heparin during receptor dimerization and activation. Proc Natl Acad Sci U S A. 1996;93:845–850. doi: 10.1073/pnas.93.2.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlodavsky I, Folkman J, Sullivan R, Fridman R, Ishai-Michaeli R, Sasse J, Klagsbrun M. Endothelial cell-derived basic fibroblast growth factor: Synthesis and deposition into subendothelial extracellular matrix. Proc Natl Acad Sci U S A. 1987;84:2292–2296. doi: 10.1073/pnas.84.8.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Tempelhoff GF, Harenberg J, Niemann F, Hommel G, Kirkpatrick CJ, Heilmann L. Effect of low molecular weight heparin (Certoparin) versus unfractionated heparin on cancer survival following breast and pelvic cancer surgery: A prospective randomized double-blind trial. Int J Oncol. 2000;16:815–824. doi: 10.3892/ijo.16.4.815. [DOI] [PubMed] [Google Scholar]
- Walicke PA. Interactions between basic fibroblast growth factor (FGF) and glycosoaminoglycans in promoting neurite outgrowth. Exp Neurol. 1988;102:144–148. doi: 10.1016/0014-4886(88)90087-8. [DOI] [PubMed] [Google Scholar]
- Weatherford DA, Sackman JE, Reddick TT, Freeman MB, Stevens SL, Goldman MH. Vascular endothelial growth factor and heparin in a biologic glue promotes human aortic endothelial cell proliferation with aortic smooth muscle cell inhibition. Surgery. 1996;120:433–439. doi: 10.1016/s0039-6060(96)80320-5. [DOI] [PubMed] [Google Scholar]
- Whalen GF, Shing Y, Folkman J. The fate of intravenously administered bFGF and the effect of heparin. Growth Factors. 1989;1:157–164. doi: 10.3109/08977198909029125. [DOI] [PubMed] [Google Scholar]
- Wong P, Hampton B, Szylobryt E, Gallagher AM, Jaye M, Burgess WH. Analysis of putative heparin-binding domains of fibroblast growth factor-1. Using site-directed mutagenesis and peptide analogues. J Biol Chem. 1995;270:25805–25811. doi: 10.1074/jbc.270.43.25805. [DOI] [PubMed] [Google Scholar]
- Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64:841–848. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]
- Yeh BK, Eliseenkova AV, Plotnikov AN, Green D, Pinnell J, Polat T, Gritli-Linde A, Linhardt RJ, Mohammadi M. Structural basis for activation of fibroblast growth factor signaling by sucrose octasulfate. Mol Cell Biol. 2002;22:7184–7192. doi: 10.1128/MCB.22.20.7184-7192.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]