

Abstract
CD45 and Fas regulate tyrosine phosphorylation and apoptotic signaling pathways, respectively. Mutation of an inhibitory wedge motif in CD45 (E613R) results in hyperresponsive thymocytes and B cells on the C57BL/6 background, but no overt autoimmunity, whereas Fas deletion results in a mild autoimmune disease on the same genetic background. In this study, we show that these two mutations cooperate in mice, causing early lethality, autoantibody production, and substantial lymphoproliferation. In double-mutant mice, this phenotype was dependent on both T and B cells. T cell activation required signaling in response to endogenous or commensal antigens, demonstrated by the introduction of a transgenic T cell receptor. Genetic deletion of B cells also prevented T cell activation. Similarly, T cells were necessary for B cell autoantibody production. However, B cells appeared to be intrinsically activated even in the absence of T cells, suggesting that they may drive the phenotype of these mice. These results reveal a requirement for careful control of B cell signaling and cell death in preventing inappropriate lymphocyte activation and autoimmunity.
In the adaptive immune system, multiple barriers minimize the likelihood that a randomly generated antigen receptor will recognize and initiate a response to self-peptide. These barriers are at work throughout the lifetime of antigen receptor–bearing cells, starting with the elimination of self-reactive cells during their development and followed by mechanisms that induce anergy, suppression, or apoptosis of any self-reactive cells escaping to the periphery (1–3). In some rare, inherited autoimmune syndromes, single-gene defects trigger a failure of one of these processes. However, for most autoimmune diseases, including lupus and arthritis, multiple interacting loci with relatively small individual effects are generally necessary for disease pathogenesis (4). Studying how genetic modifiers cooperate to cause disease is thus critical to our understanding of autoimmunity.
CD45 plays an essential role in the immune system, regulating lymphocyte development and activation through Src family kinases (SFKs) (5). CD45 maintains SFKs in a primed, quiescent state by dephosphorylating both inhibitory and activating tyrosines. These unphosphorylated SFKs assume an open conformation that can be rapidly activated by phosphorylation of the activation loop tyrosine (6). In the absence of CD45 activity, the inhibitory tyrosine is phosphorylated by the kinase Csk, and the SFK adopts a closed, inactive conformation.
One means of regulating CD45 is by spontaneous homodimerization, which inhibits its activity, preventing SFK activation and antigen receptor signaling (7, 8). The crystal structure of a related phosphatase, receptor protein tyrosine phosphatase α (RPTPα), revealed a possible mechanism for inhibition by dimerization. The membrane-proximal phosphatase domain of RPTPα crystallizes as a symmetric dimer in which a helix-turn-helix motif of one molecule forms a “wedge” and inserts into the catalytic site of the partner molecule (9). The wedge is highly conserved in CD45. Mutating the acidic amino acid at its tip prevented dimerization-mediated inhibition in vitro and, moreover, led to a lupus-like syndrome in mice when knocked into the endogenous CD45 locus (E613R) (8, 10). In a mixed C57BL/6 (B6) and 129 genetic background, the CD45 wedge mutant mice develop a pronounced lymphoproliferative disease (LPD), autoantibodies, and immune complex–mediated glomerulonephritis (10). However, as with many autoimmune phenotypes, much of the disease resolved on a homogenous B6 background (11). B6 CD45 wedge mice still develop very mild lymphoproliferation and elevated IgM levels, but autoantibodies and glomerulonephritis are absent. B cells remain hyperresponsive to various stimuli, suggesting that the CD45 wedge mutant behaves as a hypermorphic allele, but is insufficient to cause autoimmunity on the B6 background.
In this study, we ask whether the wedge mutation could act as a genetic modifier with other mutations, predisposing mice, and potentially humans, to autoimmunity. We chose to study the complement C4 knockout and a naturally occurring Fas mutation, lpr, based on their ability to induce a mild, slowly progressing autoimmunity on the B6 background. Complement is a component of the innate immune system involved in opsonization and clearance of pathogens and possibly apoptotic bodies (12, 13). Fas, when bound by Fas ligand (FasL), frequently triggers apoptosis on activated cells, and hence is part of the activation-induced cell death (AICD) pathway. In mice, the phenotypes of the C4 knockout and lpr mutation are highly dependent on strain background. Both mixed B6 × 129 C4 knockout and MRL/lpr mice develop robust autoimmune lupus-like disease, characterized by lymphocyte activation, autoantibodies against nuclear components, immune complex deposition, and glomerulonephritis (13–18). However, backcrossing both C4 and lpr to the B6 background mitigates most of their phenotype. In these mice, low levels of autoantibody are detected only in older mice, without evidence of overt organ damage (18, 19). Here, we find that combination of C4 deficiency and the CD45 wedge mutation does not alter disease compared with single-mutant mice. However, the wedge mutation and lpr act synergistically, markedly accelerating autoantibody production, lymphocyte activation, and lymphoproliferation. Thus, the CD45 wedge mutation acts as a potent modifier with the lpr mutation, but not C4 deficiency. In addressing the mechanisms behind the wedge/lpr phenotype, we further demonstrate that T cells, and specifically signaling through the TCR, and B cells are required for lymphoproliferation, suggesting the presence of a positive feedback loop between these two cell types. Moreover, B cells appear to be intrinsically activated, and therefore their activation may be the primary event driving lymphocyte activation and expansion.
RESULTS AND DISCUSSION
CD45 wedge/lpr mice die at an accelerated rate with autoantibody production
We generated wedge/lpr double-mutant mice and followed cohorts consisting of 10 mice, 5 male and 5 female, from WT, single-, and double-mutant backgrounds over the course of 1 yr. Of these cohorts, wedge/lpr double-mutant mice showed accelerated autoantibody production and mortality. 100% of wedge/lpr mice developed antinuclear antibodies (ANAs) by 2 mo of age, dsDNA antibodies by 3 mo, and died by 9 mo (Fig. 1, A–C). As previously reported, B6 Lpr mice did not develop low-titer ANAs until 6 mo of age and dsDNA antibodies until 9 mo of age (unpublished data) (18). In contrast, autoantibodies were not detected in wedge, C4, and wedge/C4 mice throughout the yearlong experiment.
Figure 1.

CD45 wedge/lpr double-mutant mice show early lethality and autoantibody production. (A) Kaplan-Meier survival curve from aging cohorts. n = 10 mice per group, 5 males and 5 females. P < 0.0005. (B) Anti-dsDNA ELISA of serum from aging mice, 6–10 mice per group. Error bars represent SEM. Analysis of variance (ANOVA): *, P < 0.05; **, P < 0.005; ***, P < 0.0005. (C) Staining of Hep2 nuclei with serum diluted 1:40 from 2-mo-old animals. Images are representative of three animals per group. Bars, 50 μm. Lymphocyte accumulation in kidney (D) and lungs (E) of 6-mo-old CD45 wedge/lpr mice. Bars, 200 μm.
In MRL/lpr mice, autoantibodies and early death are often linked to antibody deposition in the kidneys and glomerulonephritis. Proteinuria is commonly used as a marker for kidney damage in these mice, but surprisingly neither immune complex deposition nor proteinuria were detected in CD45 wedge/lpr mice as late as 9 mo of age (unpublished data). Histological examination of kidneys from 6–9-mo-old wedge/lpr mice did, however, reveal increasing amounts of perivascular lymphocyte accumulations with age. The accumulations were most prominent in kidney and lung and, to a lesser extent, the liver (Fig. 1, D and E). We speculate that progressive accumulation of lymphocytes, combined with compression of the lungs from mediastinal lymphadenopathy, contribute to the death of wedge/lpr mice from asphyxiation (Fig. S1, A–C, available at http://www.jem.org/cgi/content/full/jem.20081204/DC1). Immunohistochemical analysis revealed the cells to consist almost exclusively of CD3+ and CD4+ T cells (Fig. S1, D–G). In addition to the perivascular infiltrates, kidneys from older wedge/lpr mice appeared pale and contained enlarged, hypercellular glomeruli, which could represent early, subclinical renal ischemia (Fig. S1, H–K).
Wedge and lpr mutations cooperate to induce lymphocyte activation and expansion
On gross examination, the most striking feature of wedge/lpr mice was marked lymphadenopathy and splenomegaly, which progressed with age (Fig. S1, A and B). Both splenic weight and lymph node cellularity in wedge/lpr double-mutant mice were increased up to 10-fold compared with single-mutant and WT mice (Fig. 2, A and B). The breakdown of lymphocyte subsets is shown in Fig. 2 C. In general, the composition of lymphocytes in wedge/lpr mice is similar to that of wedge mice, which have a greater fraction of CD3 versus CD19 cells and a slightly skewed CD4 to CD8 ratio. The notable exceptions are the reduced percentage of CD4 and, more dramatically, CD8 cells as they are replaced by the substantial increase in CD3+ CD4/CD8 double-negative (DN) T cells. This is a unique subset of T cells found in lpr mice, but is much more prominent in wedge/lpr mice. The origin of these DN cells is controversial, but they are believed to represent a population of activated T cells that fail to undergo AICD as a result of the Fas mutation (17, 18). Although the percentage of some subsets was reduced, the absolute number of all lymphocyte subsets was much higher in wedge/lpr mice as a result of the overall increased cellularity (Fig. 2 D). Based on BrdU incorporation, lymphocyte expansion was a consequence of increased proliferation in vivo. In wedge/lpr mice, ∼60% of CD4+ T cells, CD8+ T cells, and CD19 B cells, had divided and incorporated BrdU over 10 d, a significantly higher percentage than wedge, lpr, and WT cells (Fig. 2 E).
Figure 2.

Lymphocyte expansion in CD45 wedge/lpr mice is accompanied by increased cell proliferation. (A) Splenic weight of 2-, 4-, and 6-mo-old mice. Mean of three to six mice per time point ± SEM. ANOVA: *, P < 0.05; **, P < 0.005; ***, P < 0.0005. (B) Total number of cells pooled from inguinal, axillary, brachial, cervical, and mesenteric lymph nodes of 2-mo-old mice. Unpaired Student's t test: ***, P < 0.0005. (C) Lymphocyte subset analysis of lymph nodes from 2-mo-old mice. CD3+ cells are separated into CD4+, CD8+, and DN (CD3+, B220+, CD4−/CD8− [DN]) cells. (D) Mean absolute number of T cell subsets and B cells in lymph nodes from 2-mo-old mice. Error bars represent SEM with two to four mice per group from three independent experiments. (E) BrdU incorporation is increased in CD4 and CD8 T cells and B cells from wedge/lpr mice. 7–8-wk-old mice were given BrdU in their drinking water for 10 d. Shown is the mean percentage of cells positive for intracellular BrdU from four mice per genotype ± SEM. Data are representative of three independent experiments. (C, D, and E) Unpaired Student's t test, P < 0.05 compared with WT, *; wedge, **; and lpr, ***.
Fitting with the observed lymphoproliferation, both peripheral T and B wedge/lpr cells were activated at a higher frequency. In 2-mo-old mice, up to 90% of CD4 and 75% of CD8 T cells in the spleen had already acquired a memory phenotype, defined by up-regulation of CD44 (Fig. 3 A and Fig. S2 A, available at http://www.jem.org/cgi/content/full/jem.20081204/DC1). An unusual population of CD4 cells found in lpr mice that have up-regulated CD44, but failed to down-regulate CD62L, was likewise expanded in wedge/lpr double mutants. The wedge/lpr memory phenotype cells showed a strong Th1 polarization. A large fraction of wedge/lpr CD4+ CD44high cells produced IFN-γ, but not IL-4 or -17, upon direct ex vivo stimulation with PMA and ionomycin (Fig. S2 C and not depicted). In addition to increased memory cell markers, wedge/lpr mice have a higher fraction of cells expressing the immediate activation marker CD69 (Fig. S2 B), which is suggestive of ongoing T cell activation. Analysis of B and myeloid cells indicates that these lineages were also activated, as measured by MHC class II and CD86 up-regulation, in wedge/lpr mice relative to controls (Fig. 3 B).
Figure 3.

T and B cells from wedge/lpr mice show signs of activation. (A) Representative flow cytometric analysis of CD4 memory phenotype cells from 2-mo-old mice. Shown are the percentage of CD3+ CD4+ T cells from lymph node and spleen that are memory (CD44high, CD62Llow/high) and naive (CD44low, CD62Lhigh) cells. Data are representative of two to four mice from three independent experiments. (B) MHC class II and CD86 expression on CD19+ B cells and CD11c+ DCs are shown as the mean MFI for two to four mice per genotype ± SEM. Data are shown as relative MFI to correct for different antibody clones and fluorophores and are representative of three independent experiments. Unpaired Student's t test P < 0.05 compared with WT, *; wedge, **; and lpr, ***.
The wedge mutation does not enhance AICD or proliferation in vitro
We next set out to understand the mechanism driving the T cell activation and expansion in the CD45 wedge/lpr double-mutant mice. Based on our observations that wedge B cells are hyperresponsive to stimulation, we hypothesized that wedge T cells might also have a reduced threshold for activation and thus be more susceptible to Fas-mediated AICD (11). Surprisingly, in vitro AICD assays did not reveal any differences in the apoptosis of wedge versus WT T cells (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20081204/DC1). lpr and wedge/lpr T cells were resistant to death in these experiments, indicating that induction of death was Fas dependent (Fig. S3 and not depicted). There was also no difference in CD3 plus CD28–induced in vitro proliferation of CD4 or CD8 T cells from young mice among the four genotypes at multiple doses and time points (unpublished data). These results raised the possibility that the wedge mutation has different effects on responses to strong in vitro stimulation versus in vivo endogenous signals.
TCR antigen recognition and B cells are required for lymphoproliferation
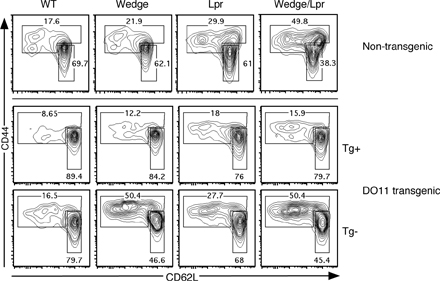
To examine the role of endogenous signaling through the TCR, we restricted the T cell repertoire to a single TCR using the ovalbumin peptide–reactive OT2 TCR transgene. The OT2 TCR transgene completely eliminated the presence of memory phenotype T cells in wedge/lpr mice (Fig. 4 B). In these mice, the small fraction of T cells escaping OT2 TCR restriction showed a slight increase in the percentage of memory cells compared with WT OT2. The degree of activation in these cells likely depends on the specific TCR transgene, the efficiency of restriction, and the strain background (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20081204/DC1). Restricting the TCR repertoire also corrected the lymphadenopathy and splenomegaly, as well as the survival defect seen in wedge/lpr mice (Fig. 4 A). None of the mice in a cohort of 6 wedge/lpr OT2 mice had died at 6 mo compared with the 50% mortality seen at this time point in nontransgenic wedge/lpr mice (unpublished data). Genetic elimination of T cells using TCRα−/− mice also prevented lymphadenopathy and splenomegaly (Fig. 4 A). Furthermore, wedge/lpr TCRα−/− mice failed to produce autoantibodies by 5 mo of age (unpublished data). Wedge/lpr B cells must therefore require T cell help to expand and secrete autoantibodies. Interestingly, B cells and DCs from wedge and wedge/lpr TCRα−/− mice still expressed higher levels of MHC class II and CD86, suggesting that B cell and DC activation is a T cell–independent effect that results mainly from the wedge mutation alone (Fig. 4 C).
Figure 4.

TCR signaling and T cells contribute to lymphocyte activation and expansion. (A) Splenic weight as an indicator of lymphoproliferation in normal, OT2 TCR transgenic (OT2), T cell-deficient (TCRα−/−), and B cell–deficient mice (Ig−/−) comparing 2-mo-old WT and wedge/lpr. Each point represents a single mouse, five to six mice per genotype. Bars represent the mean of the group. ANOVA, P < 0.0001. (B) Representative flow cytometric analysis of CD4 memory phenotype cells in 2-mo-old OT2 TCR transgenic mice. OT2 Tg+ cells were gated as CD3+ CD4+ Vα2+ Vβ5+. Tg− cells are CD3+ CD4+ Vα2− Vβ5−. Memory phenotype cells are CD44high, whereas naive cells are CD44low, CD62Lhigh. Numbers represent percentage of CD4+ cells. Data are representative of two to four mice from three independent experiments. (C) MHC class II and CD86 expression on CD19+ B cells and CD11c+ DCs from 2-mo-old TCRα−/− mice, shown as the mean MFI for 4 mice per genotype ± SEM. Data are shown as relative MFI to correct for different antibody clones and fluorophores. Data are representative of three independent experiments. Unpaired Student's t test, P < 0.05 compared with WT, *; wedge, **; and lpr, ***.
This finding led us to hypothesize that B cell activation may be responsible for initiating the lymphoproliferation in wedge/lpr mice. We therefore crossed the μMT mutation (Ig−/−) onto wedge/lpr mice to eliminate B cells and found that the extent of T cell activation was significantly reduced (Fig. 5 A). The percentage of memory phenotype T cells in wedge/lpr Ig−/− mice was similar to that of lpr Ig−/− T cells, suggesting that wedge/lpr B cells play a role in the activation of T cells. Removal of B cells also reduced the rate of BrdU incorporation in wedge/lpr T cells by 50%, which is consistent with B cells contributing to the proliferation of T cells (Fig. 5 B). In addition, the absence of B cells markedly abrogated lymphadenopathy and splenomegaly (Fig. 4 A) and rescued wedge/lpr Ig−/− mice from premature death. 6 out of 6 mice in a cohort of wedge/lpr Ig−/− mice were healthy at 6 mo (unpublished data). Thus, in contrast to the intrinsic activation of wedge/lpr B cells, T cells appear to be dependent on B cells for both activation and expansion.
Figure 5.

T cell activation and expansion is reduced in the absence of B cells. (A) Representative flow cytometric analysis of CD4 memory phenotype cells in lymph nodes from 2-mo-old B cell–deficient (Ig−/−) mice. Memory cells are CD44high, CD62Llow/high; naive cells are CD44low, CD62Lhigh. Data are representative of two to four mice from three independent experiments. (B) T cell BrdU incorporation in the presence and absence of B cells. Normal (n = 2) and B cell–deficient (Ig−/−; n = 4) mice were administered BrdU in their drinking water for 10 d. Mice were 7–8 wk old. Histograms give the mean percentage of CD4 and CD8 cells positive for intracellular BrdU ± SEM. Data are representative of three independent experiments. Unpaired Student's t test P < 0.05 compared with WT, *; wedge, **; and lpr, ***.
Based on our data, we propose the following model for the roles of CD45 and Fas in immune homeostasis and autoimmunity. CD45 regulates lymphocyte signaling through the modulation of SFK activity. SFKs control both the early activation of lymphocytes in response to foreign antigen and the low-level survival signal antigen that receptor cells produce in response to continuous exposure to endogenous antigens (6, 20). Mutation of the CD45 wedge results in a hypermorphic CD45 allele that is more competent for signaling (8, 10). Thus, instead of the low-level signal required for maintaining naive cell survival, enhanced signaling in CD45 wedge mutant cells chronically activates a subset of these cells. Several recent studies have suggested that Fas deletes such chronically activated cells. In these studies, deleted cells were activated in response to either endogenous antigens (21, 22) or persistent foreign antigens (23–25). Fas is likely to be important in deleting wedge mutant T and B cells activated by endogenous antigens as well. However, once Fas is removed, these cells survive and proliferate.
Although both T and B cells show increased activation and turnover in wedge/lpr mice, neither cell type alone is sufficient to cause lymphoproliferation. Neither T cell– or B cell–deficient wedge/lpr mice develop lymphadenopathy or splenomegaly. This phenomenon has also been previously documented for B cell deficiency in mixed background wedge mutant mice, MRL/lpr mice, and aged B6/lpr mice, which fail to develop lymphadenopathy, splenomegaly, and glomerulonephritis (11, 26–28). Together, these data suggest the presence of a positive feedback loop between B and T cells that lead to their mutual expansion (29). In wedge/lpr mice, activated APCs, and more specifically B cells, are responsible for initiating this loop. Wedge/lpr B cells express increased levels of the activation markers MHC class II and CD86, even in the absence of T cells. The elevated levels of these markers appear to be cell intrinsic and might make the B cells more effective at presenting endogenous antigens. We postulate that increased presentation of endogenous antigens then activates autoreactive T cells. These activated T cells further activate B cells, resulting in autoantibody production and proliferation of both cell types.
We have shown that the CD45 wedge mutation can act as a genetic modifier for the development of autoimmunity, but not in all contexts. When combined with the lpr mutation in Fas, the wedge mutation dramatically accelerates and exacerbates the loss of tolerance, lymphocyte activation, and lymphoproliferation. In contrast, the wedge mutation and C4 deficiency do not cooperate despite the strong predisposition of C4 deficiency to result in a lupuslike syndrome. Interestingly, genetic modifiers may be an important factor in a human disease caused by Fas mutations called autoimmune lymphoproliferative syndrome (ALPS). Even though Fas mutations are autosomal dominant, parents of ALPS patients may carry the same mutation but not display any symptoms (30). Subtle differences in the signaling threshold of lymphocytes from these individuals may account for this variable penetrance. Wedge and wedge/lpr mice may therefore serve as useful tools to begin understanding not only the pathogenesis of ALPS but also the roles of B cells and antigen receptor cell responses to endogenous antigens in other autoimmune conditions.
MATERIALS AND METHODS
Mice.
CD45 E613R mice, backcrossed at least nine generations to C57BL/6, were bred to C57BL/6 lpr and C4 knockout mice (The Jackson Laboratory). OT2 mice were a gift from M. Peters (University of California, San Francisco, San Francisco, CA). C57BL/6 TCRα−/− and μMT mice were obtained from The Jackson Laboratory. Mice were bled monthly for serum antibody analysis. All animals were housed in a specific pathogen–free barrier facility at UCSF, and experimental protocols in this research involving mice were approved by the UCSF Committee for Animal Research (CAR) according to guidelines set by the University of California San Francisco Institutional Animal Care and Use Committee and National Institutes of Health.
Autoantibody assays.
For ANA analysis, sera was diluted 1:40 in PBS/1% FBS and applied to Hep2 ANA slides (iNOVA) for 30 min at room temperature. Slides were washed and incubated with FITC anti–mouse IgG, and images were captured using a Marianis system with a Sensicam cooled charge-coupled device camera (Cooke) attached to an Axiovert microscope. Anti-dsDNA antibody titers were determined by ELISA, as previously described (10). Serum was diluted 1:100 in blocking buffer, and then diluted in twofold steps up to seven times. Pooled MRL/lpr serum was used as a positive control.
Histology.
Tissue for H&E staining was fixed in 10% formalin (Thermo Fisher Scientific) for at least 24 h, embedded in paraffin, sectioned, and stained. For immunohistochemistry, tissue was immersed in OCT, snap frozen in 1,1,1,2 tetrafluoroethane (Thermo Fisher Scientific), and cut into 5-μm sections. Slides were thawed, fixed in acetone, treated with 0.1% H2O2, and blocked according to the manufacturer's instructions (Vector Laboratories). Slides were incubated with primary antibody, followed by biotinylated secondary antibody and ABC Reagent. Slides were incubated with DAB (0.05% DAB, 0.01% H2O2, and 50 mM Tris, pH 7.4), washed, dehydrated, and mounted. Images were captured using a DM 5000B fluorescent microscope equipped with a DFC 350FX digital camera (both from Leica).
Flow cytometry and antibodies.
Pooled lymph nodes (inguinal, brachial, axillary, cervical, and mesenteric) and spleen were homogenized by grinding between frosted slides and passing through a 30-μm filter. Red blood cells were removed from spleen by ammonium chloride lysis treatment. 2 × 106 cells were stained and analyzed immediately or fixed in 1% paraformaldehyde (Electron Microscopy) for analysis the following day. Data were collected on either a FACSCalibur (BD) or CyAn ADP (Dako). Antibodies were conjugated to FITC/Alexa Fluor 488, PE, PE-Texas red, PerCP-Cy5.5, PE-Cy7, Pacific Blue, Pacific Orange, or APC/Alexa Fluor 647. Antibodies used were as follows: CD3, CD11b, CD11c, CD19, CD45R (B220), CD62L, CD69, TCR Vα2, and Vβ5 (BD); CD86 (BioLegend); CD4, CD44, and MHC class II (eBioscience); and CD8 (Invitrogen).
In vivo BrdU incorporation.
Mice were given water containing 0.8 mg/ml BrdU and 2% glucose for 10 d. Single-cell suspensions from lymph node and spleen were stained for extracellular markers with antibodies conjugated to FITC/Alexa Fluor 488, Pacific Blue, and Pacific Orange. Cells were then fixed in 70% ethanol for 30 min on ice, washed, and resuspended in 1% paraformaldehyde/PBS for 30 min at room temperature. Intracellular DNA was digested using DNase solution (50 Kunitz U/ml, 4.2 mM MgCl2, 0.15 M NaCl). Cells were washed, stained with Alexa Fluor 647–conjugated anti-BrdU (PRD-1; Invitrogen), washed again, and analyzed by flow cytometry.
Intracellular cytokine staining.
Lymph node cells were stimulated for 5 h with 20 ng/ml PMA and 1 μM ionomycin in the presence of 10 μg/ml Brefeldin A. They were washed, stained for CD3, CD4, CD8, and CD44, and fixed in 2% paraformaldehyde for 20 min at room temperature. Cells were permeabilized and stained using PE-conjugated anti–IL-4, anti–IFN-γ, anti–IL-17, and anti–IL-10 (BD) for 60 min at room temperature. Cells were analyzed by flow cytometry.
AICD assay.
Lymph node CD4 T cells were purified by MACS negative selection (Miltenyi Biotec) and stimulated for 3 d in plates coated with 5 μg/ml anti-CD3 (2C11; Harlan) and with 1 μg/ml soluble anti-CD28. At the end of 3 d, dead cells were removed with Lympholyte (Cedarlane Laboratories) according to the manufacturer's instructions. Live cells were resuspended at 0.5 × 106/ml with 50 U/ml IL-2 and incubated for 24 h. After 24 h, dead cells were again removed with Lympholyte and 0.2 × 106 live cells were added to 96-well plates coated with 0, 0.1, and 1.0 μg/ml anti-CD3. Cells were harvested 6–24 h later, stained with Annexin V and 7-AAD (BD) according to the manufacturer's instructions, and analyzed by flow cytometry.
Proliferation.
Lymph node T cells were purified by MACS negative selection (Miltenyi Biotec) and labeled with 2.5 μM CFSE. 96-well plates were coated with 0, 0.1, 1.0, and 10 μg/ml of anti-CD3 (2C11) overnight at 4°C. 0.5 × 106 cells were added to each well with or without 1 μg/ml of soluble anti-CD28. Cells were harvested 48–120 h later, stained for CD4 and CD8, and analyzed by flow cytometry.
Online supplemental material.
Fig. S1 demonstrates the extensive lymphadenopathy and abnormal kidneys in wedge/lpr mice. Fig. S2 shows the memory cell phenotype of CD8 cells in all four genotypes of mice, as well as CD69 expression and cytokine production in CD4 cells. Fig. S3 examines the AICD of WT, Wedge, and Lpr CD4 cells. Fig. S4 shows the results of analyzing wedge/lpr mice on the BALB background. The online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20081204/DC1.
Supplementary Material
Acknowledgments
We thank A. Roque for his tireless assistance with animal husbandry, T. Defranco, M. Krummel, and A. Abbas for critically reviewing the manuscript, and the Weiss laboratory for helpful comments and suggestions.
This work was supported by the National Institutes of Health (NIH) Medical Scientist Training Program and by NIH PO1 AI35297.
The authors have no conflicting financial interests.
References
- 1.Fathman, C.G., and N.B. Lineberry. 2007. Molecular mechanisms of CD4+ T-cell anergy. Nat. Rev. Immunol. 7:599–609. [DOI] [PubMed] [Google Scholar]
- 2.Goldrath, A.W., and M.J. Bevan. 1999. Selecting and maintaining a diverse T-cell repertoire. Nature. 402:255–262. [DOI] [PubMed] [Google Scholar]
- 3.Bidere, N., H.C. Su, and M.J. Lenardo. 2006. Genetic disorders of programmed cell death in the immune system. Annu. Rev. Immunol. 24:321–352. [DOI] [PubMed] [Google Scholar]
- 4.Wakeland, E.K., K. Liu, R.R. Graham, and T.W. Behrens. 2001. Delineating the genetic basis of systemic lupus erythematosus. Immunity. 15:397–408. [DOI] [PubMed] [Google Scholar]
- 5.Hermiston, M.L., Z. Xu, and A. Weiss. 2003. CD45: a critical regulator of signaling thresholds in immune cells. Annu. Rev. Immunol. 21:107–137. [DOI] [PubMed] [Google Scholar]
- 6.Palacios, E.H., and A. Weiss. 2004. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 23:7990–8000. [DOI] [PubMed] [Google Scholar]
- 7.Desai, D.M., J. Sap, J. Schlessinger, and A. Weiss. 1993. Ligand-mediated negative regulation of a chimeric transmembrane receptor tyrosine phosphatase. Cell. 73:541–554. [DOI] [PubMed] [Google Scholar]
- 8.Majeti, R., A.M. Bilwes, J.P. Noel, T. Hunter, and A. Weiss. 1998. Dimerization-induced inhibition of receptor protein tyrosine phosphatase function through an inhibitory wedge. Science. 279:88–91. [DOI] [PubMed] [Google Scholar]
- 9.Bilwes, A.M., J. den Hertog, T. Hunter, and J.P. Noel. 1996. Structural basis for inhibition of receptor protein-tyrosine phosphatase-[alpha] by dimerization. Nature. 382:555–559. [DOI] [PubMed] [Google Scholar]
- 10.Majeti, R., Z. Xu, T.G. Parslow, J.L. Olson, D.I. Daikh, N. Killeen, and A. Weiss. 2000. An inactivating point mutation in the inhibitory wedge of CD45 causes lymphoproliferation and autoimmunity. Cell. 103:1059–1070. [DOI] [PubMed] [Google Scholar]
- 11.Hermiston, M.L., A.L. Tan, V.A. Gupta, R. Majeti, and A. Weiss. 2005. The juxtamembrane Wedge negatively regulates CD45 function in B cells. Immunity. 23:635–647. [DOI] [PubMed] [Google Scholar]
- 12.Carroll, M.C. 1998. The role of complement and complement receptors in induction and regulation of immunity. Annu. Rev. Immunol. 16:545–568. [DOI] [PubMed] [Google Scholar]
- 13.Manderson, A.P., M. Botto, and M.J. Walport. 2004. The role of complement in the development of systemic lupus erythematosus. Annu. Rev. Immunol. 22:431–456. [DOI] [PubMed] [Google Scholar]
- 14.Chen, Z., S.B. Koralov, and G. Kelsoe. 2000. Complement C4 inhibits systemic autoimmunity through a mechanism independent of complement receptors CR1 and CR2. J. Exp. Med. 192:1339–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Einav, S., O.O. Pozdnyakova, M. Ma, and M.C. Carroll. 2002. Complement C4 is protective for lupus disease independent of C3. J. Immunol. 168:1036–1041. [DOI] [PubMed] [Google Scholar]
- 16.Prodeus, A.P., S. Goerg, L.M. Shen, O.O. Pozdnyakova, L. Chu, E.M. Alicot, C.C. Goodnow, and M.C. Carroll. 1998. A critical role for complement in maintenance of self-tolerance. Immunity. 9:721–731. [DOI] [PubMed] [Google Scholar]
- 17.Nagata, S., and P. Golstein. 1995. The Fas death factor. Science. 267:1449–1456. [DOI] [PubMed] [Google Scholar]
- 18.Cohen, P.L., and R.A. Eisenberg. 1991. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu. Rev. Immunol. 9:243–269. [DOI] [PubMed] [Google Scholar]
- 19.Paul, E., O.O. Pozdnyakova, E. Mitchell, and M.C. Carroll. 2002. Anti-DNA autoreactivity in C4-deficient mice. Eur. J. Immunol. 32:2672–2679. [DOI] [PubMed] [Google Scholar]
- 20.Seddon, B., and R. Zamoyska. 2002. TCR signals mediated by Src family kinases are essential for the survival of naive T cells. J. Immunol. 169:2997–3005. [DOI] [PubMed] [Google Scholar]
- 21.Chen, M., Y.H. Wang, Y. Wang, L. Huang, H. Sandoval, Y.J. Liu, and J. Wang. 2006. Dendritic cell apoptosis in the maintenance of immune tolerance. Science. 311:1160–1164. [DOI] [PubMed] [Google Scholar]
- 22.Stranges, P.B., J. Watson, C.J. Cooper, C.M. Choisy-Rossi, A.C. Stonebraker, R.A. Beighton, H. Hartig, J.P. Sundberg, S. Servick, G. Kaufmann, et al. 2007. Elimination of antigen-presenting cells and autoreactive T cells by fas contributes to prevention of autoimmunity. Immunity. 26:629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hughes, P.D., G.T. Belz, K.A. Fortner, R.C. Budd, A. Strasser, and P. Bouillet. 2008. Apoptosis regulators Fas and Bim cooperate in shutdown of chronic immune responses and prevention of autoimmunity. Immunity. 28:197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hutcheson, J., J.C. Scatizzi, A.M. Siddiqui, G.K. Haines III, T. Wu, Q.Z. Li, L.S. Davis, C. Mohan, and H. Perlman. 2008. Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity. 28:206–217. [DOI] [PubMed] [Google Scholar]
- 25.Weant, A.E., R.D. Michalek, I.U. Khan, B.C. Holbrook, M.C. Willingham, and J.M. Grayson. 2008. Apoptosis regulators Bim and Fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity. 28:218–230. [DOI] [PubMed] [Google Scholar]
- 26.Akashi, T., S. Nagafuchi, K. Anzai, D. Kitamura, J. Wang, I. Taniuchi, Y. Niho, and T. Watanabe. 1998. Proliferation of CD3+ B220- single-positive normal T cells was suppressed in B-cell-deficient lpr mice. Immunology. 93:238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan, O., and M.J. Shlomchik. 1998. A new role for B cells in systemic autoimmunity: B cells promote spontaneous T cell activation in MRL-lpr/lpr mice. J. Immunol. 160:51–59. [PubMed] [Google Scholar]
- 28.Shlomchik, M.J., M.P. Madaio, D. Ni, M. Trounstein, and D. Huszar. 1994. The role of B cells in lpr/lpr-induced autoimmunity. J. Exp. Med. 180:1295–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shlomchik, M.J., J.E. Craft, and M.J. Mamula. 2001. From T to B and back again: positive feedback in systemic autoimmune disease. Nat. Rev. Immunol. 1:147–153. [DOI] [PubMed] [Google Scholar]
- 30.Straus, S.E., M. Sneller, M.J. Lenardo, J.M. Puck, and W. Strober. 1999. An inherited disorder of lymphocyte apoptosis: the autoimmune lymphoproliferative syndrome. Ann. Intern. Med. 130:591–601. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}