

Monomeric, early-transition-metal imide complexes [M=NR], which mediate numerous catalytic and stoichiometric nitrogen-transfer reactions,[1a] can be prepared by α abstraction and extrusion of a good leaving group (e.g. alkane or amine) from a metal amide species [X–M–NHR].[1] These α-abstraction reactions are thought to be concerted,[1] but in a hypothetical stepwise process, a potential intermediate is an imidate complex [M–NR]− [Eq. (1)]. The ease with which
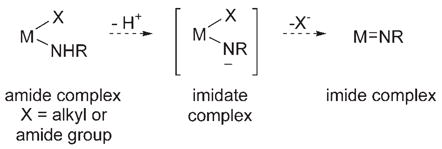 |
(1) |
such imidate species can be observed and isolated depends upon the absence of a good leaving group on the metal and on the acidity of the N–H proton. There are few reports of such deprotonation reactions in the literature;[2] the resultant imidates are either stabilized by adjacent conjugating groups[2a–c] or readily undergo subsequent reactions, thus preventing their structural characterization.[2d] We report herein the formation and structural characterization of monomeric, nonconjugatively stabilized lithium zirconocene imidate complexes.
In our ongoing studies on early-metal imide complexes, we became interested in preparing a series of substituted zirconocene methyl amide complexes as precursors to imidozirconocene complexes.[1] In systems with relatively small ancillary ligands on zirconium, such as Cp*Cp (Cp = η5-C5H5, Cp* = η5-C5Me5), methyl amide complexes such as 2a could be accessed through salt metathesis [Eq. (2)]. When [Cp*2Zr(Me)(Cl)] (1b) was subjected to the same reaction conditions, however, incomplete consumption of 1b and a mixture of two new products were observed (75% conversion based on 1b). These products were tentatively assigned as methyl amide complex 2b (48% conversion based on 1b) and
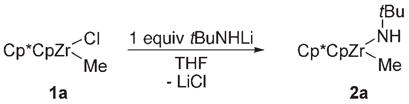 |
(2) |
lithium zirconimidate complex 3b (27% conversion based on 1b). Upon addition of a second equivalent of tBuNHLi, only complex 3 b was observed and subsequently isolated in 74% yield (Scheme 1).
Scheme 1.

Reaction of complex 1 b with tBuNHLi.
The identity and structural features of complex 3b were elucidated by NMR spectroscopy and X-ray crystallography. The 1H NMR spectrum of complex 3b lacks a diagnostic N–H peak, and the zirconium methyl signal is shifted upfield (δ = −0.75 ppm) relative to the corresponding signal in complex 2b (δ = −0.19 ppm). Further 7Li-13C and 6Li-1H NMR correlation experiments yielded measurable coupling constants (12 Hz[3] and 2.3 Hz, respectively), thus indicating that the {Me–Li} fragment is intact in solution.
X-ray crystallography revealed that the atoms comprising the metallacycle (Zr-N-Li-C) are coplanar (Figure 1).[4] The Zr–C bond length of 2.36 Å is consistent with a Zr–C single bond,[5] and the positions of the hydrogen atoms on C1 were calculated. The Zr–N bond length (rZr–N) of 1.91 Å indicates a high bond order, suggesting that zirconimidate 3b could be an intermediate structure between a zirconocene amide (rZr–N = 2.05–2.08 Å, R = alkyl)[6] and a zirconocene imide (rZr=N = 1.85–1.88 Å, R = alkyl).[1b] Both NMR spectroscopic and crystallographic data indicate a bond between the {Zr–CH3} moiety and lithium. Either σ donation from the Zr–C bond or from one or two of the C–H bonds in a rapidly rotating methyl group would be consistent with these data.
Figure 1.
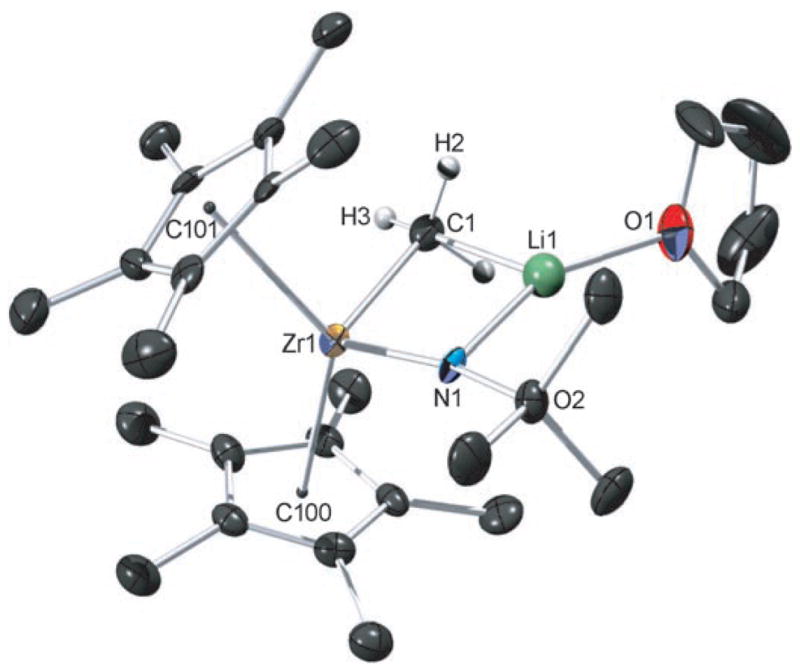
X-ray crystal structure of 3 b with thermal ellipsoids drawn at the 50% probability level; most hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Zr1–N1 1.909(4), Zr1–C1 2.358(5), C1–Li1 2.17(1), N1–Li1 2.01(1), Zr1–C100 2.3763(5), Zr1–C101 2.3589(5), Li1–O1 1.88(1), N1–C2 1.473(6); Zr1-N1-Li1 91.1(3), Zr1-C1-Li1 76.1(3), C1-Li1-N1 98.1(5), Zr1-N1-C2 167.0(3), C101-Zr1-C100 127.95(2).
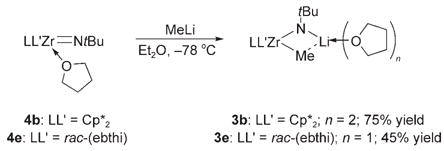
Analogous lithium zirconimidates (3c–e) were prepared by treating the corresponding methyl chlorides with 12 equiv tBuNHLi in THF solution (Scheme 2). Complexes 3 b and 3e were also obtained in comparable yields by treating the corresponding imidozirconium complexes with one equivalent of methyllithium [Eq. (3)]. Lithium zirconimidates 3b–e
Scheme 2.
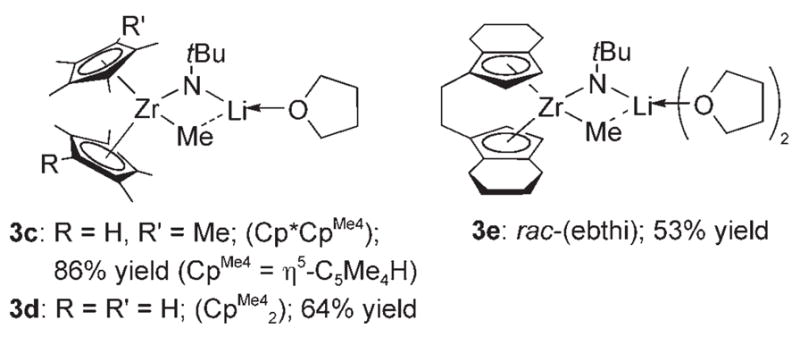
Isolated lithium zirconimidate complexes.
 |
(3) |
may therefore be thought of as either deprotonated amide species or alkyllithium adducts of zirconium imide complexes. Complexes 3b–e were thermally unstable and extremely sensitive to air and water. Efforts to isolate other zirconimidate complexes with smaller ancillary ligands or other substituents on the amide nitrogen atom were unsuccessful (see the Supporting Information).
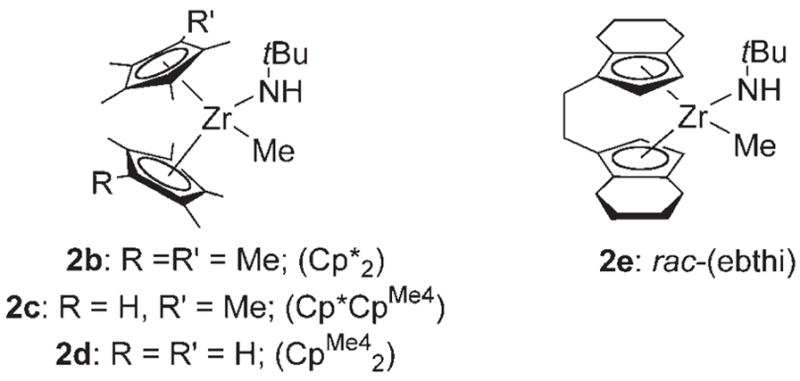
The isolation of zirconimidate complexes 3b–e presented an opportunity to examine quantitatively the acidity of the N–H proton in methyl amide complexes 2b–e (Scheme 3). We established the relative acidities of two methyl amide complexes, 2d and 2e, by a direct competition experiment (Scheme 4). Deprotonated amide complex 3d was treated with methyl amide 2 e and 0.3 equiv [12]crown-4. In the absence of [12]crown-4, the acid–base reactions were extremely slow. The mixture was allowed to attain equilibrium over three days, at which point an equilibrium constant of 12.9 ± 0.6 was measured. Unfortunately, other combinations of methyl amide and zirconimidates yielded inconclusive results owing to the thermal instability of the imidate complexes.
Scheme 3.
Methyl amide complexes 2 b–e.
Scheme 4.
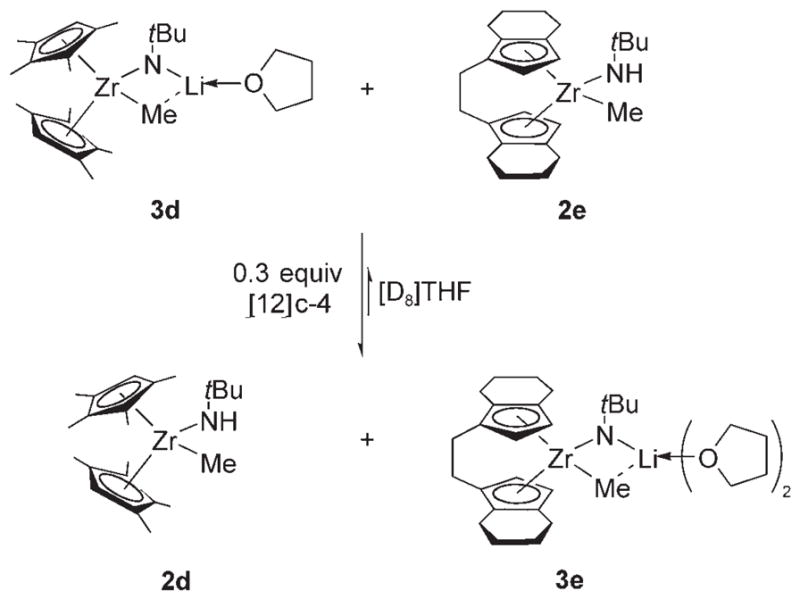
Direct competition experiment between methyl amide complexes 2 d and 2 e (Keq = 12.9 ± 0.6).
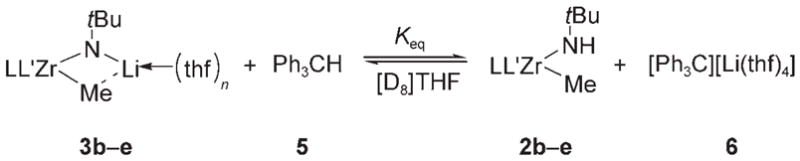
To gauge the basicities of all four zirconimidate complexes, 3b–e were treated with various Brønsted acids. When treated with [12]crown-4 and acids such as ammonium salts, phenylacetylene, or fluorene, complexes 3b–e underwent quantitative protonation to yield methyl amide complexes 2b–e. However, when complexes 3 b–e were treated with triphenylmethane and [12]crown-4, a mixture of 2–3 and 5–6 (see Table 1) was observed. These reactions were slow at room temperature, requiring two to three days to achieve equilibrium. Equivalent equilibrium constants could be obtained either by addition of 5 to 3 or by addition of 6 to 2; the composition of the equilibrium mixture was not concentration-dependent. Equivalent equilibrium constants were measured from reaction mixtures containing either 0.3 or 1.0 equiv [12]crown-4. These results indicate that the addition of [12]crown-4 to the acid–base reaction mixtures affects the rates at which the reactions proceed but does not perturb the equilibria of these reactions.
Table 1.
Acidity of methyl amide complexes 2 b–e.[a]
 | |||||
|---|---|---|---|---|---|
| Entry | Complex | Ligand | Yield [%][b] | Keq[c] | (pKa)THF |
| 1 | 2 b | Cp*2 | 33 | 0.249 ± 0.007 | 29.8 |
| 2 | 2 c | Cp*CpMe4 | 42 | 0.524 ± 0.010 | 30.1 |
| 3 | 2 d | CpMe42 | 51 | 1.05± 0.03 | 30.4 |
| 4 | 2 e | rac-(ebthi) | 22 | 0.079 ± 0.005 | 29.3 |
Reactions were run with 0.3 equiv [12]crown-4.
Yield determined by NMR spectroscopy and calculated using 1,3-dimethoxy-5-methylbenzene as an external standard.
Values are reported as the average of three runs ± standard deviation.
Given the pKa of triphenylmethane in THF (30.4),[7] pKa values for the N–H proton in methyl amide complexes 2b–e were extrapolated. A narrow range of values from 29.3 to 30.4 was observed (Table 1). The individual pKa values measured for complexes 2d and 2e are consistent with the Keq value measured in the direct competition experiment (Scheme 4). The ansa-bridged rac-(ebthi) complex 2e was the most acidic amide complex of those examined (entry 4). However, the ansa ligand in complexes 2e and 3e renders the structures of these complexes quite different from those of non-ansa complexes 2b–d and 3b–d. For example, while rac-(ebthi) zirconimidate complex 3e is solvated by two THF molecules, non-ansa zirconimidate complexes 3b–d are solvated by only one THF molecule each. It is therefore difficult to draw direct comparisons between complexes 2e and 2b–d.
Curiously, complexes with bulkier ancillary ligands possessed more acidic N–H amide protons than their smaller congeners, contrary to the expected trend based on electronic arguments alone.[8] Addition of one methyl group to the ancillary cyclopentadienyl ligands (e.g. Cp*CpMe4 to Cp*2) correlates to a three-fold increase in the acidity of the distant N–H amide proton (Table 1, entries 1–3). These results suggest that the acidity trend in methyl amide complexes 2b–d might represent a rare example of “steric acidity.”[9] That the observed pKa differences are small is consistent with the fact that the X-ray crystal structures of 3b and 3d show little structural variation between the two complexes (Figure 2).
Figure 2.
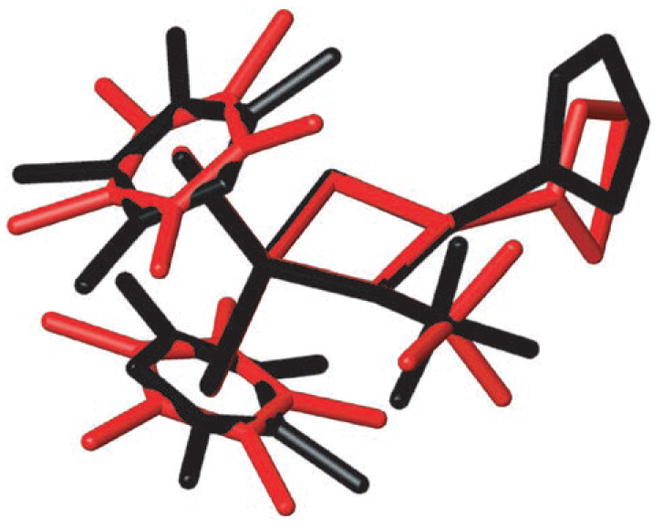
Superimposed X-ray crystal structures of 3 b (red) and 3 d (black). Hydrogen atoms are omitted for clarity.
While the protonation of complexes 3b–e with Ph3CH appears to be under thermodynamic control, we carried out an isotope exchange experiment to determine whether the deprotonation of methyl amide complexes with other ancillary ligands was disfavored due to thermodynamics or kinetics. Methyl amide complex 2g, which did not undergo observable deprotonation when treated with tBuNHLi, was treated with triphenylmethane deuterated at the methine position, triphenylmethyl(lithium), and [12]crown-4 [Eq. (4)]. No deuterium incorporation at the N–H position was observed over three days, indicating that the deprotonation
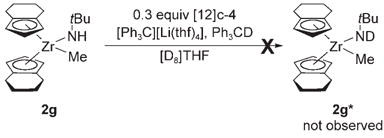 |
(4) |
of 2g has a high kinetic barrier. Thus, the deprotonation reactions of zirconium methyl amide complexes can have variable kinetic and thermodynamic profiles depending upon which ancillary ligand is coordinated to zirconium. These results demonstrate a complex relationship between ancillary ligand sterics and the acidity of the amide N–H proton.
In summary, a new class of zirconium–nitrogen species has been isolated and structurally characterized. Arising from either deprotonation of zirconium methyl amide complexes or addition of methyllithium across the zirconium imide moiety, zirconimidate complexes become decreasingly basic as their ancillary ligands become bulkier. While these basicity studies highlight the behavior of zirconimidate complexes as Brønsted basic, deprotonated amide complexes, further studies addressing the reactivity of zirconimidate complexes with other organic substrates may provide insight into their potential use as nucleophiles and nitrogen-transfer reagents.
Experimental Section
General procedure for preparing zirconimidate complexes: In an N2-filled glovebox, a 20-mL scintillation vial was equipped with a magnetic stir bar and charged with a solution of the [LL′Zr(Me)(Cl)] complex (0.2 mmol, 1 equiv) and tBuNHLi (2.4 mmol, 12 equiv) in THF (20 mL). The clear, yellow reaction mixture was stirred for 5–8 h at ambient temperature. The solvent was removed in vacuo, and the resulting residue was then extracted with pentane (3 × 4 mL). The combined pentane extracts were filtered, concentrated, and cooled to −30°C. Products were obtained as colorless or yellow crystalline solids (53–86%).
Footnotes
This work was supported by the NIH National Institute of General Medicine (GM-25459) and an NSF predoctoral fellowship to M.C. We thank Drs. Fred Hollander and Allen Oliver of the UC Berkeley CHEXRAY facility for X-ray structure determinations.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.200801463.
References
- 1.a) Duncan AP, Bergman RG. Chem Rec. 2002;2:431–445. doi: 10.1002/tcr.10039. [DOI] [PubMed] [Google Scholar]; b) Wigley DE. Prog Inorg Chem. 1994;42:239–482. [Google Scholar]; c) Hazari N, Mountford P. Acc Chem Res. 2005;38:839–849. doi: 10.1021/ar030244z. [DOI] [PubMed] [Google Scholar]; d) Fout AR, Kilgore UJ, Mindiola DJ. Chem Eur J. 2007;13:9428–9440. doi: 10.1002/chem.200701064. [DOI] [PubMed] [Google Scholar]
- 2.a) Preuss F, Becker H, Wieland T. Z Naturforsch B. 1990;45:191–198. [Google Scholar]; b) Yu X, Xue ZL. Inorg Chem. 2005;44:1505–1510. doi: 10.1021/ic0485413. [DOI] [PubMed] [Google Scholar]; c) Yu X, Cai H, Guzei IA, Xue ZL. J Am Chem Soc. 2004;126:4472–4473. doi: 10.1021/ja031899y. [DOI] [PubMed] [Google Scholar]; d) Hou Z, Breen TL, Stephan DW. Organometallics. 1993;12:3158–3167. [Google Scholar]
- 3.The {ZrMeLi} signal of the 7Li-13C HMQC spectrum contained an extraneous artifact. When the 7Li-13C correlation experiment was performed on complex 3d, no artifact and a comparable coupling constant (1J7Li-7C = 10 Hz) were observed.
- 4.a) Crystal data for 3b: ZrONC29LiH50, Mr = 526.88 gmol−1, 0.20 × 0.15 × 0.09 mm3, monoclinic, space group P21/c, a = 9.6663(8), b = 18.721(2), c = 16.278(1) Å, β = 104.103(1) °, V= 2857.0(4) Å3, Z = 4, ρcalcd = 1.225 gcm−3, μ = 4.04 cm−1, F(000) = 1128.00, 13069 reflections measured, 2565 unique [R(int) = 0.051], 2θmax = 49.4°, 293 refined parameters, R1 = 0.046 (2807 observations where I >3.00σ(I)), wR2 = 0.046, GoF = 1.46. Data were collected at 103(2) K on a Siemens SMART CCD diffractometer (Bruker SMART v.5.059, Area-Detector Software Package, Bruker AXS, Inc., Madison, WI, 1995–1999) with graphite monochromated MoKα radiation using ω scans (0.3° per 10.0-s frame). All non-hydrogen atoms were refined anisotropically except for the lithium atoms, which were refined isotropically. Hydrogen atoms were included but not refined. The maximum and minimum peaks on the final difference Fourier map corresponded to 0.59 and −0.72 eÅ−3, respectively. General methods for integration, solution, and refinement can be found in the Supporting Information. CCDC 682892 (3b) and 682893 (3d) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif (see the Supporting Information).
- 5.a) Atwood JL, Hunter WE, Hrncir DC, Samuel E, Alt H, Rausch MD. Inorg Chem. 1975;14:1757–1762. [Google Scholar]; b) Atwood JL, Barker GK, Holton J, Hunter WE, Lappert MF, Pearce R. J Am Chem Soc. 1977;99:6645–6652. [Google Scholar]
- 6.For rZr–N in Zr–NHR, where R = alkyl, see: Hoyt HM, Michael FE, Bergman RG. J Am Chem Soc. 2004;126:1018–1019. doi: 10.1021/ja0385944.Lee SY, Bergman RG. J Am Chem Soc. 1995;117:5877–5878.Harlan CJ, Tunge JA, Bridgewater BM, Norton JR. Organometallics. 2000;19:2365–2372.
- 7.Kaufman MJ, Gronert S, Streitwieser A., Jr J Am Chem Soc. 1988;110:2829–2835. [Google Scholar]
- 8.Zachmanoglou CE, Docrat A, Bridgewater BM, Parkin G, Brandow CG, Bercaw JE, Jardine CN, Lyall M, Green JC, Keister JB. J Am Chem Soc. 2002;124:9525–9546. doi: 10.1021/ja020236y. [DOI] [PubMed] [Google Scholar]
- 9.a) Lillya CP, Miller EF. Tetrahedron Lett. 1968;9:1281–1283. [Google Scholar]; b) McCoy EA, McCoy LL. J Org Chem. 1968;33:2354–2356. [Google Scholar]