

Abstract
Ventricular tachycardia (VT) in heart failure (HF) can initiate by nonreentrant mechanisms such as delayed afterdepolarizations. In an arrhythmogenic rabbit model of HF, we have shown that isoproterenol induces VT in vivo, and aftercontractions and transient inward currents in HF myocytes. To determine whether β2-adrenergic receptor (β2-AR) stimulation contributes, we performed in vivo drug infusion, in vitro myocyte and biochemical studies. Intravenous zinterol (2.5 μg/kg) led to ventricular arrhythmias including VT up to 13 beats long in 4 of 6 HF rabbits (vs 0 of 5 controls, p<0.01), an effect blocked by β2-AR antagonist ICI 118,551 (ICI, 0.2 mg/kg). In field stimulated myocytes (0.5−4 Hz, 37°C) β2-AR stimulation (1μM zinterol + 300 nM β1-AR antagonist CGP-29712A, CGP) induced aftercontractions and after-Ca-transients in 88% of HF vs 0% of control myocytes (p<0.01). β2-AR stimulation in HF (but not control) myocytes increased Ca transient amplitude (by 29%), SR Ca load (by 28%), the rate of [Ca]i decline (by 28%; n=12, all p<0.05), and PLB phosphorylation at Ser-16, but Ca current was unchanged. All of these effects in HF myocytes were blocked by ICI (100 nM). While total β-AR expression was reduced by 47% in HF rabbit LV, β2-AR number was unchanged, indicating more potent β2-AR-dependent SR Ca uptake and arrhythmogenesis in HF. Human HF myocytes showed similar β2-AR-induced aftercontractions, aftertransients, and enhanced Ca transient amplitude, SR Ca load and twitch [Ca]i decline rate. Thus, β2-AR stimulation is arrhythmogenic in HF, mediated by SR Ca overload-induced spontaneous SR Ca release and aftercontractions.
Keywords: Heart failure, β2-adrenergic stimulation, arrhythmia, sarcoplasmic reticulum, zinterol
INTRODUCTION
Heart failure (HF) is associated with significant mortality, with nearly one half of deaths occurring suddenly, primarily from ventricular tachycardia (VT) to ventricular fibrillation (VF).1,2 In three-dimensional cardiac mapping studies, both in an arrhythmogenic rabbit model of nonischemic HF3 and in the failing human heart,4 we have shown that VT initiates by a nonreentrant mechanism that may represent delayed afterdepolarizations (DAD's) due to a transient inward current (Iti), or early afterdepolarizations (EAD's).5
It is well known that arrhythmogenesis in the failing heart is enhanced by stimulation of β-adrenergic receptors (β-ARs).1,5 We recently showed that β-adrenergic stimulation with isoproterenol induced ventricular arrhythmias in vivo in HF but not control rabbits.5 Moreover, isoproterenol induced DADs and aftercontractions by increasing sarcoplasmic reticulum (SR) calcium load, leading to spontaneous SR Ca release and activation of Iti (that underlies DADs and nonreentrant VT). We found that preserved β-AR responsiveness, as may occur in HF that is not end-stage,6 is an important contributor to arrhythmogenicity in HF myocytes.5
Isoproterenol stimulates β2- as well as β1-ARs; so in HF, where there is β1-AR down-regulation but relatively preserved β2-ARs,6 some of the arrhythmogenic effects of isoproterenol could arise from β2-AR stimulation. In fact, there is evidence of enhanced β2-AR responsiveness in HF. For instance, Altschuld et al.7 have shown that β2-AR stimulation with zinterol can enhance Ca transients in HF myocytes to a greater degree than in controls (consistent with enhanced arrhythmogenesis). While the second messenger pathways of β2-AR stimulation appears to differ from that of β1-AR stimulation,8 there is evidence in failing human myocardium that β2-AR stimulation can elicit localized increases in cAMP and phosphorylation of phospholamban (PLB) that may contribute to SR Ca overload.9
To determine whether stimulation of β2-AR per se may be arrhythmogenic in HF, we assessed the response of β2-AR stimulation on arrhythmogenesis in vivo and on in intracellular Ca transients, SR Ca content and Ca current (ICa) in ventricular myocytes from control (Ctl) rabbits and from an arrhythmogenic rabbit model of HF induced by combined aortic insufficiency & aortic constriction. These studies were complemented by molecular and biochemical studies of β-ARs and phosphorylation of PLB, and were extended to human HF by selected experiments performed with isolated myocytes from human hearts. We found that in HF, β2-AR stimulation is arrhythmogenic by enhancing spontaneous SR Ca release and aftercontractions, and likely attributable to enhanced SR Ca load secondary to PLB phosphorylation.
MATERIAL AND METHODS
An expanded Methods section is available online at http://www.circresaha.org.
Rabbit heart failure model and myocyte isolation
In New Zealand White rabbits of either sex, HF was induced by aortic insufficiency and 2−4 weeks later by abdominal aortic constriction (both during isoflurane anesthesia) as previously described.3 Progression of HF was assessed by two-dimensional echocardiography.3,5,10 HF rabbits were studied when LV end-systolic dimension exceeded 1.40 cm.3,5,10 At that stage, Intravenous bolus administration of zinterol (1.0 or 2.5 μg/kg over 5 sec) ± the β2-AR blocker ICI-118,551 (0.2 mg/kg) was performed in conscious control and HF rabbits with monitoring of the surface ECG for at least 3 min. Protocols were approved by the University of Illinois at Chicago Animal Studies Committee. Rabbit left ventricular myocytes were isolated as described,5,10 with back flow across the incompetent aortic valve in HF rabbits blocked by a balloon-tipped catheter inflated in the LV outflow tract.
Contraction, [Ca]i and patch clamp
Ventricular myocytes were stored at 22°C and plated on laminin-pretreated glass-bottomed chambers. Cells were loaded with flou-3-acetoxymethyl ester and [Ca]i was measured as previously described.11 SR Ca-load was determined by rapid caffeine (10 mM) application after 20 stimulated pulses.5 Myocyte shortening was measured by video edge detection. The normal Tyrodes (NT) solution contained (in mmol/L): 140 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 5 HEPES, pH 7.4. Myocytes were field-stimulated (0.5−4 Hz, 20 beats, 37°C) followed by 10 seconds of observation for the presence of aftercontractions in the absence or presence of the β2-AR agonist zinterol (300 nM or 1 μM, generous gift from Bristol-Myers Squibb) ± the β1-AR antagonist CGP 20712A (300 nM, Sigma) or ICI 118,551 (100 nM, TOCRIS).
In other studies, ruptured patch voltage clamp was done to measure L-type ICa ± similar doses of zinterol and CGP with pipettes containing (mmol/L) CsOH 110, CsCl 20, EGTA 5, MgCl2 2, Aspartic acid 100, HEPES 5, Mg-ATP 2, Na3GTP 0.1, pH 7.2, 25°C. Membrane capacitance was measured from responses to 5-mV hyperpolarizing and depolarizing pulses.12
Phosphorylation of phospholamban
Freshly-isolated control and HF rabbit myocytes were incubated for 10 min with zinterol (1 μM), zinterol + CGP 20712A (300 nM) or zinterol + CGP + ICI 118,551 (100 nM). Cell pellets were then spun down and homogenized for protein quantification and Western blotting.
β-Adrenergic receptor assays and competitive binding studies
Saturation binding studies were carried out on LV homogenates by incubation with various concentrations (0−500 pM) of the β-AR antagonist [125I]-(−)Iodocyanopindolol as previously described.13 The equilibrium dissociation constant (Kd) and maximum binding capacity (Bmax) were determined by Scatchard analysis using GraphPad Prism. For competitive binding studies, homogenates were incubated with 500 pM 125I-CYP plus increasing dilutions of 1 μM ICI-118,551, a selective β2-AR antagonist, or 100 nM CGP 20712A, a selective β1-AR antagonist. Results were adjusted to fmol/mg protein to calculate for the specific binding, and determine the percentage of β1 and β2 receptors.
Human HF LV myocytes
Human HF LV myocytes were isolated from LV tissue obtained at the time of clinically indicated cardiac transplantation in patients with end-stage idiopathic dilated cardiomyopathy (IDCM, n=3) or ischemic cardiomyopathy (n=3) performed at Loyola University Hospital and the University of Illinois at Chicago (UIC) Hospital. This protocol has been approved by the Human Studies Committees of Loyola & UIC.
Data analysis
Data are shown as mean ± SEM and statistical significance was based on p<0.05 (Students t-test, ANOVA and chi-square).
RESULTS
HF rabbit preparation
HF was induced in 27 rabbits. After an average of 8.0 ± 1.4 months following combined aortic regurgitation and aortic constriction, rabbits developed progressive cardiac enlargement and decreased systolic function. LVEDD increased by 41% (1.51±0.03 to 2.13±0.03 cm, p<0.001) and LVESD increased by 60% (0.98±0.02 to 1.57±0.03 cm, p<0.001), while fractional shortening decreased by 25% (34.9±1.1 to 26.1±0.95 %, p<0.001).
In vivo arrhythmogenic effects of β2-AR agonist zinterol
To determine whether stimulation of β2-ARs may contribute to the arrhythmogenic effects of isoproterenol, we performed in vivo drug infusion studies in 5 control and 6 HF rabbits. Zinterol (2.5 μg/kg i.v. bolus over 5 s) did not significantly alter heart rate or mean arterial blood pressure in either control or HF rabbits. Figure 1 shows that zinterol at this dose led to ventricular arrhythmias including premature ventricular complexes (PVCs) and runs of VT (up to 13 beats long) in 4 of 6 HF rabbits (vs 0 of 5 controls, p<0.01), an effect that was blocked by the β2-AR antagonist ICI 118,551 (ICI, 0.2 mg/kg). Zinterol at a lower dose (1μg/kg i.v, n=4) did not induce ventricular arrhythmias in HF rabbits.
Figure 1.

Spontaneously-occurring ventricular arrhythmias in conscious failing rabbits and aftercontractions in isolated ventricular myocyte in response to bolus injection (2.5μg/kg) and superfusion (1 μM) of zinterol (Zin), respectively. A) Lead II ECG tracings showing sinus rhythm at baseline (left) and runs of VT with zinterol (middle) prevented by pretreatment with the β2-AR blocker ICI-118,551 (right). B) 3 Hz cell shortening at baseline conditions at 37°C (left) and aftercontractions (ACs) induced by zinterol (middle) prevented by ICI (right).
In vitro arrhythmogenic effects of zinterol
To determine the underlying cellular mechanism of ventricular arrhythmias induced by zinterol, we measured contractions in isolated LV myocytes from control and HF rabbits. Field stimulation (0.5−4 Hz) of fluo-3 loaded myocytes (37°C) demonstrated that zinterol (1 μM) induced aftercontractions (AC's) in 7 of 7 HF myocytes, compared to 0 of 6 controls (p<0.01), an effect that was blocked by ICI-118,551 (100 nM, Fig.1, bottom). These ACs were associated with spontaneous SR Ca release or aftertransients. To verify that zinterol's effects were due to stimulation of β2-ARs, additional β2-AR stimulation studies were performed with 1 μM zinterol plus 300 nM of the β1-AR blocker CGP-20712A (CGP). Zinterol + CGP induced AC's (associated with aftertransients, Fig.2) in 7 of 8 HF myocytes, compared to 0 of 7 controls (p<0.01), an effect that was blocked by 100 nM ICI-118,551. Similar results were found in HF and control myocytes on glass slides without laminin, ruling out potential effects of laminin on β2-AR signaling.14 Lower concentrations of Zinterol (300nM) with CGP failed to induce aftertransients in 4 HF myocytes, consistent with a dose-dependent specific effect.
Figure 2.
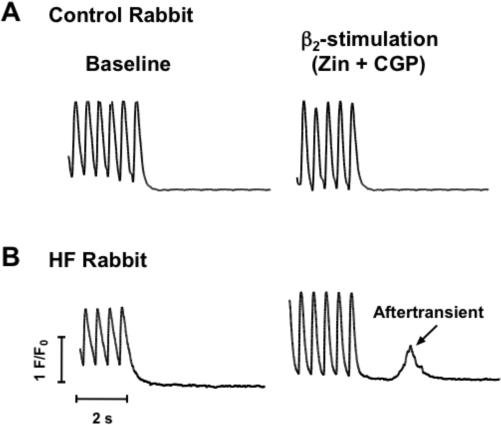
β2-stimulation (zinterol + CGP) induces after-Ca-transients in HF myocytes. A) Ca transients (2 Hz) of myocytes from control rabbits before (left) and after zinterol + CGP. B) Baseline Ca transients (2 Hz) of myocytes from failing rabbit (left) and induction of after-transients with zinterol + CGP (right).
Effects of β2-AR stimulation on cell shortening, Ca transients and SR Ca load
Zinterol + CGP had no effects on cell shortening in control myocytes (at 1 or 3 Hz; Fig 3C). However, this β2-AR stimulation significantly increased cell shortening in HF myocytes (42% and 41% increase at 1 and 3 Hz, respectively, n=7, p<0.05; Fig 4C).
Figure 3.

Ca transients in control rabbit myocytes are not altered by β2-stimulation (zinterol + CGP). A) 1 Hz (upper) & 3 Hz (lower) Ca transients and caffeine-induced Ca transient records at baseline (left) and after zinterol + CGP (right). B) mean values (± SEM) of Ca transient amplitude and SR Ca content at 1 Hz (left, n=14) and 3 Hz (right, n=6) at baseline and after perfusion with zinterol + CGP (Zin+CGP). C) Cell shortening at 1 (left, n=7) and 3 Hz (right, n=4) and during rapid caffeine application at baseline and with zinterol + CGP.
Figure 4.

Ca transients in HF rabbit myocytes are enhanced by β2-stimulation (zinterol + CGP), and reversed by the β2-blocker ICI. A) 1 and 3 Hz Ca transients and SR Ca content records at baseline (left) and with zinterol + CGP (right) in HF rabbit myocytes. B) Average values of Ca transient amplitude and SR Ca content at 1 Hz (left) and 3 Hz (right) at baseline (Base, n= 12), increase after superperfusion with zinterol + CGP (p< 0.05), and reversed with ICI (grey bars, p<0.05). C) Cell shortening at 1 Hz (left, n=7) and 3 Hz (right, n=6) and during rapid caffeine application at baseline, increase with zinterol + CGP (p< 0.05), and reversed with ICI (p<0.05). *denotes p < 0.05.
In control rabbit myocytes, β2-AR stimulation (zinterol + CGP) did not increase Ca transient amplitude or SR Ca load (assessed by caffeine contractures) at either 1 or 3 Hz stimulation frequency (Fig 3A-B). However, in HF myocytes β2-AR stimulation significantly increased Ca transient amplitude and SR Ca load at 1 Hz (1.28 ± 0.05 vs 0.99 ± 0.06 and 1.82 ± 0.06 vs 1.42 ± 0.06 Δ F/Fo, n=12, p<0.05, Figs. 4A-B). Similar effects were seen at 3 Hz (Fig 4A-B). All these effects were blocked by the β2-AR blocker ICI-118,551 (Fig 4B). Thus, β2-AR stimulation enhances SR Ca content, Ca transients and contractions in HF, but not control myocytes, and this enhancement of SR Ca load and spontaneous SR Ca release may be arrhythmogenic in HF by enhancing aftercontractions and afterdepolarizations.5
To assess the effects of β2-AR stimulation on SERCA and NCX function, we measured the rate of [Ca]i decline during twitch and caffeine-induced Ca transients, respectively (Fig 5). Zinterol + CGP had no effect on tau of twitch [Ca]i decline in control myocytes, but significantly accelerated twitch [Ca]i decline in HF myocytes (156 ± 11 vs 199 ± 12 msec at 1 Hz, 104 ± 8 vs 125 ± 8 msec at 3 Hz, n=12,12, p<0.05). Zinterol + CGP had no effect on the tau of Ca decline of the caffeine transient either HF or control myocytes, suggesting unaltered NCX (Fig 5).
Figure 5.

β2-stimulation (zinterol + CGP) increases SERCA, but does not alter NCX in HF rabbit myocytes (or either in control). A) Superimposed normalized 1 Hz Ca transients at baseline and with β2-AR show no difference in control rabbit myocytes (left) but decline faster with zinterol +CGP in HF rabbit myocytes (right). B) Superimposed normalized caffeine-induced Ca transient ± zinterol +CGP show similar decay in control (left) and HF (right) rabbit myocytes. C) Mean values of exponential decay (τ) of twitch Ca transients and caffeine-induced Ca transients at baseline and with zinterol + CGP in nonfailing rabbit myocyte (left, n=11) and HF rabbit myocytes (right, n=12). * denotes p < 0.05.
β2-AR effects on Ca current in HF
Figure 6A shows that in control myocytes, zinterol (1 μM) increased ICa (e.g. by ∼40% for Em between −10 and +20 mV). This is much smaller than the 300% increase in ICa that we previously observed with 1 μM isoproterenol (combined β1- and β2-AR activation)5 in this same HF myocyte preparation. However, when we repeated the ICa measurements in control myocytes upon zinterol challenge in the presence of the β1-AR antagonist CGP, zinterol did not change ICa. This implies that the modest ICa stimulation by zinterol alone in control myocytes in Fig 6A is likely due to a minor crosstalk from the extremely potent β1-AR mediated effect on ICa. In HF myocytes, ICa was unaltered by zinterol (Fig. 6B). We conclude that β2-AR does not appreciably alter ICa in either control or HF rabbit myocytes.
Figure 6.

Zinterol effects on ICa on control and HF rabbit myocytes. A) L-type ICa at 0 mV (left) and mean I-V curve (right) in control rabbit myocytes ± zinterol (Zin) (n=6). B) ICa at 0 mV (left) and I-V curve (right) in HF rabbit myocytes ± zinterol (n=5). * denotes p < 0.05.
β2-AR effects on phospholamban phosphorylation
We assessed the effects of β2-AR stimulation on PLB phosphorylation at Ser-16 (protein kinase A site) and Thr-17 (CaMKII site). At baseline, HF myocytes showed decreased Ser-16 and increased Thr-17 phosphorylation, as we have previously demonstrated.15 Control and HF rabbit myocytes were exposed to 1 μM zinterol + 300 nM CGP 20712A in the presence or absence of 100 nM ICI118,551. HF (but not control) myocytes exhibited a 78% increase in PLB phosphorylation at the Ser-16 (PKA) site (p<0.05) but no significant change at the Thr-17 (CaMKII) site which was already elevated in HF (n= 5,4, Fig. 7A). The slight tendency for zinterol-induced increase in Thr-17 phosphorylation in HF (not significant) could be secondary to the zinterol-induced enhancement of Ca transients, which was seen only in HF myocytes.
Figure 7.

PLB phosphorylation and β-AR density. A) Effects of β2-AR stimulation of PLB phosphorylation at Ser-16 (PKA) and Thr-17 (CaMKII) sites. Representative Western blots (above) and summarized data (below) for freshly-isolated control and HF rabbit myocytes at baseline or after 10 min exposure to zinterol (1 μM) + CGP 20712A (300nM) in the absence or presence of 100nM ICI 118,551 (n= 4 or 5). B) β-AR density in LV homogenates, indicating total β-AR density and 125I-CYP affinity (left) and amounts of β1- and β2-AR, based on the component sensitive to either 100 nM CGP-20712A (β1-AR) or 1 μM ICI-118,551 (β2-AR), and the percent β2-AR (right).
β2-AR density and subtype distribution in HF
Based on β-AR binding assays, HF rabbits LV exhibit a 47% downregulation of total β -ARs (p<0.05, Fig. 7B), and our results here in LV homogenates are nearly identical to those reported using LV membrane preps from a similar HF rabbit model16 as well as in the failing human heart.6 In control rabbit LV, β 2-ARs represent only ∼8% of total β -ARs (similar to Brodde et al,17). With HF, there is a 50% downregulation of β1-AR, but the amount of β2-ARs is unaltered in HF. This implies that the prominent β2-AR-mediated SR Ca uptake and arrhythmogenic effects are due to altered coupling of β2-AR to the PKA-dependent SR effects, rather than increased β2-AR number or effects on ICa.
Human HF LV myocytes
Studies in rabbit myocytes were validated by experiments in human myocytes from six patients with end-stage HF (3 ischemic and 3 nonischemic) at the time of cardiac transplantation. Human HF hearts exhibited severely depressed LV systolic function with a mean LV ejection fraction of 18 ± 5%. Four of the 6 HF patients had an implantable defibrillator for documented ventricular tachycardia and 3 were known to be taking β-AR blockers. No apparent differences were seen among myocytes from the different HF hearts, so data from these 6 failing hearts were pooled.
In HF human myocytes, zinterol (1 μM) + CGP (300 nM; 1 Hz, 37°C) significantly increased cell shortening (10.7 ± 3.0 vs 4.2 ± 0.9, n=6, p<0.05), and induced aftercontractions in 6 of 6 cells (e.g. see Fig 8A middle panel). In human HF myocytes loaded with fluo-3 (37°C; Fig 8), zinterol + CGP significantly increased both Ca transient amplitude (3.34 ± 0.72 vs 1.48 ± 0.18 ΔF/Fo, n=6, p<0.05, Fig 8B) and SR Ca load (assessed by caffeine Ca transient, 4.82 ± 0.60 vs 3.94 ± 0.44 ΔF/Fo, n=5, p<0.05, Fig 8B). Moreover, zinterol + CGP enhanced the rate of twitch [Ca]i decline (τ of 306 ± 26 vs 436 ± 85 msec, n=6, p<0.05, Fig 8C) consistent with enhanced SERCA activity, while the rate of [Ca]i decline of the caffeine contracture was unchanged (indicating unaltered NCX function after β2-AR). All of these effects of zinterol + CGP were reversed by the β2-AR antagonist ICI 118,551.
Figure 8.
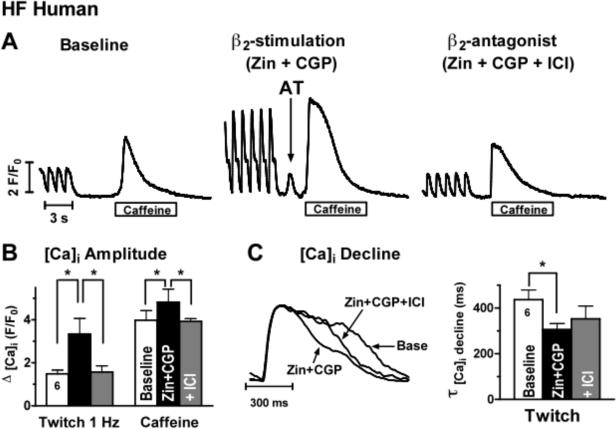
Human HF myocytes and β2-stimulation (zinterol + CGP) induce aftertransients, and increase Ca transient amplitude, Ca transient decline and SR Ca load in HF human myocytes (effects reversed by ICI). A) 1 Hz Ca transients and caffeine-induced Ca transient at baseline, with zinterol+CGP showing a spontaneous aftertransient (AT) before caffeine application (middle) and after perfusion with ICI. B) Mean values of Ca transient amplitude and caffeine-induced Ca transient at baseline, which increase with zinterol + CGP (n=6, p<0.05 vs baseline), and reverse with ICI (gray bar, p< 0.05 vs Zin+CGP). C) Superimposed 1 Hz normalized Ca transient records at baseline, showing faster decline with β2-AR stimulation, and reversal with ICI. D) mean values of exponential decay of 1 Hz Ca transient at baseline, with β2-AR activation (n=6, p<0.05 vs baseline), and with ICI addition.
DISCUSSION
We found that β2-AR stimulation is arrhythmogenic in the failing heart. In conscious HF rabbits (but not controls) infusion of the β2-AR agonist zinterol induces ventricular arrhythmias that are prevented by the β2-AR antagonist ICI 118,551. In HF (but not control) rabbit myocytes, zinterol plus the β1-AR antagonist CGP 29712A induces aftercontractions as well as increased Ca transient amplitude, SR Ca load, and PLB phosphorylation. This increase of SR Ca content in HF is not due to changes on ICa or NCX activity but increased SR Ca pump rate (indicated by acceleration of twitch Ca decline). Similar results were found in human failing myocytes. Thus, β2-AR stimulation enhances arrhythmogenesis in HF by increasing SR Ca load and consequent spontaneous SR Ca release (presumably via DADs).
In vivo arrhythmogenesis
We assessed the in vivo arrhythmogenic effects of β2-AR stimulation in our well-characterized arrhythmogenic rabbit HF model.3,5,10 This HF model of combined aortic insufficiency and aortic constriction is ideally suited for these studies for a number of reasons. We previously found that HF rabbits exhibit spontaneously-occurring VT in vivo that initiates by a nonreentrant mechanism,3 and that these arrhythmias are enhanced by β-AR stimulation with isoproterenol.5 Moreover, HF rabbits have an ∼10% incidence of sudden cardiac death.5,10
Zinterol infusion led to spontaneous ventricular arrhythmias and VT in HF but not control rabbits. While zinterol has predominantly β2-AR agonist effects, at high doses it might stimulate β1-ARs. However, we think these effects are β2-AR mediated because there were no changes in heart rate (as expected for β1-AR effects) and we were able to block the enhancement of VT by zinterol in the HF rabbits with the β2-AR antagonist ICI 118,551.
These findings are, to our knowledge, the first demonstration that β2-AR stimulation is arrhythmogenic in the failing heart, although β2-AR stimulation has been shown to be arrhythmogenic in other pathophysiologic setting (e.g. dogs with increased sympathetic stimulation (from exercise) and acute coronary occlusion develop VF that is prevented by the β2-AR antagonist ICI).18
Arrhythmogenicity in vitro (aftercontractions and after-transients)
We have previously shown in HF rabbit myocytes that isoproterenol (β1- and β2-AR activation) induced DADs, aftercontractions, spontaneous SR Ca release and activation of Iti that is mediated by NCX (that is upregulated two-fold).5 Here we report that β2-AR stimulation in HF (but not control) myocytes induced aftercontractions and spontaneous SR Ca release (after-transients). This was associated with increased SR Ca load but no change in NCX or ICa, and suggests that the arrhythmogenicity of β2-AR stimulation is mediated through the increased SR Ca load that likely activates SR Ca release and Iti. We previously showed how Iti induction depends critically on SR Ca content in this model.5
We found that β2-AR stimulation enhanced SR Ca uptake in both HF rabbit and HF human myocytes, based on an enhanced rate of twitch [Ca]i decline in the absence of altered NCX function. This strongly suggests enhanced SERCA function, which is further substantiated by our findings that β2-AR stimulation specifically enhanced PLB phosphorylation the Ser-16 (PKA) site in HF but not control myocytes. This is in line with findings of Kaumann et al.9 in failing human LV myocytes. With a trend for enhanced PLB phosphorylation at the Thr-17 (CaMKII) site in our quiescent myocytes, we cannot rule out that activation of CaMKII may contribute to increasing SR Ca load. Our findings of decreased PLB phosphorylation at Ser-16 and increased phosphorylation at Thr-17 with HF are similar to what we have previously reported.15 While β2-AR stimulation could potentially decrease the threshold level of SR Ca load at which spontaneous SR Ca release occurs in HF myocytes, we previously showed that isoproterenol did not alter this threshold level in HF rabbit myocytes5 making the enhanced uptake rate and load by a PKA- (and possibly CaMKII-) mediated phosphorylation of phospholamban a more likely mechanism. In these rabbit HF myocytes, RyR phosphorylation by CaMKII (but not PKA) might also increase the diastolic SR Ca leak which may be arrhythmogenic;15 so some contribution of RyR phosphorylation to the observed β2-AR-induced arrhythmias cannot be excluded. However, since SR Ca content is elevated (vs. lowered) in that setting, we infer that the SR Ca loading effect of β2-AR activation is the predominant arrhythmogenic basis here. That is, if RyR activation were predominant, β2-AR activation should lower SR Ca load.
Validation in failing human myocytes
Focused parallel studies in human HF myocytes gave results comparable to those in HF rabbits, with aftercontractions as well as enhanced Ca transient amplitude, SR Ca load, and rate of Ca decline in response to β2-AR stimulation (and block of all of these effects by ICI). Our findings, which are consistent with those of del Monte et al,19 validate our results in the HF rabbit model and further support its applicability to the electrophysiologic substrate of the failing human heart. These findings also suggest that even at end-stage HF, when overall β-AR responsiveness is diminished,6 preserved β2-AR responsiveness can contribute to the altered electrophysiologic substrate that underlies sudden death in patients with end-stage HF.1
Increased β2-AR responsiveness in HF
Our findings of increased arrhythmogenicity (and contractile response) with β2-AR stimulation in HF further supports work by others7,9 that HF may be characterized by increased β2-AR responsiveness. For example, β2-AR stimulation with zinterol can enhance Ca transients in HF myocytes to a greater degree than in controls in a way that could potentially be arrhythmogenic.7 We began to explore the basis for this increased β2-AR responsiveness. As in failing human LV,6 HF rabbit LV exhibits a decrease in the total amount of expressed β-ARs, which is due to downregulation of β1-ARs but with β2-ARs being unaltered. The unaltered β2-AR expression here, but dramatic enhancement of β2-AR effects on SR Ca uptake and arrhythmogenic SR Ca release events, suggest altered coupling to G proteins (e.g. preserved Gsα-protein coupling in HF)9 and/or changes in other downstream signaling pathways.8,20 Recent studies have delineated some of the complexity of the β2-AR signal transduction pathway, including functional compartmentalization of signaling mediated by Gi and PI3K.8,21 and alterations of β2-AR phosphorylation state.22 Membrane microdomains such as caveolae or lipid rafts may play an important role in localizing β2-AR response and downstream signaling, especially in the failing heart. A recent report that disrupting of caveolae led to an increase in responsiveness of β2-AR stimulation further supports this.23
Some aspects of signaling appear to be species-specific. For instance, β2-AR stimulation elicits positive inotropic effects in control rat and dog myocytes that are mediated by an increase in Ica (with action potential prolongation)24 but no lusitropic effect and no significant phosphorylation of PLB.25,26 However, in human HF the situation appears to be quite different, with β2-AR stimulation leading to both inotropic & lusitropic effects, including PLB phosphorylation.9 Our findings suggest that β2-AR pathways in rabbit heart (HF and control) may more closely resemble that of humans. The enhanced SR Ca uptake and inotropic state induced by β2-AR stimulation in HF would be potentially beneficial, but the increased arrhythmogenic propensity may limit this functional benefit. This raises the challenge of therapeutically enhancing SR Ca function without enhancing arrhythmogenesis.
Implications of Arrhythmogenic effects of β2-AR stimulation
The β2-AR is being targeted as a potential therapeutic target for the treatment of HF, and approaches to increase β2-AR number by transgenic and adenoviral gene transfer27,28 have yielded enhancement of cardiac function that is likely due to enhancement of SR Ca load. There may be a therapeutic window in which β2-AR stimulation would lead to increased contractility with much additional arrhythmogenic risk. However, the results of the present study suggest that this approach in failing myocardium will need to be balanced by the potential for enhanced arrhythmogenesis from SR Ca overload in an environment that makes myocytes more prone to developing triggered arrhythmias (e.g. upregulated NCX which can mediate the arrhythmogenic Iti, enhanced β2-AR-induced SR Ca loading, and down-regulated inward rectifying current IK1 which could destabilize resting potential and enhance depolarizing effects of Iti).5 It will thus be important to examine the effects of these approaches on the electrophysiologic substrate of the failing heart.
While “cardioselective” β1-AR blockers such as metoprolol have been used to treat HF patients and decrease mortality and sudden cardiac death,29 there is evidence that nonselective (β1-+β2-AR) blockers such as carvedilol may have greater efficacy in preventing sudden death.30 Our results suggest that “nonselective” β-AR blockade could offer therapeutic advantages by limiting catecholamine-mediated increases in SR Ca load mediated by stimulation of the β2-AR and thereby further stabilize the arrhythmogenic substrate of the failing heart. While there is a potential concern that β2-AR could have negative inotropic effects, these have not been borne out in clinical trials.30
Limitations
Zinterol has been used for many β2-AR studies,7,21,24-26 but at high doses (e.g. 10−5M) zinterol may also stimulate β1-AR.31 We therefore performed most of these studies with zinterol in the presence of the β1-AR antagonist CGP 20712A (this was more difficult for in vivo studies where we found β1-AR blockade was not always well tolerated hemodynamically in rabbits with HF). In some in vitro studies, we also used zinterol alone and found results comparable to zinterol + CGP. This suggests that at the doses used, zinterol's effects were predominantly due to stimulation of β2-ARs,21,24-26 (with the exception that zinterol could slightly increase ICa via β1-AR). Overall, the ability to reverse zinterol's effects by the specific β2-AR blocker ICI-118,551 supports our conclusions about β2-AR-induced effects. Wang et al14 reported that myocytes on laminin-coated cover slips exhibited greater β2-AR responsiveness to zinterol. Our findings of enhanced arrhythmogenicity with β2-AR stimulation in vivo as well as in vitro (whether on laminin-coated cover slips or on glass cover slips without laminin) suggest that the laminin was not a confounding issue. Moreover, laminin may help simulate the extracellular protein environment in intact tissue that involves interactions with laminin and integrins.14
Conclusion
We found that β2-AR stimulation is arrhythmogenic in the failing rabbit and human heart by enhancing spontaneous SR Ca release and aftercontractions (secondary to enhanced SR Ca uptake and load). Notably, β2-AR activation is more arrhythmogenic in the failing vs. nonfailing heart. Thus, β2-AR blockade may offer therapeutic advantages to stabilize the altered electrophysiologic substrate of failing myocardium and help decrease the incidence of sudden death in HF patients.
ACKNOWLEDGEMENTS
This work was supported by NIH grant HL46929 and HL 73966 (to SMP) and HL30077 and HL-64724 (to DMB). The authors would like to thank Jodi Jeanes and Theresa Dahl for technical assistance.
Footnotes
This is an un-copyedited author manuscript accepted for publication in Circulation Research, copyright The American Heart Association. This may not be duplicated or reproduced, other than for personal use or within the “Fair Use of Copyrighted Materials” (section 107, title 17, U.S. Code) without prior permission of the copyright owner, The American Heart Association. The final copyedited article, which is the version of record, can be found at http://circres.ahajournals.org/. The American Heart Association disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties.
REFERENCES
- 1.Packer M. Sudden unexpected death in patients with congestive heart failure: a second frontier. Circulation. 1985;72(4):681–685. doi: 10.1161/01.cir.72.4.681. [DOI] [PubMed] [Google Scholar]
- 2.Kjekshus J. Arrhythmias and mortality in congestive heart failure. Am J Cardiol. 1990;65:421–481. doi: 10.1016/0002-9149(90)90125-k. [DOI] [PubMed] [Google Scholar]
- 3.Pogwizd SM. Nonreentrant mechanisms underlying spontaneous ventricular arrhythmias in a model of nonischemic heart failure in rabbits. Circulation. 1995;92:1034–1048. doi: 10.1161/01.cir.92.4.1034. [DOI] [PubMed] [Google Scholar]
- 4.Pogwizd SM, McKenzie JP, Cain ME. Mechanisms underlying spontaneous and induced ventricular arrhythmias in patients with idiopathic dilated cardiomyopathy. Circulation. 1998;98:2404–2414. doi: 10.1161/01.cir.98.22.2404. [DOI] [PubMed] [Google Scholar]
- 5.Pogwizd SM, Schlotthaurer K, Li L, Weilong Y, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 6.Bristow MR, Ginsburg R, Umans V, et al. β1- and β2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: Coupling of both receptor subtypes to muscle contraction and selective β1-receptor down-regulation in heart failure. Circ. Res. 1986;59:297–309. doi: 10.1161/01.res.59.3.297. [DOI] [PubMed] [Google Scholar]
- 7.Altschuld RA, Starling RC, Hamlin RL, et al. Response of failing canine and human heart cells to β2-adrenergic stimulation. Circulation. 1995;92:1612–1618. doi: 10.1161/01.cir.92.6.1612. [DOI] [PubMed] [Google Scholar]
- 8.Xiao RP, Cheng H, Zhou Y-Y, Kuschel M, Lakatta EG. Recent advances in cardiac β2-adrenergic signal transduction. Circ Res. 1999;85:1092–1100. doi: 10.1161/01.res.85.11.1092. [DOI] [PubMed] [Google Scholar]
- 9.Kaumann A, Bartel S, Molenaar P, et al. Activation of β2-adrenergic receptors hastens relaxation and mediates phosphorylation of phospholamban, troponin I, and C-protein in ventricular myocardium from patients with terminal heart failure. Circulation. 1999;99:65. doi: 10.1161/01.cir.99.1.65. [DOI] [PubMed] [Google Scholar]
- 10.Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na/Ca exchanger expression and function in an arrhythmogenic rabbit model of nonischemic cardiomyopathy. Circ Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- 11.DeSantiago J, Maier LS, Bers DM. Phospholamban is required for CaMKII-dependent recovery of Ca transients and SR Ca uptake during acidosis in cardiac myocytes. J Mol Cell Cardiol. 2004;36:67–74. doi: 10.1016/j.yjmcc.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 12.Delbridge LM, Bassani JWM, Bers DM. Steady-state twitch Ca2+ fluxes and cytosolic Ca2+ buffering in rabbit ventricular myocytes. Am J Physiol. 1996;270:C192–199. doi: 10.1152/ajpcell.1996.270.1.C192. [DOI] [PubMed] [Google Scholar]
- 13.Xiao RP, Zhang SJ, Chakir K, Avdonin P, Zhu W, Bond RA, Balke W, Lakatta EG, Cheng H. Enhanced Gi signaling selectively negates β2-adrenergic receptor (AR) – but not β1-AR-mediated positive inotropic effect in myocytes from failing rat hearts. Circulation. 2003;108:1633–1639. doi: 10.1161/01.CIR.0000087595.17277.73. [DOI] [PubMed] [Google Scholar]
- 14.Wang YG, Samarel AM, Lipsius SL. Laminin binding to β1-integrins selectively alters beta-1 and beta-2 adrenergic signaling in cat atrial myocytes. J. Physiol. 2000;527:3–9. doi: 10.1111/j.1469-7793.2000.t01-2-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–22. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 16.Gilson N, el Houda Bouanani N, Corsin A, Crozatier B. Left ventricular function and beta-adrenoceptors in rabbit failing heart. Am J Physiol. 1990;258:H634–41. doi: 10.1152/ajpheart.1990.258.3.H634. [DOI] [PubMed] [Google Scholar]
- 17.Brodde O-E, Leifert F-J, Krehl H-J. Coexistence of β1- and β2-adrenoceptors in the rabbit heart: Quantitative analysis of the regional distribution by (−)-3H-dihydroalprenolol binding. J Cardiovasc Pharmacol. 1982;4:34–43. doi: 10.1097/00005344-198201000-00007. [DOI] [PubMed] [Google Scholar]
- 18.Billman GE, Castillo LC, Hensley J, Hohl CM, Altschuld R. β2-Adrenergic receptor antagonists protect against ventricular fibrillation. In vivo and in vitro evidence for enhanced sensitivity to β2-adrenergic stimulation in animals susceptible to sudden death. Circulation. 1997;96:1914–1922. doi: 10.1161/01.cir.96.6.1914. [DOI] [PubMed] [Google Scholar]
- 19.Del Monte F, Kaumann AJ, Poole-Wilson PA, et al. Coexistence of functioning β1- and β2-adrenoceptors in single myocytes from human ventricle. Circulation. 1993;88:854–863. doi: 10.1161/01.cir.88.3.854. [DOI] [PubMed] [Google Scholar]
- 20.Steinberg SF. The molecular basis for distinct β-adrenergic receptor subtype action in cardiomyocytes. Circ Res. 1999;85:1101–1111. doi: 10.1161/01.res.85.11.1101. [DOI] [PubMed] [Google Scholar]
- 21.Jo SH, Leblais V, Wang PH, et al. Phosphatidylinositol 3-Kinase functionally compartmentalizes the concurrent Gs Signaling during β2-adrenergic stimulation. Circ Res. 2002;91:46–53. doi: 10.1161/01.res.0000024115.67561.54. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, De Arcangelis V, Gao X, Ramani B, Jung Y, Xiang Y. Norepinephrine- and epinephrine-induced distinct β2-adrenoceptor signaling is dictated by GRK2 phosphorylation in cardiomyocytes. J Biol Chem. 2008;283:1799–1807. doi: 10.1074/jbc.M705747200. [DOI] [PubMed] [Google Scholar]
- 23.Calaghan S, White E. Caveolae modulate excitation-contraction coupling and β2-adrenergic signaling in adult rat ventricular myocytes. Cardiovas Res. 2005;69:816–824. doi: 10.1016/j.cardiores.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 24.Xiao RP, Lakatta Beta-1 adrenoceptor stimulation and beta-2 adrenoceptor stimulation differ in teir effects on contraction, cytosolic Ca2+, and Ca2+ current in single rat ventricular cells. Circ Res. 1993;73:266–300. doi: 10.1161/01.res.73.2.286. [DOI] [PubMed] [Google Scholar]
- 25.Xiao RP, Hohl C, Altschuld R, et al. Beta 2-adrenergic receptor-stimulated increase in cAMP in rat heart cells is not coupled to changes in Ca2+ dynamics, contractility, or phospholamban phosphorylation. J. Biol. Chem. 1994;269:19151–6. [PubMed] [Google Scholar]
- 26.Kuschel M, Zhou YY, Spurgeon HA, Bartel S, Karczewski P, Zhang SJ, Krause EG, Lakatta EG, Xiao RP. β2-adrenergic cAMP signaling is uncoupled from phosphorylation of cytoplasmic proteins in canine heart. Circulation. 1999;99:2458–2465. doi: 10.1161/01.cir.99.18.2458. [DOI] [PubMed] [Google Scholar]
- 27.Milano CA, Allen LF, Rockman HA, Dolber PC, McMinn TR, Chien KR, Johnson TD, Bond RA, Lefkowitz RJ. Enhanced myocardial function in transgenic mice overexpressing the β2-adrenergic receptor. Science. 1994;264:582–586. doi: 10.1126/science.8160017. [DOI] [PubMed] [Google Scholar]
- 28.Shah AS, Lilly E, Kypson AP, et al. Intracoronary adenovirus-mediated delivery and overexpression of the β2-adrenergic receptor in the heart. Prospects for molecular ventricular assistance. Circulation. 2000;101:408–414. doi: 10.1161/01.cir.101.4.408. [DOI] [PubMed] [Google Scholar]
- 29.MERIT-HF Study Group Effects of metoprolol CR/XL in chronic heat failure: Metoprolol CR/XL randomized intervention trial in congestive heart failure (MERIT-HF). Lancet. 1999;353:2001–2007. [PubMed] [Google Scholar]
- 30.Poole-Wilson PA, Swedberg K, Cleland JGF, Di Lenarda A, Hanrath P, Komajda M, Lubsen J, Lutiger B, Metra M, Remme WJ, Torp-Pedersen C, Scherhag A, Skene A. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomized control trial. Lancet. 2003;362:7–13. doi: 10.1016/S0140-6736(03)13800-7. [DOI] [PubMed] [Google Scholar]
- 31.Laflamme MA, Becker PL. Do β2-adrenergic receptors modulate Ca2+ in adult rat ventricular myocytes? Am J Physiol. 1998;274:H1308–H1314. doi: 10.1152/ajpheart.1998.274.4.H1308. [DOI] [PubMed] [Google Scholar]