

A promising approach to the functionalization of unactivated hydrocarbons (RH) involves a 1,2-RH-addition across metal–heteroatom multiple bonds [MX] (M =Groups 4–6, X = NR,[1,2] CR2,[3] CR,[4] O[5]) to afford products of general structure [M(XH)(R)]. Extensive selectivity studies have been performed on Group 4 imido [M =NR′] systems that activate hydrocarbons under reversible conditions.[1l–m] However, isolation of kinetically controlled product ratios has not yet been achieved owing to the reverse reaction: competitive extrusion of RH from the resulting [M(NHR′)(R)] products. One process requiring kinetic control of the product distribution is diastereoselective activation of a C–H bond, a desirable transformation wherein kinetic selectivity will play a critical role. Herein, we report the first Zr=NR system capable of generating kinetic product distributions in the selective C–H bond activation of unactivated sp2 and sp3 hydrocarbons. The results provide important information regarding the selectivity and mechanism of the 1,2-RH-addition event.
In an ongoing study in our laboratory,[1a–e] zirconium complexes bearing Cp*Cp ligands emerged as ideal candidates for the investigation of kinetic control in C–H bond activation (Cp* =η5-C5Me5, Cp =η5-C5H5). Specifically, imido precursors [Cp*CpZr= NCMe3(thf)](1) and [Cp*CpZr(Me)(NHCMe3)] (4) extrude THF or methane, respectively, on thermolysis to form the transient imido complex [Cp*CpZr= NCMe3](2) [Eq. (1) and (2)].[6a] We found that the transient species 2 reacted with benzene to produce the C–H activation product [Cp*CpZr(Ph)-(NHCMe3)] (3 f) in both cases. In contrast to most Lewis base (LB) complexes of the type [LnZr=NR(LB)], the use of complex 1 resulted in the activation of benzene at a lower temperature than did its corresponding methyl amide complex 4.[7] That methyl amide 4 appeared more thermally stable than imide 1 suggested that the analogous hydrocarbyl amide products of C–H activation (e.g., 3 f) may form irreversibly from 1 at 45°C.
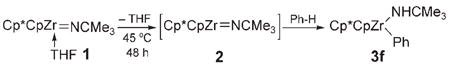 |
(1) |
 |
(2) |
Given the unique reactivity of 1, kinetic control would require that products [Cp*CpZr(R)(NHCMe3)] (3-R) form irreversibly under the reaction conditions. Complex 1 reacted with tert-butylacetylene and (E)-tert-butylethylene at ambient temperature to generate products 3a and 3d, respectively, whereas gentle heating (45°C) was required to activate other substrates depicted in Table 1 (forming 3b–c,[6b] 3e–k).[6c] These preliminary data suggested that the hydrocarbon substrate was involved in the rate-determining step of the reaction.
Table 1.
Relative kinetic (kRH/kR′H) and thermodynamic (KRH/R′H) selectivity of C–H bond activation for substrates RH by complex 1.
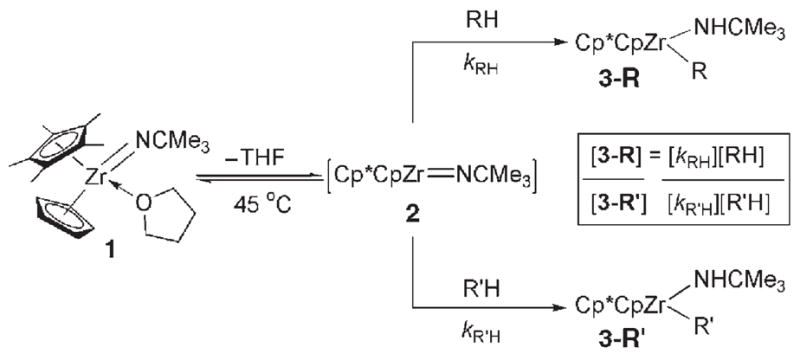 | |||||||
|---|---|---|---|---|---|---|---|
| 3-R[a] | RH | kRH/kR′H[b] | KRH/R′H[c] | 3-R[a] | RH | kRH/kR′H[b] | KRH/R′H[c] |
| 3 a |
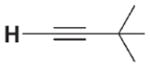
|
>106 | n.d.[d] | 3 f |
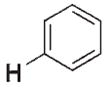
|
17 | 170 |
| 3 b |
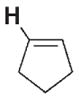
|
700 | 370 | 3 g |
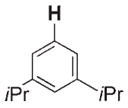
|
16 | 160 |
| 3 c |
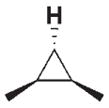
|
280 | 40 | 3 h |
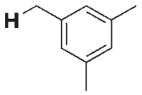
|
15 | 30 |
| 3 d |
|
220 | 750 | 3 i |
|
1.7 | 1 |
| 3 j |
|
1.5 | n.d.[d] | ||||
| 3 e |
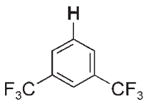
|
36 | 26 000 | 3 k |
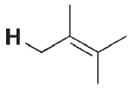
|
1 | 0.6[e] |
Products 3-R formed from 1 and the explicitly drawn C–H bond in the corresponding substrate RH.[8]
Relative rates (kRH/kR′H) for RH activation by 1 calculated per reactive RH bond at 45°C; kR′H = kRH for 2,3-dimethyl-2-butene.
Relative thermodynamic stability (KRH/R′H) of products 3-R relative to 3 i measured at 150°C from 1 and calculated per reactive RH bond.
Determination of thermodynamic selectivity was unsuccessful owing to decomposition of 3-R. [e] Thermodynamic selectivity measured at 105°C.
To determine the thermal stability of C–H activation products 3a–k, these complexes were thermolyzed with 10 equivalents of an imido trapping agent. Upon extrusion of RH from complexes 3a–k, intermediate 2 was generated and trapped irreversibly with di-p-tolylacetylene to produce metallacycle 5, which subsequently rearranged to cyclometal-lated complex 6 upon extended heating [Eq. (3)].[8] Elevated temperatures (75–150°C) were generally required for extrusion of RH from 3-R.[8,9a] Given the high thermal stability of these products, we presume that C–H bond activation from 1 is effectively irreversible at 45°C. This confirms that C–H activation occurred under kinetic control at 45°C.
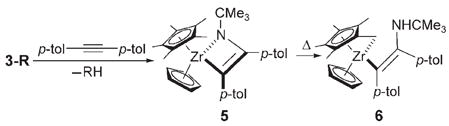 |
(3) |
To gain mechanistic insight into this process, we performed a crossover labeling study to determine whether the 1,2-RH-addition event proceeded in an inter- or intramolec-ular fashion. Reaction of complex 1 with a mixture of [D0]mesitylene/[D6]benzene solely furnished products 3h/ [D6]- f3. The lack of crossover products ([D1]-3h/[D5]-3 f) indicated that 1,2-RH(D)-addition to 2 placed the R and H(D) groups from the reacting hydrocarbon on the same metal center in the final product: an intramolecular process.
The nature of the transition state was examined by performing kinetic isotope effect (KIE) studies. Measured at 45°C, KIE values for reaction of 1 with [D0]/[D6]benzene, [D0]/[D12]n-pentane,[9c] [D0]/[D12]mesitylene, and [D0]/[D1]-(E)-neohexene were kH/kD = 7.4, 8.9, 8.8, and 6.9, respectively.[1n] These large positive values indicated a primary KIE consistent with a direct C–H bond-breaking event in the rate-determining step of the reaction. Most likely, this occurred through a four-center transition state, wherein transfer of H from R to N is relatively linear.[10] These values contrasted with the equilibrium isotope effect for [D0]/[D6]benzene at 150°C, KH/KD = 1.02, confirming that we have been able toselect independent thermodynamic conditions by varying the reaction temperature.
Given that the hydrocarbon substrate underwent concerted, rate-determining 1,2-RH-addition to the Zr=N bond of intermediate 2, intermolecular competition studies were conducted under kinetic conditions (45°C) such that the observed product ratios ([3-R]/[3-R′]) reflected the relative rates (kRH/kR′H) of RH activation (Table 1). Generally, substrates bearing C–H bonds with a greater degree of s character formed products 3-R with the highest relative rates:sp (3a) >alkene sp2 (3b,3d) ≈cPr (3c) >are-ne sp2 (3e–g) >sp3 (3h–k).[9b] Less sterically hindered substrates reacted up to 15 times faster than larger substrates bearing similar electronic properties (3b >3d; 3h >3k).Arenes bearing electron-withdrawing substituents (e.g., 1,3-bis(tri-fluoromethyl)benzene to form 3e) reacted only twice as fast as other arenes, even in cases where the thermodynamic stabilities of the products were substantially different (see below).
The high relative rate for activation of tert-butylacetylene led us to speculate that 1 may activate alkynes by a mechanism distinct from that of direct 1,2-RH-addition via transition state 7 (Scheme 1, path a). In analogy to a Ti=O system,[5] alkynes may undergo rate-determining metalla-cycle formation (Scheme 1, kmet, path b) with 2 to form intermediate metallacycle 8, followed by rearrangement (krearr) to provide 3a. In a KIE competition study to form 3a/[D1]-3a, kH/kD was found to be 0.8. In contrast with the large primary KIE values determined for other substrates, this small, inverse value indicated that a C–H bond was not broken on or before the rate-determining step. Thus, we propose that alkynes follow path b with an initial rate-determining metallacycle-forming step, whereas other unsaturated substrates react either analogously to path a or via reversible metallacycle (kmet/kmet −1) formation followed by a rate-determining rearrangement reaction (krearr).
Scheme 1.
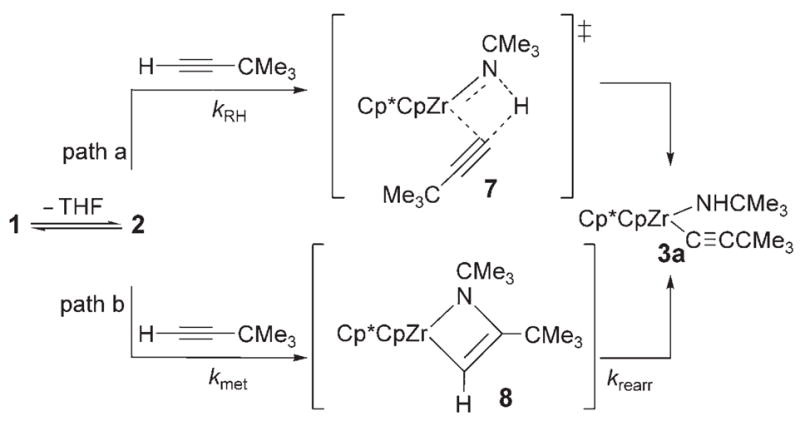
Mechanistic pathways for activation of tert-butylacetylene.
To compare kinetic selectivity data to the relative thermodynamic stability (KRH/R′H) of products 3-R, intermolecular competition studies were performed at higher temperatures (150°C) in which interconversion of the products takes place: 3e ≫ 3d, 3b >3 f, 3g >3c, 3h >3i, 3k (Table 1).[8] With modest exceptions, substrates reacting with the highest relative rates generally formed the most thermodynamically stable products. The unusually high stability of 3e may be attributed to electronic stabilization imparted by the arene substitutents.
In summary, mixed-ring Cp*Cp complex 1 provided the first example of the isolation of kinetic product ratios in sp2 and sp3 C–H bond activation with Group 4 M=NR complexes. This feature of the Cp*Cp system allowed selectivity and mechanistic experiments to probe the 1,2-RH-addition event. Hybridization of reacting C–H bonds generally determined the relative rate of RH activation, whereas electronic factors and substrate size were responsible for more subtle differences. Substrates that formed the most thermodynamically stable products generally reacted most rapidly. KIE values indicated that alkyne substrates likely undergo rate-determining metallacycle formation followed by rearrangement, whereas the RH bond was likely broken directly in the rate-determining step for other hydrocarbons. Continuing work focuses on designing complexes capable of diastereoselective C–H bond activation as well as on determining the factors responsible for reactivity differences promoted by various ancillary Cp-based ligands.
Footnotes
This work was supported by the NIH through grant no. GM-25459, We thank Dr. Herman van Halbeek for assistance with 2D NMR spectroscopy experiments.
Supporting information for this article (including full experimental procedures) is available on the WWW under http://www.ange-wandte.org or from the author.
References
- 1.a) Walsh PJ, Hollander FJ, Bergman RG. J Am Chem Soc. 1988;110:8729. [Google Scholar]; b) Walsh PJ, Hollander FJ, Bergman RG. Organometallics. 1993;12:3705. [Google Scholar]; c) Lee SY, Bergman RG. J Am Chem Soc. 1995;117:5877. [Google Scholar]; d) Duncan AP, Berg-man RG. Chem Rec. 2002;2:431. doi: 10.1002/tcr.10039. [DOI] [PubMed] [Google Scholar]; e) Hoyt HM, Michael FE, Bergman RG. J Am Chem Soc. 2004;126:1018. doi: 10.1021/ja0385944. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Cummins CC, Baxter SM, Wolczanski PT. J Am Chem Soc. 1988;110:8731. [Google Scholar]; g) Cummins CC, Schaller CP, Van Duyne GD, Wolczanski PT, Chan AWE, Hoffmann R. J Am Chem Soc. 1991;113:2985. [Google Scholar]; h) Schaller CP, Wolczanski PT. Inorg Chem. 1993;32:131. [Google Scholar]; i) de With J, Horton AD. Angew Chem. 1993;105:958. [Google Scholar]; Angew Chem Int Ed Engl. 1993;32:903. [Google Scholar]; j) Bennett JL, Wolczanski PT. J Am Chem Soc. 1994;116:2197. [Google Scholar]; k) Schaller CP, Bonanno JB, Wolczanski PT. J Am Chem Soc. 1994;116:2179. [Google Scholar]; l) Schaller CP, Cummins CC, Wolczanski PT. J Am Chem Soc. 1996;118:591. [Google Scholar]; m) Bennett JL, Wolczanski PT. J Am Chem Soc. 1997;119:10696. [Google Scholar]; n) Schafer DF, II, Wolczanski PT. J Am Chem Soc. 1998;120:4881. [Google Scholar]; o) Royo P, Sanchez-Nieves J. J Organomet Chem. 2000;597:61. [Google Scholar]
- 2.Activation of HC–CR: Blake RE, Jr, Antonelli DM, Henling LM, Schaefer WP, Hardcastle KI, Bercaw JE. Organometallics. 1998;17:718.Polse JL, Andersen RA, Bergman RG. J Am Chem Soc. 1998;120:13405.
- 3.a) van der Heijden H, Hessen B. J Chem Soc Chem Commun. 1995:145. [Google Scholar]; b) Coles MP, Gibson VC, Clegg W, Elsegood MRJ, Porelli PA. Chem Commun. 1996:1963. [Google Scholar]; c) Cheon J, Rogers DM, Girolami GS. J Am Chem Soc. 1997;119:6804. [Google Scholar]; d) Pamplin CB, Legzdins P. Acc Chem Res. 2003;36:223. doi: 10.1021/ar0202215. and references therein. [DOI] [PubMed] [Google Scholar]; e) Wada K, Pamplin CB, Legzdins P, Patrick BO, Tsyba I, Bau R. J Am Chem Soc. 2003;125:7035. doi: 10.1021/ja0349094. [DOI] [PubMed] [Google Scholar]; f) Blackmore IJ, Legzdins P. Organometallics. 2005;24:4088. [Google Scholar]; g) Tsang JYK, Buschhaus MSA, Legzdins P, Patrick BO. Organometallics. 2006;25:4215. [Google Scholar]
- 4.a) Bailey BC, Fan H, Baum EW, Huffman JC, Baik M, Mindiola DJ. J Am Chem Soc. 2005;127:16016. doi: 10.1021/ja0556934. [DOI] [PubMed] [Google Scholar]; b) Bailey BC, Huffman JC, Mindiola DJ. J Am Chem Soc. 2007;129:5302. doi: 10.1021/ja0684646. [DOI] [PubMed] [Google Scholar]
- 5.Activation of HC–CR: Polse JL, Andersen RA, Bergman RG. J Am Chem Soc. 1995;117:5393.
- 6.Zuckerman RL, Krska SW, Bergman RG. J Am Chem Soc. 2000;122:751. doi: 10.1021/ja992869r. b) Only one diastereomer of 3c was detected in solution[8]; c) On a preparative scale, products 3-R were obtained (83–99%) on thermolysis of 4 in hydrocarbon solvents RH.[8]
- 7.Reaction of 4 requires elevated temperatures (compared with 1) to extrude CH4.
- 8.See the Supporting Information for further details.
- 9.a) Product 3j slowly extrudes n-pentane at 45°C. b) The kinetic selectivity for n-pentane remained constant up to 50% conversion. c) KIE measured at 20% conversion.
- 10.a) Cundari TR, Klinckman TR, Wolczanski PT. J Am Chem Soc. 2002;124:1481. doi: 10.1021/ja016248l. [DOI] [PubMed] [Google Scholar]; b) Slaughter LM, Wolczanski PT, Klinckman TR, Cundari TR. J Am Chem Soc. 2000;122:7953. [Google Scholar]