

Abstract
We have developed a general strategy for creating littermates bearing either a tissue-specific point mutation or deletion in any target gene, and used the method to dissect the roles of Brg, the ATPase subunit of the chromatin-remodeling Brg-associated factor (BAF) complex, in early thymocyte development. We found that a point mutation that inactivates the Brg ATPase recapitulates multiple defects previously described for Brg deletion (Chi, T.H., M. Wan, P.P. Lee, K. Akashi, D. Metzger, P. Chambon, C.B. Wilson, and G.R. Crabtree. 2003. Immunity. 19:169–182). However, the point mutant helps reveal unexpected roles of Brg in CD25 repression and CD4 activation. Surprisingly, CD4 activation occurs independently of the Brg ATPase and is perhaps mediated by physical interactions between Brg and the CD4 locus. Our study thus suggests that the BAF complex harbors novel activities that can be necessary and even sufficient for stimulating transcription from an endogenous chromatin template in the absence of Brg-dependent remodeling of that template. We conclude that conditional point mutants, rarely used in mammalian genetics, can help uncover important gene functions undetectable or overlooked in deletion mutants.
Intrathymic development of T lymphocytes bearing αβ antigen receptor (TCRαβ) is one of the best-defined ontogenetic systems in vertebrates (1). The earliest T cells express neither CD4 nor CD8 and are known as double-negative (DN) cells. DN cells pass through four successive stages (DN1–4), as defined by differential expression of CD25 and CD44. Cells at the DN3 stage recombine the TCRβ gene to express a pre-TCR complex that propels DN3 cells into the DN4 stage. DN4 cells activate CD8 transcription to become immature single-positive (ISP) cells that subsequently derepress CD4 to become CD4+CD8+ double-positive (DP) cells (2). DP cells displaying proper TCRαβ extinguish either CD4 or CD8 expression and mature into CD8 or CD4 SP cells, respectively, before their export to the periphery (3).
The execution of transcriptional programs during development relies on precise temporal- and spatial-specific regulation of gene expression, which in turn requires the modulation of chromatin structure of target genes. An important class of enzymes capable of manipulating chromatin structure is the “chromatin-remodeling complexes,” the multisubunit molecular motors that use energy derived from ATP hydrolysis to physically disrupt histone–DNA contacts (4). The best-known chromatin remodeler in mammals is the Brg-associated factor (BAF) complex related to the yeast Swi–Snf complex (5). The BAF complex contains ∼10 subunits, including the ATPase Brg or its homologue Brm. In addition to Brg/Brm, several other subunits of the BAF complex are encoded by gene families, thus leading to the combinatorial assembly and generation of perhaps hundreds of complexes with divergent functions (6). The BAF complex plays diverse roles in the immune system (7) and is essential for early T cell development, because Brg deletion in DN2, DN3, and DN4 cells impairs their survival, proliferation, and developmental transitions, and causes premature CD4 expression on these cells (8, 9).
Although the BAF complex can remodel chromatin, it might have functions in gene regulation beyond classical ATP-dependent remodeling, as suggested by the following data. Specifically, the ATPase activity of Brg and, therefore, chromatin remodeling by the BAF complex is critically dependent on a lysine residue (K785) located in the nucleotide-binding pocket of the ATPase domain of Brg (10, 11). Remarkably, although the ATPase-dead Brg mutant Brg (K785R) fails to activate the mouse mammary tumor virus promoter integrated into the genome of a Brg-deficient cell line, it efficiently stimulates the same promoter in transient transfection assays (12). In contrast to the stably integrated promoter that is organized into nucleosomes, the transiently transfected promoter is nucleosome free (13), which apparently circumvents the need for ATP-dependent remodeling, thus revealing the potential of the BAF complex to activate transcription independently of Brg ATPase activity, although the data interpretation is confounded by the fact that Brg (K785R) was overexpressed in that study. The existence of the putative novel activities of the BAF complex unrelated to Brg-catalyzed remodeling is consistent with the fact that although the BAF complex consists of 10 subunits, 4 subunits (including Brg) are fully sufficient for nucleosome disruption in vitro (14), which suggests that the remaining “dispensable” subunits may harbor the novel activities. In addition, Brg itself may also harbor such activities in regions outside its ATPase domain.
These data raise the question of whether the phenotype seen in Brg-deleted thymocytes specifically reflects defects in chromatin remodeling. Brg is essential for the integrity of the BAF complex (15), for tethering the BAF complex to nuclear matrix (15), for recruiting the BAF complex to certain target genes via interaction with sequence-specific transcription factors (16), and presumably for stable association of the BAF complex with chromatin via its bromodomain and a region rich in lysines and arginines (17, 18). Thus, Brg deletion may have completely eliminated the functions of the entire BAF complex, suggesting that the phenotype in Brg-deleted cells reflects both ATPase-dependent and -independent roles of the BAF complex in gene regulation.
To test whether Brg has ATPase-independent activity and to dissect the complex phenotype seen in Brg deletion mutants, we generated mice bearing a thymocyte-specific Brg (K785R) point mutation. These mice were produced within the same litters as mice lacking Brg in thymocytes, allowing for direct phenotypic comparison. We found that Brg ATPase is generally required for Brg function, but Brg does have an unexpected, ATPase-independent role in CD4 regulation that is obscured in the Brg deletion mutant. Brg point mutation also reveals a role of Brg in CD25 regulation not detected in the Brg deletion mutant.
RESULTS
Generation of littermates bearing a thymocyte-specific Brg point mutation or Brg deletion
We first generated a bifunctional Brg allele (BrgO) that is normally a null allele but can be conditionally turned into the point mutant allele expressing the Brg (K785R) protein. Specifically, we introduced K/R point mutation into the exon that encodes part of the ATP-binding site in the Brg ATPase domain (Fig. 1 A). At the same time, we placed, downstream of the mutant exon, a removable promoter-trapping cassette consisting of a splice acceptor (SA) and the neomycin resistance gene (Neo). Upon splicing, the promoter-trapping cassette captures the mutant exon, leading to the production of a fusion protein between the N terminus (aa 1–813) of Brg and Neo. This fusion protein is unstable and, therefore, does not act as a dominant negative to interfere with the function of the WT Brg protein (see Fig. 1 H), but it can be converted into the full-length Brg (K/R) protein upon Cre-mediated excision of the gene-trapping cassette and subsequent restoration of correct splicing between the mutant exon and the downstream exon (Fig. 1 A). We then bred mice bearing the BrgO allele, the “traditional” floxed Brg (BrgF) allele (8, 19), and the transgene expressing Cre in thymocytes from the LCK-proximal promoter (LCK-Cre; Fig. 1 B) (20). In these mice, nonthymocytes express Brg (1–813)–Neo fusion and WT Brg from the BrgO and BrgF alleles, respectively, whereas in thymocytes, the fusion is converted into the Brg (K/R) point mutant and WT Brg is eliminated. These mice can be generated within the same litters as BrgF/F; LCK-Cre mice by crossing BrgF/F; LCK-Cre with BrgF/O, thus allowing direct comparison of the phenotypes caused by Brg point mutation versus Brg deletion (Fig. 1 B).
Figure 1.

Generation of conditional Brg (K785R) point mutation. (A) A removable promoter-trapping cassette is used to generate a bifunctional Brg allele that is normally a null allele but is converted into the point mutant allele expressing Brg(K785R) upon Cre expression. (B) Brg (K785R) is specifically expressed in thymocytes in mice bearing the bifunctional allele (BrgO) in combination with the traditional floxed Brg allele (BrgF) and LCK-Cre (left). The phenotypes are then compared with that seen in traditional conditional Brg knockout mice (right) to reveal a potential ATPase-independent function of the BAF complex during T cell development. (C) Targeting strategy. Four relevant exons in the Brg locus are depicted, arbitrarily designated E1–4. A floxed promoter-trapping cassette—consisting of an adenoviral SA, the promoterless neomycin resistant gene (Neo), IRES, a destabilized version of GFP, and an SV40 polyadenylation signal (pA)—is inserted between E2 and E3. The primers for screening ES cells (a–e) and for detecting Cre-mediated recombination (f and g) are indicated. Primers (h and i) are used for detecting the BrgF allele in thymocytes (see Fig. 2 B; and Fig. 3, E and F). Also indicated are the two exons (E3 and 4) floxed in the BrgF allele. The asterisks indicate Brg K/R point mutations. (D–H) Analysis of ES cells. The integration of left and right arms was screened by RT-PCR (D) and PCR (E), respectively. Cre-mediated recombination was confirmed by PCR (F). The targeting was confirmed with Southern blotting (G), where the genomic DNA was digested with Sca I and Not1 cutting outside and within the targeting vector, respectively, thus releasing a 5.5-kb fragment encompassing the Neo sequence. The Brg-Neo fusion protein was analyzed by Western blotting (H). Primer pairs used for RT-PCR were indicated to the right of the gel images in D–F. The asterisk in D denotes the PCR product amplified from the mRNA expressed from the bifunctional allele before E2-Neo splicing, and/or from the contaminating genomic DNA sequence. Unmodified ES cells (Ctrl) are used as controls where indicated.
The targeting vector is depicted in Fig. 1 C. The floxed promoter-trapping cassette contains the promoterless Neo–internal ribosome entry site (IRES)–GFP sequence downstream of an adenoviral SA. This cassette is flanked by Brg genomic sequences, the left arm being a 1.8-kb genomic fragment containing the exon (E2) bearing K/R point mutation, whereas the right arm is a 4.6-kb fragment containing another exon (E3). The vector was electroporated into embryonic stem (ES) cells, and a total of 72 colonies emerged after G418 selection, 22 of which were analyzed. Expectedly, most of the clones express RNA in which E2 is spliced to Neo, as detected by RT-PCR using primers b and c recognizing E2 and Neo, respectively (Fig. 1 D, top). To determine whether the Brg locus has been targeted, RT-PCR was performed using primers a and c, the former recognizing an exon (E1) not present in the targeting vector. Clones #5 and #14 expressed RNA that contains E1 spliced to Neo, indicating that the left arm is correctly recombined into the Brg locus in these two clones (Fig. 1 D, bottom). To check the integration of the right arm, DNA from these two clones was subjected to PCR using primers d and e, the latter recognizing an exon (E4) not present in the vector. An amplicon of the expected size (∼5 kb) was obtained, indicating correct recombination of the right arm (Fig. 1 E). To determine whether Cre can indeed excise out the Neo-IRES-GFP cassette from the targeted locus, a Cre-expressing plasmid was electroporated into the ES cells, and the resultant DNA was analyzed by PCR using primers f and g recognizing the 5′ LoxP site and a Brg genomic sequence downstream of the 3′ LoxP site. Excision of the floxed promoter-trapping cassette would shorten the distance between the two primers, leading to the amplification of 170 bp DNA. Indeed, rearrangement was specifically detected in the transfected ES cells (Fig. 1 F).
Southern blotting using a Neo probe was performed, which confirmed that the gene-trapping cassette was correctly targeted and that there is no secondary integration of concatemers, as the latter would have generated an ∼13-kb band, the size of the linearized targeting vector (Fig. 1 G).
Western blotting using an antibody against the N terminus of Brg was performed to assess the level of Brg (1–813)–Neo fusion protein in the ES clones. This fusion is structurally similar to the Brg-Neo fusion expressed by the constitutive Brg null allele (Brg−), the latter known to be unstable and undetectable by Western blotting (21). Indeed, the antibody detected full-length Brg but not the fusion protein expressed from the BrgO allele that should migrate at 120 kD (Fig. 1 H), suggesting that the fusion protein is unstable and thus should not interfere with the function of WT Brg protein, although the protein must be present at some level to confer resistance to neomycin for the selection of ES cells. Alternatively, the protein may be present at a high level but still be undetectable because of masking of the Brg epitope, but this seems less likely because the BrgO allele behaves as a null allele rather than a dominant-negative allele (see Fig. 2 A). In addition, there is no evidence for alternative splicing between E2 and E3 in BrgO RNA, consistent with the fact that the mRNA bearing E2 spliced to E3 is twofold less abundant in BrgO/+ cells as compared with WT cells (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20080938/DC1). Collectively, these data indicate that the BrgO allele is a true null allele before the excision of the gene-trapping cassette.
Figure 2.

Effects of Brg point mutation and Brg deletion on thymocyte development. (A) Thymocytes were resolved into various subsets based on CD3, CD25, and CD44 expression, and the CD4/CD8 expression profile of each subset was then determined. The genotypes of the mice are indicated (far right). Mice in rows A–C were analyzed at a different time than mice in rows D–F. The total cell number of each strain (±SD) is indicated inside the FACS plots in column 1. Red circles mark significantly changed populations; green borders are shown to facilitate comparisons. Note that in Brg point mutants, CD25 expression in post-DN3 cells is elevated, and therefore the post-DN3 gates are correspondingly enlarged (row E, column 2). Percentages of cells are shown. Data are representative of three independent experiments. (B) PCR analysis of the BrgO and BrgF alleles. DNA was isolated from thymocytes pooled from multiple mice to avoid sampling errors. Asterisks indicate internal controls for PCR (see Materials and methods for details). (C) Quantification of cellularity. (top) Comparison of the abundance of total, DN3, and post-DN3 thymocytes in the Brg mutants relative to those in WT mice, set as 100. (bottom) Comparison of the percentages of various components of the post-DN3 pool in the Brg mutants. Data represent the means from three mice.
GFP in the BrgO allele was intended for marking the cells that have undergone Cre-mediated excision; those cells should lose GFP expression. As GFP has a long half-life (24 h), we fused GFP to the protein degradation sequence PEST to reduce its half-life. Unfortunately, none of the ES colonies that emerged after G418 selection had visible GFP expression (unpublished data), suggesting that the rapid degradation has eliminated GFP protein (see Discussion). Mice bearing the BrgO allele were then generated, and their thymocytes were analyzed as described below.
BrgO/+ mice recapitulate the thymocyte defects in Brg+/− mice
We first determined the thymic phenotypes in BrgO/+ mice bearing the BrgO allele together with the WT Brg allele. Developmental stages of thymocytes are usually defined by a CD4/CD8 expression pattern, but such a definition is invalid if the coreceptors are misregulated, as in the case of Brg deletion that derepresses CD4 while impairing CD8 expression (8, 22). We therefore used CD25/CD44, together with CD3, as maturation markers, as previously described (8, 22). In brief, total thymocytes are first resolved into mature (CD3hi) and immature (CD3−/lo) subsets based on CD3 expression (Fig. 2 A, column 1). The immature thymocytes are then resolved into two major subsets, namely DN3 (CD25+CD44−) and post-DN3 (CD25−CD44−; Fig. 2 A, column 2) before each population is analyzed for CD4/CD8 expression (columns 3–5). In WT mice, the DN3 cells are, by definition, CD4−CD8− (Fig. 2 A, row A, column 3). The post-DN3 compartment in WT mice consists of DN4 (CD4−CD8−), ISP (CD4−CD8+), and DP cells; the DN4 and ISP subsets are enriched and detectable only within the “post-DN3 blasts” population, the most immature cells in the post-DN3 pool marked by high forward scatter and a complete lack of CD3 expression (FShiCD3−; Fig. 2 A, row A, column 5).
We have previously shown that in Brg+/− mice, the only detectable phenotype is impairment in CD8 expression in early thymocytes, which is manifested as a triad of concurrent developmental defects within post-DN3 blasts: loss of ISP cells, emergence of a novel CD4+CD8− population, and reduction in CD8 expression in early DP cells (22). These defects are recapitulated in BrgO/+ mice (Fig. 2 A, row B, column 5), as expected from the fact that the BrgO allele is a null allele by default. In addition, as in the case of Brg+/− mice, there is no other obvious thymocyte defect in BrgO/+ mice. In particular, these mice contain normal numbers of cells in the DN4 compartment, reinforcing the notion that deleting one allele of Brg is not sufficient to derepress CD4 (22).
Loss of DN4 cells in BrgO/+; LCK-Cre+/− mice
We next examined the effects of Brg point mutation on T cell development by introducing the LCK-Cre transgene into BrgO/+ mice. There is no gross defect in T cell development in BrgO/+; Cre+/− mice, although the total thymocyte number is mildly reduced (from ∼136 to ∼98 million; Fig. 2 A, row C, column 1), largely because of a decrease in DP cells. Neither is there a significant change in the cellularity of the peripheral CD4 and CD8 cells (unpublished data). As in BrgO/+ mice, CD8 expression in early thymocytes is impaired, resulting in the loss of ISP and emergence of CD4+CD8− cells, but the CD8 expression level on DP cells seems less affected (Fig. 2 A, row C, column 5). Surprisingly, DN4 cells are completely missing in BrgO/+; Cre+/− mice (red circles). As DN4 cells are an essential intermediate in T cell development, these cells must be present but have somehow become phenotypically undetectable (see Discussion).
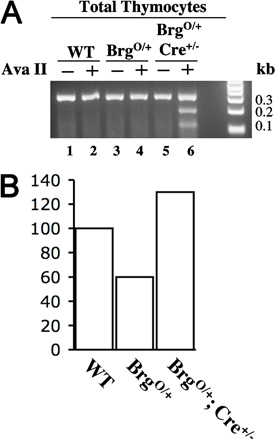
We then analyzed the status of the BrgO allele in various cell types isolated from the BrgO/+; Cre+/− mice. The BrgO allele appears to be fully converted to the Brg (K/R) allele in DP cells and in peripheral CD4 and CD8 cells, as indicated by the loss of the BrgO allele, whereas DN3 cells show little conversion (Fig. 2 B, top). The loss of the BrgO allele was accompanied by the emergence of the Brg (K/R) allele, as expected (unpublished data). This temporal-specific conversion is consistent with the progressive deletion of the BrgF allele by the LCK-Cre transgene in BrgF/+; Cre+/− mice, where deletion is marginal in DN2 and DN3 cells but complete by the DN4 stage (8).
We conclude that the BrgO→Brg (K/R) conversion is induced by the LCK-Cre transgene in BrgO/+; Cre+/− mice, but there is no gross perturbation of thymocyte development, suggesting that one allele of Brg is largely sufficient to mask the effects of Brg point mutation on thymocytes. Of note, this scenario is not generally true for other cell types, because we could not breed BrgO/+; Actin-Cre mice carrying a transgene ubiquitously expressing Cre, apparently because of embryonic lethality (unpublished data).
Phenotypic differences between BrgF/O; LCK-Cre+/− and BrgF/F; LCK-Cre+/− mice
To fully expose the effects of Brg point mutation on T cell development, we generated BrgF/O; LCK-Cre+/− mice in which Cre not only induces BrgO→Brg (K/R) conversion in the BrgO allele but also deletes the BrgF allele that is masking the effects of Brg point mutation. BrgF/F; LCK-Cre+/− mice were used as littermate controls.
We have previously reported that in BrgF/−; LCK-Cre+/− mice bearing the BrgF allele together with the constitutive Brg null allele, BrgF is hardly deleted in DN3 but completely deleted in DN4 cells, and Brg-deleted DN4 cells rapidly die by apoptosis before they can further develop (8). Similarly, in BrgF/O; Cre+/− mice, thymocyte numbers are severely reduced (∼13-fold) because of a loss of post-DN3 cells, and there are few CD4/CD8 SP cells in the thymus or periphery, whereas DN3 cellularity is unaffected (Fig. 2 A, row E, columns 1, 2, and 6; and Fig. 2 C, top). This indicates that Brg ATPase is important for thymocyte development. However, a small number of post-DN3 cells (∼5 million) manage to develop in BrgF/O; Cre+/− mice, whereas post-DN3 cells are virtually absent in BrgF/−; LCK-Cre+/− mice, suggesting that Brg (K/R) retains the residual ability to support post-DN3 production. In addition, the presence of two loxP sites in the BrgO allele might impair BrgF deletion via competition for the Cre recombinase, thus contributing to the generation of the post-DN3 cells. Interestingly, a similar number of post-DN3 cells is also generated in BrgF/F; Cre+/− mice, which presumably also represents a “leaky” phenotype resulting from incomplete BrgF deletion (Fig. 2 A, row F, columns 1 and 2; and Fig. 2 C, top). Importantly, the post-DN3 cells in the two Brg mutant strains differ in two ways, and the difference apparently cannot be ascribed to the leakiness in the phenotype in BrgF/F; Cre+/− mice. First, these cells aberrantly express low to intermediate levels of CD25 only in BrgF/O; Cre+/− mice (Fig. 2 A, rows D–F, column 2). The second difference lies in the composition of post-DN3 cells. Specifically, BrgF/O; Cre+/− mice harbor a conspicuous CD4+CD8− population that constitutes, on average, 38% of the total post-DN3 pool, whereas this population makes up only 14% in BrgF/F; Cre+/− mice (Fig. 2 A, rows E and F, column 4; and Fig. 2 C, bottom). On the other hand, a CD4−CD8+ subset, analogous to ISP cells, is seen only in BrgF/F; Cre+/− mice (Fig. 2 A, rows E and F, column 4; and Fig. 2 C, bottom). It is not clear whether this subset is bona fide ISP cells, because the majority of this subset has small cell size, whereas ISP cells in WT mice are large blasts.
We then isolated major thymocyte subsets and analyzed the status of the Brg alleles. As expected, in BrgF/O; Cre+/− mice, BrgO→Brg (K/R) conversion and BrgF deletion are each minimal/undetectable in DN3 cells but appear to be complete in CD4+CD8− and DP cells (Fig. 2 B, middle, lanes 2–4). Similarly, in BrgF/F; Cre+/− mice, BrgF deletion is minimal/undetectable in DN3 and virtually complete in post-DN3 (i.e., CD4+CD8−, CD4−CD8+, and DP) cells (Fig. 2 B, bottom, lanes 2–5); the presence of residual amounts of the BrgF allele in post-DN3 cells, revealed by faint PCR bands in lanes 3–5, is consistent with the aforementioned leaky phenotype in these mice. We conclude that Brg point mutation produces similar but not identical defects as Brg deletion.
Phenotypic differences in Bcl-xL-rescued thymocytes
Although the post-DN3 cells in the two Brg mutant strains show phenotypic differences, these cells are scarce and are undergoing apoptosis, thus confounding interpretations and hampering mechanistic studies. To circumvent these problems, we introduced a transgene expressing Bcl-xL under the control of the LCK-proximal promoter (23). This transgene, when introduced into BrgF/−; Cre+/− mice, was able to rescue Brg-deleted DN4 cells from apoptosis, leading to their accumulation (8). The DN4 cells do not accumulate because they hyperproliferate: these cells are in fact completely cell-cycle arrested, and most of them are atrophic. The accumulation instead results from a blockade in DN4→DP differentiation, based on the fact that DP cells are virtually absent even though DN4 cells are abundant in BrgF/−; Cre+/−; Bcl-xL+/+ mice. In addition, Brg-deleted DN3 cells, which are hardly detectable in BrgF/−; Cre+/− mice, are also rescued from death and subsequently accumulate. The accumulation similarly results from a blockade in differentiation (into DN4 cells) rather than hyperproliferation, because the rescued DN3 cells are cell-cycle arrested. Furthermore, a subset of Brg-deleted, Bcl-xL–rescued DN3 and DN4 cells undergo premature CD4 derepression (8).
The phenotype of BrgF/F; Cre+/−; Bcl-xL+/+ mice (hereafter called Brg Δ) is similar to that of BrgF/−; Cre+/−; Bcl-xL+/+ mice, although the former contains threefold more thymocytes. Specifically, total thymocyte cellularity is increased almost 10-fold, from 9 million in BrgF/F; Cre+/− mice to 83 million in Brg Δ mice (Fig. 3 A, row C). As in the case of BrgF/−; Cre+/−; Bcl-xL+/+ mice, this increase results largely from the accumulation of CD4− DN4 and CD4+ DN4 cells (8), i.e., the CD4−CD8− and CD4+CD8− populations in the post-DN3 pool; these cells are mostly atrophic and, thus, detectable not only within post-DN3 blasts but also in the total post-DN3 pool (Fig. 3 A, row C, columns 3 and 4). DP cells are present in Brg Δ mice, but their cellularity relative to DN4 cells is dramatically reduced as compared with that in WT mice, indicating a severe defect in the DN4→DP transition (Fig. 3 A, row A vs. C, column 3). Interestingly, the CD4−CD8+ subset, conspicuous in BrgF/F; Cre+/− mice, is absent from Brg Δ mice (Fig. 3 A, row C, column 3). Brg-deleted DN3 cells are similarly rescued, and they accumulate and undergo partial CD4 derepression as DN4 cells (Fig. 3 A, row C, column 2), and both DN3 and DN4 cells are cell-cycle arrested (Fig. 3 B).
Figure 3.

Phenotypic analysis of Bcl-xL–rescued thymocytes. (A) CD4/CD8 expression pattern of various thymocyte subsets. Cells were stained and analyzed as in Fig. 2 A. The total cell number (±SD) for each strain is indicated within the plots in column 1. Red circles mark differences in CD4 expression in the indicated subset between two mutants. The blue-shaded region in column 4 indicates the CD8− cells whose CD4 expression was analyzed in Fig. 4 A. In Brg* mice, CD25 is elevated in post-DN3 cells, and the post-DN3 gate is enlarged accordingly (row B, column 1). Percentages of cells are shown. Data are representative of three independent experiments. (B) Histograms comparing BrdU incorporation efficiencies of DN3 and 4 cells. BrdU was injected into mice and thymocytes were analyzed 15 min later. The numbers within the plots indicate the percentages of BrdU+ cells. At least two mice in each group were analyzed. (C) CD25 expression in immature T cells in A was analyzed by histograms. The vertical line within the plot marks the boundary between DN3 and post-DN3 cells, with the DN3 cells located to the right of the line. (D) Bar graph comparing the percentages of various components of the post-DN3 pool in the Brg mutants. Data represent the means from three mice. (E–G) Analysis of BrgO conversion and BrgF deletion at DNA (E and F) and protein (G) levels. Brg* and Brg Δ denote BrgF/O; Cre+/−; Bcl-xL+/+ and BrgF/F; Cre+/−; Bcl-xL+/+ mice, respectively. The PCR assays are identical to those in Fig. 2 B. Cell subsets were sorted from thymocytes pooled from multiple mice to avoid sampling errors.
The phenotype of the BrgF/O; Cre+/−; Bcl-xL+/+ (hereafter called Brg*) mice is similar to that of Brg Δ mice in four ways: total thymocyte cellularity is significantly increased (∼8-fold) as compared with BrgF/O; Cre+/− mice, largely because of an increase in post-DN3 cells (Fig. 3 A, column 1); DP cellularity relative to DN4 cells is much reduced (column 3); DN3 cells also accumulate and partially derepress CD4 (column 2); and finally, rescued cells are growth arrested (Fig. 3 B). Thus, Brg point mutation impairs thymocyte survival, proliferation, and developmental transition, as does the Brg deletion. However, there are two differences between the Brg mutant strains. First, in Brg* mice, as in BrgF/O; Cre+/− mice, CD25 expression is aberrantly elevated in post-DN3 cells (Fig. 3 A, column 1; and Fig. 3 C), whereas this aberrance is not seen in Brg Δ mice. Second, the composition of post-DN3 cells differs: CD4−DN4 cells constitute 25% of the total post-DN3 pool in Brg Δ but only 9% in Brg* mice cells, whereas CD4+DN4 cells constitute 39% in Brg Δ but 52% in Brg* mice (Fig. 3 D). Thus, CD4−DN4 cells are enriched in Brg Δ but depleted in Brg* mice, whereas the opposite is seen for CD4+DN4 cells. The two differences between the Brg mutant strains cannot be ascribed to potential leakiness in the phenotype of Brg Δ mice, because the same differences are observed when comparing Brg* and BrgF/−; Cre+/−; Bcl-xL+/+ mice. These differences seem to have emerged in BrgF/O; Cre+/− and BrgF/F; Cre+/− mice (Fig. 2), but the situation there is complicated by cell death.
We then determined the status of the Brg alleles in DN3 and post-DN3 cells. In Brg Δ mice, as in BrgF/−; Cre+/−; Bcl-xL+/+ mice (8), BrgF is almost completely deleted and Brg protein is undetectable (Fig. 3 F; and Fig. 3 G, lanes 5–7), whereas in Brg* thymocytes, BrgF is deleted and BrgO is converted, concomitantly with the expression of the Brg (K/R) protein (Fig. 3 E; and Fig. 3 G, lanes 3 and 4).
DN2 (CD25+CD44+) cells in Brg Δ and Brg* mice also accumulate and undergo partial CD4 derepression, but the extent of accumulation and CD4 derepression are rather variable in both strains (unpublished data), presumably because of inefficient and variable Cre-mediated deletion at this early stage. We conclude that Brg point mutation recapitulates the defects in cell survival, proliferation, and developmental transitions seen in Brg-deleted thymocytes, but a major difference between the two mutants lies in the unique ability of Brg point mutation to deplete CD4− DN4 cells, suggesting that Brg point mutation and deletion differentially affect CD4 transcription.
Brg deletion decreases the probability of CD4 transcription in DN4 cells
CD4 transcription is driven by the CD4 promoter and enhancer, together with a locus control region (LCR) necessary for suppressing the position-effect variegation in certain transgenic models (1, 24). Overall, these positive regulatory elements are constitutively active; the stage-specific fluctuation in CD4 expression depends on the CD4 silencer that overrides the CD4 enhancer/promoter, because CD4 silencer deletion leads to ectopic CD4 expression in DN and CD8 cells (25–27). The CD4−DN4 cells have been previously observed in BrgF/−; Cre+/−; Bcl-xL+/+ mice, constituting ∼37% of the post-DN3 population, and we have postulated that the CD4−DN4 cells arise because Brg deletion cannot completely inactivate the CD4 silencer, thus leading to CD4 derepression only in a subset of DN4 cells (8). Paradoxically, Brg point mutation, presumably less effective in silencer inactivation, is able to deplete CD4−DN4 cells, which prompted us to revisit the mechanism underlying the generation of CD4−DN4 cells.
We noticed that although ∼8% of cells in Brg* mice fall within the CD4−CD8− compartment, these CD4−DN4 cells appear to express low levels of CD4 and blend into the CD4+CD8− pool, whereas the CD4−DN4 cells in Brg Δ mice appear to truly lack CD4 expression, and in many cases, form a discrete population that can be readily discerned (Fig. 3 A, column 3, red circles). This trend is upheld in post-DN3 blasts (Fig. 3 A, column 4). The data suggest that Brg point mutation uniformly derepresses CD4 in DN4 cells and turns them into CD4+DN4 cells, whereas Brg deletion leads to variegated CD4 derepression, with only a subset of DN4 cells expressing CD4. To explore this possibility, we used histograms to quantify CD4 expression in the CD8− cells in Fig. 3 A, i.e., the blue-shaded population in post-DN3 blasts and the corresponding cells in the total post-DN3 pool. In both post-DN3 blasts and the total post-DN3 pool, there is only a single, CD4+ population in Brg* mice, but Brg Δ mice harbor a second population whose CD4 expression level is, surprisingly, even lower than that on DN4 cells from WT (BrgF/F) mice (Fig. 4 A, left). WT DN4 cells are known to marginally express CD4; although DN4 cells are by definition CD4−, these cells have actually begun to turn on CD4 transcription and express some CD4 protein on the cell surface (28). Presumably, a developmentally regulated, temporal-specific mechanism has partially inactivated the CD4 silencer in DN4 cells, thus triggering the onset of CD4 transcription. The complete lack of CD4 expression on CD4−DN4 cells therefore suggests that Brg deletion blocks the onset of CD4 transcription in a subset of DN4 cells, thus revealing a positive, ATPase-independent role of Brg in CD4 transcription. Interestingly, in contrast to DN4 cells, the CD4 expression pattern on DN3 cells is indistinguishable between the two Brg mutants, suggesting that Brg is dispensable for CD4 transcription at this stage (Fig. 4 A, right).
Figure 4.

Brg promotes uniform CD4 expression in DN4 cells. (A) Surface CD4 expression levels on the CD8− cells in post-DN3 pool (left) and on DN3 cells (right). The data are derived from the plots in Fig. 3 A. The CD8− cells within the post-DN3 blast (left) are located in the blue-shaded region in Fig. 3 A, column 4. (B) CD4 mRNA levels in various subsets of thymocytes analyzed by semiquantitative (top) and quantitative (bottom) RT-PCR in two independent experiments. The quantitative RT-PCR was done in triplicates, the values were averaged and normalized to that of actin mRNA, and the normalized values were plotted relative to those in BrgF/F DP cells, with the latter set as 100. UD, undetectable. (C) Effects of CD4 silencer deletion on CD4 expression in Brg Δ mice. CD8− cells from blastic and total post-DN3 cells were analyzed for CD4 expression (left), as were DN3 cells (right). Vertical lines are shown to facilitate the comparison of fluorescent intensities. Data are representative of two independent experiments. (D) Chromatin immunoprecipitation analysis of Brg binding to CD4 regulatory elements. A 5-million-cell equivalent of chromatin was added to each immunoprecipitation reaction using either rabbit serum (IgG) or an anti-Brg antibody (Brg), and 40% of precipitated DNA was analyzed by semiquantitative PCR. At least two independent sources of cells were used for chromatin preparation. Asterisks indicate an amplicon from the GAPDH gene. The sizes of the amplicons (bp) are indicated (left). DN, thymocytes from Rag2−/− mice, consisting mostly of DN3 cells; DP, total thymocytes from TCRα−/− mice; DN4, thymocytes at the DN stages from Brg mutants, 90% of which were DN4 cells (see Materials and methods). (E) Restriction enzyme accessibility of the CD4 enhancer and LCR. Cells were partially digested with Dde I or BseR1 (red arrows), followed by DNA isolation and complete digestion with a second enzyme (Bam H1 or Alu1) that generated normalization controls. The cleaved ends were detected by ligation-mediated PCR. The cells types used were the same as those in D. Note that CD4 enhancer is equally accessible in DN and DP cells (but less so in B cells), consistent with the fact that the enhancer is poised for activation in DN cells (reference 38). The restriction enzyme cleavage sites are depicted at the bottom, where the numbers refer to the nucleotide positions of the sites. The sizes of the PCR amplicons are indicated at the left of the gels. Data are representative of at least three independent experiments.
To confirm that Brg deletion prevents the onset of CD4 transcription in CD4−DN4 cells, we performed semiquantitative RT-PCR analysis (Fig. 4 B, top). As expected, in WT (BrgF/F) mice, CD4 mRNA is undetectable in DN3 cells but becomes detectable in DN4 cells, although the expression level is lower than DP cells (lanes 1–3). Importantly, CD4 mRNA is indeed undetectable in CD4−DN4 cells from Brg Δ mice (lane 7). As controls, we show that CD4 mRNA is present in CD4+ DN3/4 (but not CD4−DN3) cells in Brg Δ mice (lanes 4–6). Quantitative RT-PCR analysis confirmed that CD4 mRNA is detectable in WT but not mutant DN4 cells (Fig. 4 B, bottom).
The defect in CD4 transcription in Brg-deleted DN4 cells suggests that Brg is required for the functions of the CD4 promoter/enhancer/LCR. However, this “transcription defect” might be an artifact resulting from a failure to inactivate the silencer. In this scenario, the CD4 silencer somehow remains active in Brg-deleted CD4−DN4 cells, thus blocking CD4 expression. To exclude this possibility, we deleted the CD4 silencer in Brg Δ mice by crossing Brg Δ mice with silencer KO mice (25). Silencer deletion significantly enhances CD4 expression in DN4 cells from WT (BrgF/F) mice, as expected, but indeed fails to rescue CD4 expression in CD4−DN4 cells from Brg Δ mice (Fig. 4 C, left). Of note, silencer deletion induces CD4 expression on CD4−DN3 cells in Brg Δ mice (compare Fig. 4 C, left, with Fig. 4 A, right), indicating that the CD4−DN3 cells are generated simply because Brg deletion cannot completely inactivate the silencer, and confirming that Brg is dispensable for CD4 expression in DN3 cells.
Collectively, the data show that Brg uses an ATPase-independent activity to increase the probability of CD4 expression in DN4 cells; in the absence of Brg, CD4 transcription in DN4 cells becomes variegated, with only a subset of DN4 cells expressing CD4. Furthermore, this variegated expression pattern, once established, seems epigenetically stable, surviving in vitro culture for at least 3 d (unpublished data).
Brg binds the CD4 enhancer and LCR
We have previously shown that Brg binds the CD4 silencer, suggesting that Brg directly regulates CD4 silencer function (22). To explore how Brg stimulates CD4 expression, we analyzed Brg binding to the positive regulatory elements at the CD4 locus. Chromatin from various cell types was immunoprecipitated with an anti-Brg antibody and the DNA was analyzed by two sets of PCR. The first is a multiplex PCR that coamplifies sequences from the CD4 enhancer, promoter, and silencer together with a fragment from the GAPDH gene, which serves as an internal control (29). The second is a duplex PCR coamplifying sequences from the CD4 LCR and GAPDH genes. As expected, Brg binds the CD4 silencer in DN (mostly DN3) cells from Rag2−/− mice, and in DN (mostly DN4) cells from Brg* mice (Fig. 4 D, top, lanes 1 and 4). Remarkably, in these cells, Brg also binds the CD4 enhancer and LCR, although binding to the CD4 promoter is marginal (Fig. 4 D, lanes 1 and 4). Interestingly, in DP cells from WT mice, Brg binds all four CD4 regulatory elements, including the promoter (Fig. 4 D, lane 2). The binding is specific because the signals were undetectable in DN cells from Brg Δ mice or in B cells from WT mice (Fig. 4 D, lanes 3, 5, and 6). These data suggest that Brg facilitates CD4 transcription in DN4 cells by directly acting on CD4 enhancer/LCR.
Effects of Brg mutations on CD4 enhancer/LCR accessibility
We have begun to explore the molecular mechanism whereby Brg stimulates CD4 transcription. Our genetic data indicate that Brg uses a novel, ATPase-independent activity to stimulate CD4 transcription, which predicts that Brg-dependent remodeling of the CD4 enhancer/LCR, if any, is not relevant to the functions of these elements. To confirm this, we used restriction enzymes to probe the accessibility of the CD4 regulatory elements. Brg mutations strongly reduce the silencer accessibility (unpublished data), as expected from the essential role of the Brg ATPase in CD4 silencer function. In contrast, Brg mutations do not affect the LCR accessibility, suggesting that Brg-dependent remodeling does not play any role in LCR function (Fig. 4 E, right). Although Brg mutations do moderately reduce CD4 enhancer accessibility, the extent of decrease in CD4−DN4 cells is comparable to that in CD4+ DN4 cells, suggesting that the accessibility decrease cannot explain the selective loss of CD4 expression in CD4−DN4 cells (Fig. 4 E, left). These data are consistent with the idea that ATP-dependent remodeling is dispensable for Brg to stimulate CD4 transcription.
DISCUSSION
We have developed a method whereby mice with a thymocyte-specific point mutation in the Brg ATPase domain are generated within the same litters as mice deleting Brg in thymocytes. Brg point mutation recapitulates multiple defects produced by Brg deletion but unexpectedly alters CD25 expression and is far more effective in derepressing CD4 than Brg deletion. Further analysis indicates that Brg not only represses CD4 in DN cells but also ensures the onset of CD4 transcription during DN→DP transition; the latter effect, obscured in the Brg deletion mutant, is ATPase independent and perhaps mediated by physical interactions between Brg and the CD4 locus. This is the first demonstration that the ATPase-dead Brg point mutant, at a physiological concentration, is able to activate an endogenous gene. Our study thus proves that the BAF complex indeed harbors a novel activity in gene regulation distinct from ATP-dependent remodeling, the hallmark function of the BAF complex. Furthermore, it reveals that this activity is sufficient to regulate at least one target gene in the absence of Brg-dependent remodeling, which is counterintuitive given that endogenous genes are chromatinized. Our data highlight the complexity in the mechanisms of gene regulation in vivo, and raise the exciting possibility that the novel activities of the BAF complex might be widely involved in the regulation of BAF target genes, although by themselves such activities in general appear insufficient for gene regulation without concomitant Brg-dependent remodeling. Collectively, our study illustrates the power of conditional point mutations, rarely used in mammalian genetics, for dissecting and discovering gene functions. The strategy for simultaneous generation of deletion and point mutants is generally applicable to other genes.
Deletion versus point mutation
Protein functions are typically studied by deleting the gene encoding the protein. This approach has several limitations. A protein often has multiple functions. Deletion abolishes all the functions, which may produce complex phenotypes, thus obscuring certain defects, such as the positive role of Brg in CD4 regulation. On the other hand, some defects may be masked by the redundant action of a related protein. Such a scenario could potentially explain why Brg point mutation (but not Brg deletion) enhances CD25 expression, assuming that the Brg homologue Brm could compensate for Brg to repress CD25.
These limitations in deletion analysis may be overcome by introducing point mutations into relevant regions of the gene, because point mutations are more specific, and the mutant protein may act as a dominant negative to prevent the related protein from masking the phenotypes. Point mutations may help uncover protein functions via other mechanisms. For example, a point mutation at serine 276 in the p65 subunit of NF-κb, which prevents p65 phosphorylation but does not affect its DNA binding, produces developmental defects distinct from those seen in p65 knock-out mice, thus providing unique insights into the biological functions of p65 (30). As in the case of deletion mutations, it is often desirable to introduce point mutations in a tissue-specific manner, but the technology for making such point mutations has not been well established. Before this work, three approaches were proposed to generate such mutations. The first two rely on Cre-mediated excision of an inverted, mutated exon that turns a WT allele into the mutant allele (31, 32). In each case, mutant LoxP sites that enable unidirectional recombination are used. An advantage of these methods is that the mutation can be introduced anywhere in the gene. The disadvantages are that inserting the inverted exon may interfere with the expression of the WT gene, and that Cre-mediated recombination of the mutant LoxP sites may not be as efficient as WT loxP sites (33, 34). So far, no publications describing the successful use of these methods for generating conditional point mutant mice have appeared. The third strategy has been successfully used to introduce mutations into the 3′ end of the GABAA-R gene (35). In this method, the 3′ portion of the GABAA-R gene that encompasses the site to be mutated is floxed, and a mutated version of this portion is duplicated and placed downstream of the endogenous locus. This mutant segment is transcriptionally silent until unmasked by Cre-mediated excision of the WT segment, where the mutant segment replaces the WT counterpart in the targeted gene. Whether this method can be used to introduce mutations elsewhere in the gene remains to be demonstrated.
Our method has several useful features. First, mutations can be introduced anywhere in the gene. Second, the status of the bi-functional allele can theoretically be monitored using the GFP engineered in the gene-trapping cassette, which should facilitate phenotypic analysis. Although we failed to detect GFP expression in the targeted Brg allele, we believe this is caused by the use of the destabilized version of GFP. Indeed, we have introduced a gene-trapping cassette expressing a regular GFP into the BAF57 gene encoding another subunit of the BAF complex and were able to visualize GFP in the targeted ES clones (unpublished data). The last feature is that our strategy can generate deletion and point mutants within the same litters, allowing for the direct comparison of phenotypes. A drawback of our strategy is that two different alleles (i.e., a floxed and a bifunctional allele) must be generated to reveal the effects of the point mutation, as opposed to the aforementioned methods that only need one conditional allele. Finally, the genes being targeted must be haplosufficient for embryogenesis. However, given that traditional floxed alleles have already been made for numerous genes, many of which are haplosufficient for embryogenesis, our strategy should be widely useful.
Dual role of the BAF complex in CD4 regulation
We and others previously showed that Brg deletion disrupts CD4 repression in DN cells (8, 9). Unexpectedly, Brg point mutation is more effective in derepressing CD4 than the Brg deletion, which led to the discovery of the dual roles of Brg in CD4 regulation, as proposed in the model in Fig. 5. In DN1–3 cells, the CD4 promoter is completely inhibited by the fully active silencer. In DN4 cells, the silencer is presumably beginning to be inactivated by a physiological mechanism not involving Brg, thus initiating CD4 transcription that culminates in full-blown CD4 expression in DP cells. Brg is required for CD4 silencer function in DN3 and 4, and presumably also in DN1 and 2 cells. Brg also increases the probability of CD4 transcriptional initiation in DN4 cells. Thus, Brg deletion produces conflicting effects in DN4 cells: it facilitates CD4 derepression by helping inactivate the CD4 silencer while stochastically blocking the onset of CD4 transcription, leading to a variegated CD4 expression pattern that seems epigenetically stable once established. CD4 repression by Brg requires its ATPase, whereas an ATPase-independent activity is used to promote CD4 transcription.
Figure 5.

A model for the roles of Brg in CD4 regulation. T cells develop from DN to DP cells. CD4 expression (red dots), first detected in DN4 cells, is controlled by the silencer (S), CD4 promoter (P), enhancer (E), and LCR (L). For simplicity, the silencer is depicted to be located upstream of, and to act on, the CD4 promoter (top), but the exact mechanism of repression is unknown. Brg plays a dual role in CD4 regulation in DN cells by acting on both the silencer and enhancer/LCR, using distinct biochemical activities (bottom). Brg binds these elements, but it remains to be seen whether this interaction is functionally relevant.
How does Brg repress CD4? Brg binds the CD4 silencer, where it uses the ATPase to remodel the chromatin and facilitates the entry of Runx1, a transcription repressor essential for CD4 inhibition in DN cells, and this effect might be responsible for CD4 repression (unpublished data). The mechanism for positive CD4 regulation is unclear. Brg binds the CD4 enhancer and LCR, suggesting that the BAF complex acts directly on these elements, but future experiments are needed to prove this; it is possible that Brg binding at the CD4 enhancer/LCR is functionally irrelevant (Fig. 5). For example, Brg may act instead by stimulating the expression of a transcription activator required for efficient CD4 expression. Interestingly, Brg is dispensable for CD4 expression in DN3 cells, indicating that a distinct mechanism is used to ensure uniform CD4 expression in these cells. Brg also seems dispensable for CD4 expression in DP cells, because DP cells in Brg Δ mice show uniform, high level CD4 expression. The role of Brg in CD4 regulation in ISP cells is unknown; ISP cells are undetectable in Brg Δ mice apparently because they have lost CD8 expression.
Finally, we note that the effectiveness of Brg point mutation in CD4 derepression may explain the “loss” of DN4 cells in BrgO/+; Cre +/− mice. Presumably, Brg (K/R) has interfered with WT Brg protein, leading to premature CD4 expression on DN4 cells, which subsequently leave the DN4 compartment and blend into the CD4+CD8− pool.
MATERIALS AND METHODS
DNA construct, ES cell targeting, and mice breeding.
To make the construct described in Fig. 1 B, we first constructed a gene-trapping cassette consisting of the following: (a) a 120-bp fragment from pCEM MINX containing the adenovirus major late transcript splicing acceptor sequence from the intron 1/exon 2 boundary (a gift from S. Berget, Baylor College of Medicine, Houston, TX) (36, 37); (b) the neomycin resistant gene; (c) IRES; (d) the hr GFP (Stratagene) fused to the PEST sequence; and (e) the SV40 poly A sequence. We then floxed this cassette by inserting it into pEasy Lox (a gift from K. Rajewsky, Harvard University, Cambridge, MA), replacing the PGK-Neo–poly A–LoxP sequence in the original vector. Finally, we inserted PCR-amplified Brg genomic sequences upstream and downstream of the loxP sites to create the left and right arms, respectively. The left arm contains the exon (E2 in Fig. 1 C) encoding lysine 785, and this residue was mutated to arginine. The cloned fragments were sequenced in their entirety and no PCR-introduced error was found. The construct was electroporated into 129/SV ES cells and correctly targeted ES cells identified by RT-PCR. An ES clone was injected into blastocysts from C57B/6 mice to generate chimeras carrying the bifunctional allele. The founders were crossed with mice bearing the BrgF allele, and the LCK-Cre and LCK–Bcl-xL transgenes (gifts from C. Wilson, University of Washington, Seattle, WA) to generate mice with various genotypes, as described in Fig. 2. The CD4 silencer knockout mice were provided by D. Littman (New York University, New York, NY). All the mice were on mixed genetic backgrounds, and experiments were performed according to the guidelines from Yale University's Institutional Animal Care and Use Committee.
Flow cytometry.
Flow cytometrical analysis was performed as previously described (8), except that the samples were analyzed on LSRII (BD) at Yale University, which necessitated minor modifications in the staining schemes. In brief, to analyze CD4/CD8 expression in various subsets of thymocytes, cells were stained with anti-CD4–allophycocyanin (APC), anti-CD8–PE-Cy7, anti-CD25–FITC, anti-CD44–PE, and anti-CD3–Cy5, and the cells were analyzed as described in Results. To assay cell proliferation, BrdU was injected intraperitoneally and mice were killed 15 min afterward, as previously described (8). Cells were stained with anti-CD4–Alexa Fluor 680, anti-CD8–PE-Cy7, anti-CD25–APC, anti-CD44–PE, and anti-BrdU–FITC before analysis.
PCR, Western blotting, and RT-PCR analysis of thymocytes.
The BrgO and BrgF alleles in thymocytes were monitored by PCR using primer pairs b/c and h/i, respectively, as depicted in Fig. 1 C; these primers can only recognize the parental allele and not the recombination products. For detecting the BrgO allele, a fragment from the CD4 enhancer was coamplified as an internal control. For detecting the BrgF allele in BrgF/F; Cre; (Bcl-xL) mice, a fragment from the CD4 silencer was coamplified as an internal control. However, for detecting the BrgF allele in BrgF/O; Cre; (Bcl-xL) mice, the h/i primer pair alone is sufficient, because the primer pair, which binds the regions in the Brg gene flanking the 3′ LoxP site (placed downstream of E4), not only detects the BrgF allele but also the BrgO allele before or after its conversion, with the latter therefore serving as an internal control because its amplification efficiency is not affected by Cre-mediated conversion. Western blotting was performed using an anti-Brg antibody from Millipore. For semiquantitative RT-PCR, total RNA was extracted using the RNAeasy kit (QIAGEN) and analyzed using a one-step RT-PCR master mix (Invitrogen). For quantitative RT-PCR, the mRNA was first reverse-transcribed into cDNA and then quantified using Quantitect qPCR mix (QIAGEN). DN3, DN4, and DP cells from the Brg mutants were directly sorted from total thymocytes. DN3 and DN4 cells from BrgF/F mice (Fig. 4 B) were first enriched by depleting CD8+ cells with magnetic beads, followed by their purification via cell sorting. The purity of the sorted cells exceeded 90% (not depicted).
Chromatin immunoprecipitation.
Thymocytes at the DN stage from Brg* mice were isolated by negative selection with biotin–anti-CD8/CD3 antibodies and streptavidin magnetic beads (BD), which removed DP and residual SP cells. The majority (>90%) of the isolated cells were DN4 cells, with the remaining being DN1–3 cells. It is not feasible to deplete the DN2 and 3 cells using anti-CD25 beads, because the DN4 cells in Brg* mice express low to intermediate levels of CD25 (Fig. 3 A). Thymocytes at the DN stage from Brg Δ mice were similarly isolated except that the CD4+ and CD4− cells were further separated using anti-CD4 beads; these cells were also predominantly at the DN4 stage. B cells were magnetically purified from WT (CD1) mice, with purity >90%. DP cells were from TCRα−/− mice. Chromatin was fixed, sheared, precleared, immunoprecipitated, and analyzed by PCR essentially as previously described (29); the Brg antibody was from Millipore.
Restriction enzyme accessibility assay.
The restriction enzyme accessibility assay was performed as previously described (38). In brief, 0.2 million lymphocytes were digested with restriction enzymes in 10 μl 1× NEB digestion buffer II in the presence of 0.1% NP-40, which permeabilizes the membrane. After the digestion, the reaction was heated to 80°C for 20 min to inactivate the enzyme. 1 μl proteinase K (10 μg) was added and the sample was digested overnight at 50°C. The sample was then heated to 80°C for 20 min, and PMSF was added to 0.1 mM to completely inactivate proteinase K. DNA was directly digested with 0.3 μl of a second enzyme for 3 h, and a mixture of linkers recognizing the first and second cuts was added together with 0.3 μl NEB T4 DNA ligase. The reaction was incubated at room temperature for 12 h before PCR analysis using 1 μl of ligated DNA. The amplicons were detected by primer extension using radiolabeled, nested primers. The amplicons were resolved on sequencing gels, and the intensity of the short relative to the long amplicons was taken as a measure of accessibility.
Online supplemental material.
Fig. S1 A shows that alternative splicing in the BrgO allele is undetectable. Fig. S1 B shows that Brg mRNA level is reduced in BrgO/+thymocytes but restored in BrgO/+; Cre thymocytes. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20080938/DC1.
Supplementary Material
Acknowledgments
We thank Drs. C.B. Wilson for LCK-Cre mice, D. Littman and Y. Zou for CD4 silencer KO mice, S. Berget for the pCEM MINX plasmid, K. Rajewsky for the pEasy Lox plasmid, and P. Soriano for advice on gene trapping.
This work is funded by the National Institutes of Health (grant R01AI063554-02 to T. Chi), the Anna Fuller Foundation (to A. Jani), and the Intramural Research Program of the National Heart, Lung, and Blood Institute/National Institutes of Health (K. Zhao).
The authors have no conflicting financial interests.
Abbreviations used: BAF, Brg-associated factor; DN, double negative; DP, double positive; ES, embryonic stem; IRES, internal ribosome entry site; ISP, immature SP; LCR, locus control region; SA, splice acceptor; SP, single positive.
M. Wan, J. Zhang, and K. Cui contributed equally to this paper.
J. Zhang's present address is Institute for Cell Engineering, Johns Hopkins University School of Medicine, Baltimore, MD 21205.
References
- 1.Ellmeier, W., S. Sawada, and D.R. Littman. 1999. The regulation of CD4 and CD8 coreceptor gene expression during T cell development. Annu. Rev. Immunol. 17:523–554. [DOI] [PubMed] [Google Scholar]
- 2.von Boehmer, H., I. Aifantis, O. Azogui, J. Feinberg, C. Saint-Ruf, C. Zober, C. Garcia, and J. Buer. 1998. Crucial function of the pre-T-cell receptor (TCR) in TCR beta selection, TCR beta allelic exclusion and alpha beta versus gamma delta lineage commitment. Immunol. Rev. 165:111–119. [DOI] [PubMed] [Google Scholar]
- 3.Guidos, C.J. 1996. Positive selection of CD4+ and CD8+ T cells. Curr. Opin. Immunol. 8:225–232. [DOI] [PubMed] [Google Scholar]
- 4.Narlikar, G.J., H.Y. Fan, and R.E. Kingston. 2002. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 108:475–487. [DOI] [PubMed] [Google Scholar]
- 5.Wang, W. 2003. The SWI/SNF family of ATP-dependent chromatin remodelers: similar mechanisms for diverse functions. Curr. Top. Microbiol. Immunol. 274:143–169. [DOI] [PubMed] [Google Scholar]
- 6.Wu, J.I., J. Lessard, I.A. Olave, Z. Qiu, A. Ghosh, I.A. Graef, and G.R. Crabtree. 2007. Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron. 56:94–108. [DOI] [PubMed] [Google Scholar]
- 7.Chi, T. 2004. A BAF-centred view of the immune system. Nat. Rev. Immunol. 4:965–977. [DOI] [PubMed] [Google Scholar]
- 8.Chi, T.H., M. Wan, P.P. Lee, K. Akashi, D. Metzger, P. Chambon, C.B. Wilson, and G.R. Crabtree. 2003. Sequential roles of Brg, the ATPase subunit of BAF chromatin remodeling complexes, in thymocyte development. Immunity. 19:169–182. [DOI] [PubMed] [Google Scholar]
- 9.Gebuhr, T.C., G.I. Kovalev, S. Bultman, V. Godfrey, L. Su, and T. Magnuson. 2003. The role of Brg1, a catalytic subunit of mammalian chromatin-remodeling complexes, in T cell development. J. Exp. Med. 198:1937–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khavari, P.A., C.L. Peterson, J.W. Tamkun, D.B. Mendel, and G.R. Crabtree. 1993. BRG1 contains a conserved domain of the SWI2/SNF2 family necessary for normal mitotic growth and transcription. Nature. 366:170–174. [DOI] [PubMed] [Google Scholar]
- 11.de la Serna, I.L., K.A. Carlson, and A.N. Imbalzano. 2001. Mammalian SWI/SNF complexes promote MyoD-mediated muscle differentiation. Nat. Genet. 27:187–190. [DOI] [PubMed] [Google Scholar]
- 12.Trotter, K.W., and T.K. Archer. 2004. Reconstitution of glucocorticoid receptor-dependent transcription in vivo. Mol. Cell. Biol. 24:3347–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Archer, T.K., P. Lefebvre, R.G. Wolford, and G.L. Hager. 1992. Transcription factor loading on the MMTV promoter: a bimodal mechanism for promoter activation. Science. 255:1573–1576. [DOI] [PubMed] [Google Scholar]
- 14.Phelan, M.L., S. Sif, G.J. Narlikar, and R.E. Kingston. 1999. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol. Cell. 3:247–253. [DOI] [PubMed] [Google Scholar]
- 15.Zhao, K., W. Wang, O.J. Rando, Y. Xue, K. Swiderek, A. Kuo, and G.R. Crabtree. 1998. Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell. 95:625–636. [DOI] [PubMed] [Google Scholar]
- 16.Kadam, S., and B.M. Emerson. 2003. Transcriptional specificity of human SWI/SNF BRG1 and BRM chromatin remodeling complexes. Mol. Cell. 11:377–389. [DOI] [PubMed] [Google Scholar]
- 17.Hassan, A.H., P. Prochasson, K.E. Neely, S.C. Galasinski, M. Chandy, M.J. Carrozza, and J.L. Workman. 2002. Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell. 111:369–379. [DOI] [PubMed] [Google Scholar]
- 18.Bourachot, B., M. Yaniv, and C. Muchardt. 1999. The activity of mammalian brm/SNF2alpha is dependent on a high-mobility-group protein I/Y-like DNA binding domain. Mol. Cell. Biol. 19:3931–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sumi-Ichinose, C., H. Ichinose, D. Metzger, and P. Chambon. 1997. SNF2beta-BRG1 is essential for the viability of F9 murine embryonal carcinoma cells. Mol. Cell. Biol. 17:5976–5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee, P.P., D.R. Fitzpatrick, C. Beard, H.K. Jessup, S. Lehar, K.W. Makar, M. Perez-Melgosa, M.T. Sweetser, M.S. Schlissel, S. Nguyen, et al. 2001. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 15:763–774. [DOI] [PubMed] [Google Scholar]
- 21.Bultman, S., T. Gebuhr, D. Yee, C. La Mantia, J. Nicholson, A. Gilliam, F. Randazzo, D. Metzger, P. Chambon, G. Crabtree, and T. Magnuson. 2000. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell. 6:1287–1295. [DOI] [PubMed] [Google Scholar]
- 22.Chi, T.H., M. Wan, K. Zhao, I. Taniuchi, L. Chen, D.R. Littman, and G.R. Crabtree. 2002. Reciprocal regulation of CD4/CD8 expression by SWI/SNF-like BAF complexes. Nature. 418:195–199. [DOI] [PubMed] [Google Scholar]
- 23.Chao, D.T., and S.J. Korsmeyer. 1997. BCL-XL-regulated apoptosis in T cell development. Int. Immunol. 9:1375–1384. [DOI] [PubMed] [Google Scholar]
- 24.Adlam, M., and G. Siu. 2003. Hierarchical interactions control CD4 gene expression during thymocyte development. Immunity. 18:173–184. [DOI] [PubMed] [Google Scholar]
- 25.Zou, Y.R., M.J. Sunshine, I. Taniuchi, F. Hatam, N. Killeen, and D.R. Littman. 2001. Epigenetic silencing of CD4 in T cells committed to the cytotoxic lineage. Nat. Genet. 29:332–336. [DOI] [PubMed] [Google Scholar]
- 26.Siu, G., A.L. Wurster, D.D. Duncan, T.M. Soliman, and S.M. Hedrick. 1994. A transcriptional silencer controls the developmental expression of the CD4 gene. EMBO J. 13:3570–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sawada, S., J.D. Scarborough, N. Killeen, and D.R. Littman. 1994. A lineage-specific transcriptional silencer regulates CD4 gene expression during T lymphocyte development. Cell. 77:917–929. [DOI] [PubMed] [Google Scholar]
- 28.Petrie, H.T., M. Pearse, R. Scollay, and K. Shortman. 1990. Development of immature thymocytes: initiation of CD3, CD4, and CD8 acquisition parallels down-regulation of the interleukin 2 receptor alpha chain. Eur. J. Immunol. 20:2813–2815. [DOI] [PubMed] [Google Scholar]
- 29.Yu, M., M. Wan, J. Zhang, J. Wu, R. Khatri, and T. Chi. 2008. Nucleoprotein structure of the CD4 locus: implications for the mechanisms underlying CD4 regulation during T cell development. Proc. Natl. Acad. Sci. USA. 105:3873–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong, J., E. Jimi, H. Zhong, M.S. Hayden, and S. Ghosh. 2008. Repression of gene expression by unphosphorylated NF-kappaB p65 through epigenetic mechanisms. Genes Dev. 22:1159–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schnutgen, F., N. Doerflinger, C. Calleja, O. Wendling, P. Chambon, and N.B. Ghyselinck. 2003. A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat. Biotechnol. 21:562–565. [DOI] [PubMed] [Google Scholar]
- 32.Zhang, Z., and B. Lutz. 2002. Cre recombinase-mediated inversion using lox66 and lox71: method to introduce conditional point mutations into the CREB-binding protein. Nucleic Acids Res. 30:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolb, A.F. 2001. Selection-marker-free modification of the murine beta-casein gene using a lox2272 [correction of lox2722] site. Anal. Biochem. 290:260–271. [DOI] [PubMed] [Google Scholar]
- 34.Wang, Z., P. Engler, A. Longacre, and U. Storb. 2001. An efficient method for high-fidelity BAC/PAC retrofitting with a selectable marker for mammalian cell transfection. Genome Res. 11:137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skvorak, K., B. Vissel, and G.E. Homanics. 2006. Production of conditional point mutant knockin mice. Genesis. 44:345–353. [DOI] [PubMed] [Google Scholar]
- 36.Robberson, B.L., G.J. Cote, and S.M. Berget. 1990. Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol. Cell. Biol. 10:84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friedrich, G., and P. Soriano. 1991. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 5:1513–1523. [DOI] [PubMed] [Google Scholar]
- 38.Yu, M., M. Wan, J. Zhang, J. Wu, R. Khatri, and T. Chi. 2008. Nucleoprotein structure of the CD4 locus: implications for the mechanisms underlying CD4 regulation during T cell development. Proc. Natl. Acad. Sci. USA. 105:3873–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}