

Metal catalyzed allylic alkylations increase in importance as a synthetic method as the ability to expand their scope increases. A major feature of this method is its applicability for formation of a broad array of bond types including carbon-carbon and carbon-heteroatom bonds. The importance of nitrogen containing bioactive molecules1 directs special attention to the formation of carbon-nitrogen bonds.2 The recent revelation of bromopyrroles as a growing family of bioactive natural products represented by the manzacidines, axinellamine A, dibromophakellstatin, and palau’amine, typically derived from marine organisms3, led us to consider the use of pyrroles as nucleophiles in AAA (asymmetric allylic alkylation) reactions. The agelastatins (1), a family of four tetracyclic compounds (see Fig. 1), possess nanomolar activity against several cancer cell lines.4 Furthermore, agelastatin A inhibits glycogen synthase kinase-3β (GSK-3β), a behavior that may provide an approach for the treatment of Alzheimer’s disease.4a In this paper, we report the use of pyrroles as nucleophiles in the Pd AAA and the use of such a process for a facile asymmetric synthesis of agelastatin A.
Figure 1.
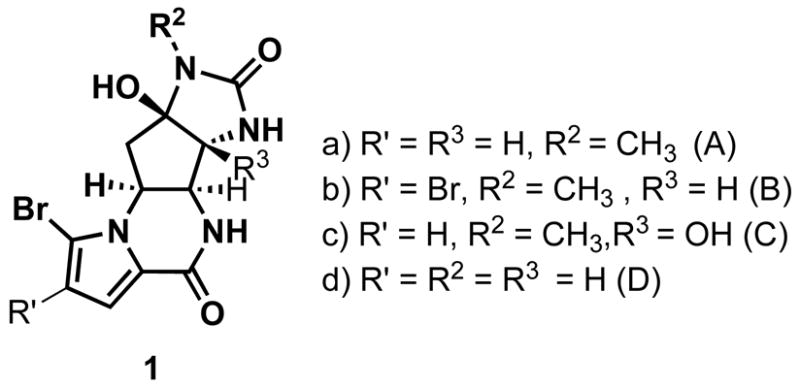
Agelastatins
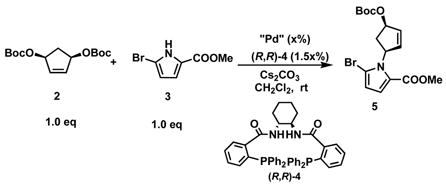
Initial studies examined the reaction between the Boc-activated cyclopentene-1,4-diol 2 and methyl 5-bromo-pyrrole-2-carboxylate 3 (eq.1). After a general screening, Cs2CO3 and DCM proved to be the base and best solvent combination.
 |
(1) |
The yield and enantioselectivity were optimized by varying the palladium source and loading, base loading, and concentration (Table 1). From these studies emerged the most practical set of conditions as shown in entry 6 which gives the N-alkyl pyrrole 5 in 83% yield and 92% ee. Direct transformation of the carboxylate ester 5 to the N-methoxyamide 6 failed,5 but a two-step process (hydrolysis, condensation) gave a high yield (Scheme 1). Although the chiral ligand was not necessary for cyclization to piperazinone 7, the intramolecular Pd catalyzed AAA with the N-methoxy amide as the nucleophile gave a higher yield when (R, R)-4 was used as a ligand (91%) compared to dppp (70%). At this point the absolute configuration was assigned by analogy to other reactions of substrate 2.
Table 1.
Selected Optimization studies:
| Entry | Pd source (mol%) | Cs2CO3(eq.) | Conc.(M) | Yield(%)a | ee(%)b |
|---|---|---|---|---|---|
| 1 | Pd2(dba)3CHCl3(5) | 1.0 | 0.02 | 90 | 84 |
| 2 | Pd2(dba)3CHCl3(5) | 0.7 | 0.02 | 75 | 92 |
| 3 | Pd2(dba)3CHCl3(2.5) | 1.0 | 0.02 | 89 | 66 |
| 4 | Pd2(dba)3CHCl3(1.25) | 1.0 | 0.08 | 75 | 85 |
| 5 | [Pd(C3H5)Cl]2(2) | 1.0 | 0.17 | 88–93 | 87 |
| 6 | [Pd(C3H5)Cl]2(1.25) | 1.0 | 0.08 | 83 | 92 |
Isolated yield.
Enantioselectivities were determined by chiral HPLC.
Scheme 1.

Piperazinone synthesis
a) LiOH (1N), THF/water = 3/1, 48hr, rt; b) oxalyl chloride, cat. DMF in THF then, NH2Ome·HCl, K2CO3, and H2O, rt.
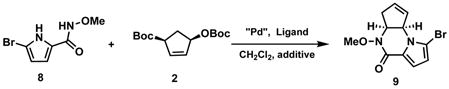
With success of both the pyrrole and the N-methoxyamide as nucleophiles respectively and considering that the nitrogen on the N-methoxyamide is more nucleophilic than the one on the pyrrole, we designed a cascade reaction to further extend this methodology. Theoretically, piperazinone 9 could be synthesized in one-pot from successive alkylations with the N-methoxyamide 86 as nucleophile. Surprisingly, almost no reaction occurred when base was present (see Table 2). We hypothesized that after
Table 2.
Synthesis of piperazinone 9a
| Entry | Pd source
(mol%) |
Ligand
(mol%) |
Additive
(mol%) |
Temp.
(°C) |
Yield
(%)b |
ee
(%)c |
|---|---|---|---|---|---|---|
| 1 | [Pd(C3H5)Cl]2(2) | (R,R)- 4 (6) | none | rt | trace | NA |
| 2 | [Pd(C3H5)Cl]2(2) | (R,R)- 4 (6) | Cs2CO3 (100) | rt | 0 | NA |
| 3 | [Pd(C3H5)Cl]2(5) | (R,R)- 4 (15) | HOAc (10) | rt | traced | NA |
| 4 | Pd2(dba)3CHCl3(5) | (R,R)- 4 (15) | HOAc (10) | rt | 51 | NA |
| 5 | Pd2(dba)3CHCl3(5) | (R,R)- 4 (15) | BSA (100) | rt | 50 | NA |
| 6 | Pd2(dba)3CHCl3(5) | (R,R)- 4 (15) | HOAc (10) | 0 to rt | 65e | 89 |
| 7 | Pd2(dba)3CHCl3(5) | rac- 4 (15) | HOAc (10) | rt | 88e | NA |
| 8 | Pd2(dba)3CHCl3(5) | (R,R)- 4 (15) | HOAc (10) | 0 to rt | 80e,f | 96 |
| 9 | Pd2(dba)3CHCl3(5) | (R,R)- 4 (15) | HOAc (10) | 0 to rt | 82e,g | 97.5 |
Unless otherwise indicated, all reaction were performed with 1.0 eq. 2 and 1.0 eq. 8 at 0.2 M in DCM;
Isolated yield.
Enantioselectivities were determined by chiral HPLC;
Single alkylation product was the main product.
The reaction was performed with 1.5 eq. 2 and 1.0 eq. 8;
Another portion of Pd2(dba)3CHCl3 (5 mol%), (R,R)-4 (15 mol%) was added after 1h.
Another portion of Pd2(dba)3CHCl3 (5 mol%), Rac-4 (15 mol%) was added after 3.5 h.
 |
(2) |
deprotonation, 8 could act as a good bidentate ligand for palladium, and the first ionization might be inhibited. Based on this hypothesis, 10 mol% HOAc was added to the reaction. To our delight, piperazinone 9 was obtained in 51% yield when Pd2(dba)3CHCl3 was used as the palladium source. After optimization, piperazinone 9 could be obtained in up to 82% yield, 97.5% ee (entry 9). Thus, by proper choice of pyrrole nucleophiles in the Pd catalyzed AAA, access to either piperazinone regioisomer is possible.
For agelastatin A4, 7 (Scheme 2) starting with piperazinone 7, we envisioned aziridination of the double bond followed by transformation to the required urea. The aziridination which we anticipated to be difficult led us to explore the N-heterocyclic carbene complex 1 48 which, to our knowledge, has not previously been explored for aziridination. Indeed, this catalyst performed well for this difficult rather electron deficient cyclopentene. Hydrolytic ring opening of 10 occurs best upon heating in a microwave. Dess-Martin oxidation then gives α-amino ketone 1 2. A more efficient direct oxidative opening with DMSO, for which few cases previously existed,9 was explored. While following the previously reported thermal protocol proved inefficient, heating N-tosyl aziridine 10 in the presence of 0.7 eq. In(OTf)310 in DMSO at 80 °C provides the α-amino ketone 12 in excellent yield. Finally, addition of methyl isocyanate to 12, followed by SmI2 mediated cleavage of N-OMe and N-Ts, completed the total synthesis of (+)-agelastatin A (1).11 This completion also established the absolute configuration of the Pd AAA as shown in Scheme 1.
Scheme 2.

Total synthesis of (+)-agelastatin A
a) catalyst 14 (0.5 eq.), PhI=NTs (5 eq.), 4Å M.S., benzene, 0 °C to rt; b) TFA (10 eq.), microwave, dioxane/water = 3/2, 150 °C, 2.5 hr; c) DMP, DCM, rt; d) In(OTf)3 (0.7 eq.), DMSO, 80 °C, 6h; e) CH3NCO(1.2 eq.), Cs2CO3 (0.2 eq.), DCM, 0 °C to rt; f) SmI2 (10 eq.), THF, 0 °C to rt.
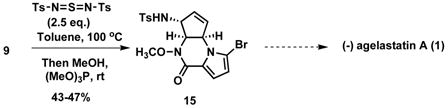
Access to the natural (−)-enantiomer simply requires use of the S,S- ligand in eq. 1. Alternatively, the product of the one pot annulation 9 could also provide access to the (−)-enantiomer based upon the work of Weinreb.7a To explore this prospect, piperazinone 9 was subjected to allylic amination12 as shown in eq. 3. A single regio– and diastereomer was obtained which, by analogy to other reactions of this reagent, is assigned as 15. Given Weinreb’s synthesis, it is reasonable to propose that (−)-1 could be accessed from 15.
 |
(3) |
In conclusion, we have developed new classes of nucleophiles, pyrroles and N-alkoxyamides, for palladium-catalyzed AAA reactions. By varying the functional groups at the 2-position of pyrroles, we can efficiently and enantioselectively access either regioisomer of the piperazinones. Using one regioisomer, we completed the total synthesis of (+)-agelastatin A in a short and concise way (10 steps total), during the course of which we developed a new copper catalyst for aziridination, and an In(OTf)3-DMSO system to oxidatively open an N-tosyl aziridine. We further show the prospect to access (−)-agelastatin A using the same enantiomer of the chiral catalyst in the Pd AAA by using the other piperazinone regioisomer.
Supplementary Material
Experimental procedures and characterization data for all new compounds (PDF). This material is available free of charge via the internet at http://pubs.acs.org.
Acknowledgments
We thank the National Science Foundation and the National Institutes of Health (GM13598) for their generous support of our programs. Mass spectra were provided by the Mass Spectrometry Regional Center for the University of California-San Francisco, supported by the NIH Division of Research Resources. We are indebted to Johnson-Matthey who generously provided palladium salts.
References
- 1.Cordell GA, editor. The Alkaloids. Chemistry and Biology. Academic Press; San Diego: 2005. p. 62. and earlier volumes in the series. Hesse M. Alkaloids: Nature’s Curse or Blessing. Wiley-VCH; New York: 2002.
- 2.For a recent review: Trost BM, Crawley ML. Chem Rev. 2003;103:2921. doi: 10.1021/cr020027w. Also see Trost BM. Chem Pharm Bull. 2004;50:1. doi: 10.1248/cpb.50.1.
- 3.Gribble GW. J Nat Prod. 1992;55:1353.Also see Faulkner DJ. Nat Prod Rep. 2002;19:1. doi: 10.1039/b009029h.Berlinck RGS, Kosuga MH. Nat Prod Rep. 2005;22:516. doi: 10.1039/b209227c. and references therein.
- 4.For the isolation and biological activities of (−)-agelastatin A: D’Ambrosio M, Guerriero A, Debitus C, Ribes O, Pusset J, Leroy S, Pietra F. J Chem Soc, Chem Comm. 1993;1305D’Ambrosio M, Guerriero A, Chiasera G, Pietra F. Helv Chim Acta. 1994;1895;77D’Ambrosio M, Guerriero A, Ripamonti M, Debitus C, Waikedre J, Pietra F. Helv Chim Acta. 1996;79:727.Maijer L, Thunnissen AM, White AW, Garnier M, Nikolic M, Tsai LH, Walter J, Cleverley KE, Salinas PC, Wu YZ, Biernat J, Mandelkov EM, Kim SH, Pettit GR. Chem Bio. 2000;2:51. doi: 10.1016/s1074-5521(00)00063-6.
- 5.Reagents that have been tried: AlMe3, ClMgiPr, KCN, MgCl2, Zr(OBut)4, an N-heterocyclic carbene, Sn[N(TMS)2]2, etc.
- 6.See supporting information for the synthesis of 8.
- 7.For previous total syntheses: Stien D, Anderson GT, Chase CE, Koh Y, Weinreb SM. J Am Chem Soc. 1999;121:9574.Feldman KS, Saunders JC. J Am Chem Soc. 2002;124:9060. doi: 10.1021/ja027121e.Domostoj MM, Irving E, Scheinmann F, Hale KJ. Org Lett. 2004;6:2615. doi: 10.1021/ol0490476.Davis FA, Deng J. Org Lett. 2005;7:621. doi: 10.1021/ol047634l.
- 8.(a) Fructos MR, Belderrain TR, Nicasio MC, Nolan SP, Kaur H, Díaz-Requejo MM, Pérez PJ. J Am Chem Soc. 2004;126:10846. doi: 10.1021/ja047284y. [DOI] [PubMed] [Google Scholar]; (b) Fructos MR, Belderrain TR, de Frément P, Scott NM, Nolan SP, Kaur H, Díaz-Requejo MM, Pérez PJ. Angew Chem Int Eng. 2005;44:5284. doi: 10.1002/anie.200501056. [DOI] [PubMed] [Google Scholar]; (c) Gawley RE, Narayan S. Chem Comm. 2005;40:5109. doi: 10.1039/b509958g. [DOI] [PubMed] [Google Scholar]
- 9.For N-aroylaziridines: Heine HW, Newton T. Tetrahedron Lett. 1967;8:1859.For N-alkoxycarbonylaziridines: Fujita S, Hiyama T, Nozaki H. Tetrahedron Lett. 1969;10:1677.Fujita S, Hiyama T, Nozaki H. Tetrahedron. 1970;26:4347. We thank J. Du Bois, and K. Guthikonda for drawing our attention to application of this thermal method for opening trichloroethoxysulfamoylaziridines.
- 10.Yadav JS, Reddy BV, Subba Kumar G, Mahesh Murthy ChVSR. Syn Comm. 2002;32:1797. and earlier references therein. [Google Scholar]
- 11.The spectroscopic date are identical to the reported data except for [α]D20 +53.2° (c = 0.13, MeOH), while [α]D20 for (−)-agelastatin A is −59.3° (c = 0.13, MeOH) given by Hong TW, Jimenez DR, Molinski TFJ. Nat Prod. 1998;61:158. doi: 10.1021/np9703813.
- 12.(a) Sharpless KB, Hori T. J Org Chem. 1976;41:176. [Google Scholar]; (b) Bussas R, Kresze G. Liebigs Ann Chem. 1980:629. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and characterization data for all new compounds (PDF). This material is available free of charge via the internet at http://pubs.acs.org.